
Method Article
Färbung und hochauflösende Bildgebung von dreidimensionalen Organoid- und Sphäroidmodellen
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Hier stellen wir detaillierte, robuste und komplementäre Protokolle zur Verfügung, um Färbung und subzelluläre Bildgebung von fixierten dreidimensionalen Zellkulturmodellen im Bereich von 100 μm bis zu mehreren Millimetern durchzuführen und so die Visualisierung ihrer Morphologie, Zelltypzusammensetzung und Wechselwirkungen zu ermöglichen.
Zusammenfassung
Dreidimensionale (3D) In-vitro-Zellkulturmodelle wie Organoide und Sphäroide sind wertvolle Werkzeuge für viele Anwendungen, darunter Entwicklung und Krankheitsmodellierung, Wirkstoffforschung und regenerative Medizin. Um diese Modelle voll auszuschöpfen, ist es entscheidend, sie auf zellulärer und subzellulärer Ebene zu untersuchen. Die Charakterisierung solcher In-vitro-3D-Zellkulturmodelle kann jedoch technisch anspruchsvoll sein und erfordert spezifisches Fachwissen, um effektive Analysen durchzuführen. In diesem Artikel werden detaillierte, robuste und komplementäre Protokolle zur Durchführung von Färbungs- und subzellulärer Bildgebung von fixierten In-vitro-3D-Zellkulturmodellen im Bereich von 100 μm bis zu mehreren Millimetern vorgestellt. Diese Protokolle sind auf eine Vielzahl von Organoiden und Sphäroiden anwendbar, die sich in ihren Ursprungszellen, ihrer Morphologie und ihren Kulturbedingungen unterscheiden. Von der 3D-Strukturgewinnung bis zur Bildanalyse können diese Protokolle innerhalb von 4-5 Tagen abgeschlossen werden. Kurz gesagt, 3D-Strukturen werden gesammelt, fixiert und können dann entweder durch Paraffineinbettung und histologische/immunhistochemische Färbung oder direkt immunmarkiert und für die optische Klärung und 3D-Rekonstruktion (200 μm Tiefe) durch konfokale Mikroskopie vorbereitet werden.
Einleitung
In den letzten Jahrzehnten haben Fortschritte in der Stammzellbiologie und In-vitro-3D-Kulturtechnologien eine Revolution in Biologie und Medizin eingeläutet. Zellmodelle mit höherer Komplexität in 3D sind sehr beliebt geworden, da sie es Zellen ermöglichen, zu wachsen und mit einem umgebenden extrazellulären Gerüst zu interagieren und Aspekte lebender Gewebe einschließlich ihrer Architektur, Zellorganisation und -interaktionen oder sogar Diffusionseigenschaften genau zu rekapitulieren. So können 3D-Zellkulturmodelle einzigartige Einblicke in das Verhalten von Zellen in sich entwickelnden oder erkrankten Geweben in vitro liefern. Organoide und Sphäroide sind beide mehrzellige 3D-Strukturen, die von einigen Mikrometern bis zu Millimetern reichen und die prominentesten In-vitro-3D-Strukturen sind. Beide können in einem Trägergerüst kultiviert werden, einschließlich (i) Hydrogele, die aus Tieren (Basalmembranextrakt, Kollagen), Pflanzen (Alginat / Agarose) gewonnen oder aus Chemikalien synthetisiert werden, oder (ii) inerte Matrizen, die Poren enthalten, um die Zellproliferation und das Zellwachstum zu fördern.
Organoide und Sphäroide können sich auch ohne das Vorhandensein eines Stützgerüsts entwickeln, indem sie sich auf Zellen verlassen, um sich selbst zu Clustern zusammenzusetzen. Dies beruht auf verschiedenen Techniken, wie z. B. der Verwendung von nicht adhäsiven Materialien zur Hemmung der Zellanhaftung, der Oberflächenspannung und der Gravitationskraft (z. B. Hanging-Drop-Techniken) oder der konstanten kreisförmigen Rotation von Gefäßen (z. B. Spinnerkultur). In allen Fällen erleichtern diese Techniken Zell-Zell- und Zell-Matrix-Interaktionen, um die Einschränkungen der traditionellen einschichtigen Zellkulturzu überwinden 1. Die Begriffe "Organoide" und "Sphäroide" wurden in der Vergangenheit synonym verwendet, aber es gibt wesentliche Unterschiede zwischen diesen beiden 3D-Zellkulturmodellen. Organoide sind in vitro 3D-Zellcluster, die von pluripotenten Stammzellen oder gewebespezifischen Stammzellen abgeleitet sind, in denen sich Zellen spontan in Vorläuferzellen und differenzierte Zelltypen organisieren und die zumindest einige Funktionen des interessierenden Organs rekapitulieren2. Sphäroide umfassen ein breiteres Spektrum von mehrzelligen 3D-Strukturen, die unter nicht-adhärenten Bedingungen gebildet werden und aus einer großen Vielfalt von Zelltypen wie immortalisierten Zelllinien oder Primärzellen entstehenkönnen 3. Daher haben Organoide aufgrund ihrer intrinsischen Stammzellherkunft eine höhere Neigung zur Selbstorganisation, Lebensfähigkeit und Stabilität als Sphäroide.
Dennoch handelt es sich bei diesen beiden Modellen im Wesentlichen um 3D-Strukturen, die aus mehreren Zellen bestehen, und die Techniken, die zu ihrer Untersuchung entwickelt wurden, sind daher sehr ähnlich. Zum Beispiel sind leistungsfähige bildgebende Ansätze auf der Ebene der Einzelzellauflösung notwendig, um die zelluläre Komplexität sowohl von Organoiden als auch von Sphäroiden zu untersuchen. Durch die Zusammenfassung der Expertise dieser Gruppe und der führenden Unternehmen auf dem Gebiet der Organoide4 beschreibt dieser Artikel detaillierte Verfahren zur Durchführung von zweidimensionalen (2D) und 3D-Whole-Mount-Färbungen, Bildgebung und Analysen der zellulären und subzellulären Zusammensetzung und der räumlichen Organisation von Organoiden und Sphäroiden im Bereich von 100 μm bis zu mehreren Millimetern. In der Tat stellt dieses Verfahren zwei verschiedene und komplementäre Arten der Färbung und Bildgebung dar, um eine Vielzahl von Größen und Arten von In-vitro-3D-Zellkulturmodellen zu analysieren. Die Verwendung des einen (3D-Whole-Mount-Analyse) oder des anderen (2D-Schnittanalyse) hängt vom untersuchten Modell und den gesuchten Antworten ab. Die 3D-Whole-Mount-Analyse mittels konfokaler Mikroskopie kann beispielsweise eingesetzt werden, um Zellen in 3D-Kulturen bis zu 200 μm Tiefe zu visualisieren, unabhängig von der Gesamtgröße der 3D-Struktur, während die Analyse von 2D-Schnitten Einblicke in Proben beliebiger Größe ermöglicht, wenn auch auf 2D-Ebene. Dieses Verfahren wurde erfolgreich auf eine Vielzahl von Organoiden4,5 und Sphäroide angewendet, die aus menschlichen und murinen Zellen stammen und aus verschiedenen embryonalen Keimblättern stammen. Die Übersicht über das Verfahren ist in Abbildung 1 dargestellt. Die wichtigsten Phasen, die Beziehungen zwischen ihnen, entscheidende Schritte und das erwartete Timing werden angegeben.

Abbildung 1: Schematische Übersicht des Verfahrens. In-vitro-3D-Zellkulturmodelle werden gesammelt und fixiert, dann entweder für die 3D-Ganzkörperfärbung vorbereitet (Option a) oder in Paraffin eingebettet (Option b). Bei 3D-Whole-Mount-Färbeexperimenten werden fixierte 3D-Strukturen nach dem Fixierungsschritt immunmarkiert. Ein optionaler optischer Clearing-Schritt kann durchgeführt werden, um die Abbildungsqualität und -tiefe der optischen Mikroskopie zu verbessern, indem die Lichtstreuung während der Bildverarbeitung reduziert wird. Die Bilder werden mit einem inversen konfokalen Mikroskop oder einem konfokalen High-Content-System aufgenommen und mit der entsprechenden Software analysiert. Für die Paraffineinbettung werden 3D-Strukturen direkt verarbeitet (Option b.1 für große Strukturen ≥ 400 μm) oder in ein Gel (b.2; kleine Strukturen ≤ 400 μm) zur Dehydratisierung und Paraffineinbettung aufgenommen. Anschließend werden Paraffinblöcke geschnitten und gefärbt (histologische oder immunchemische Färbung). Bilder von 2D-Schnitten werden auf einem digitalen Objektträgerscanner oder einem aufrechten Mikroskop erhalten und auf einer Bildanalyseplattform mittels schneller digitaler quantitativer Analyse analysiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
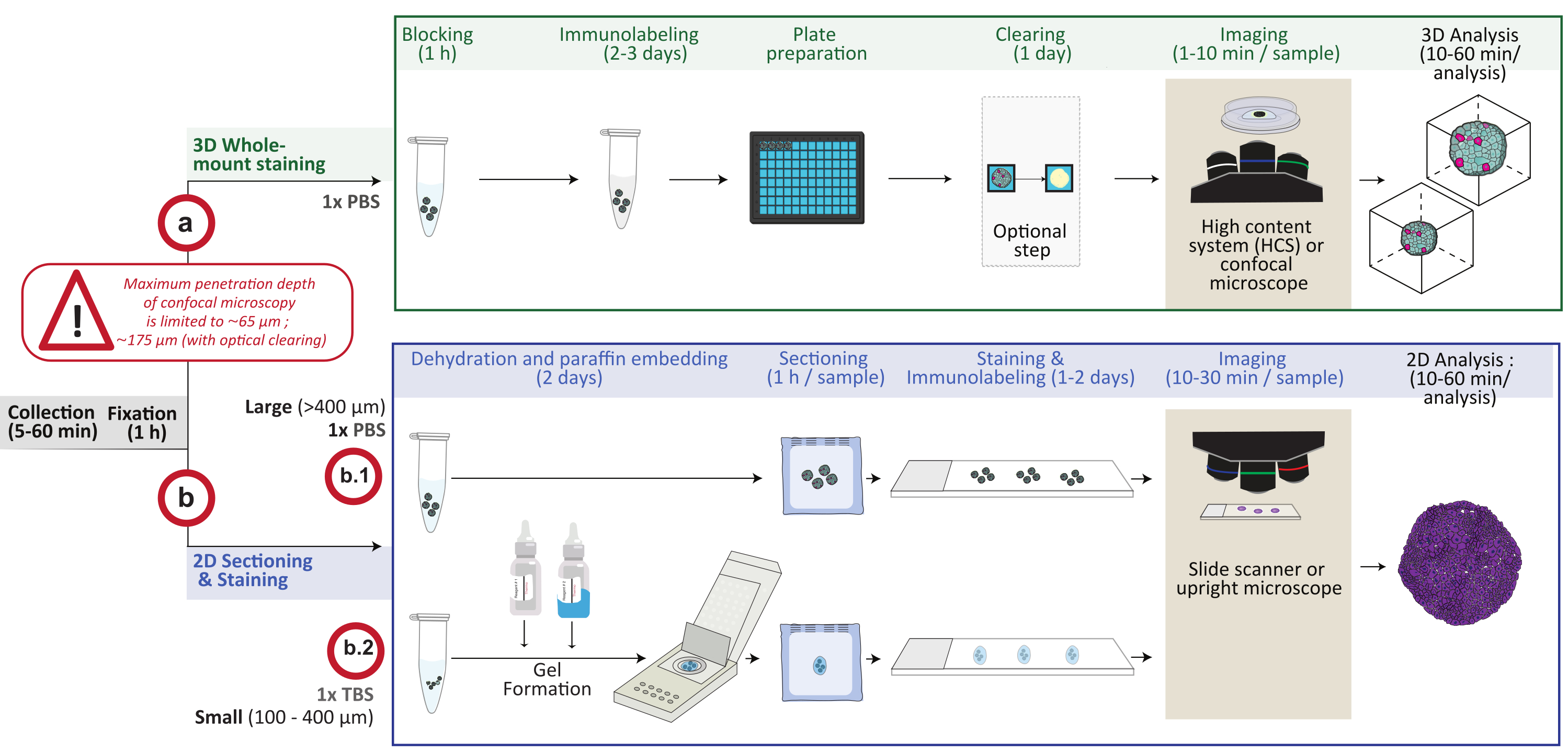{kind=link}
Protokoll
HINWEIS: Bei den Schritten des Reagenzienwechsels und des Waschens im folgenden Verfahren ist mit einem Verlust von ≤25 % der ursprünglichen Anzahl von 3D-Strukturen zu rechnen. Planen Sie eine endgültige Anzahl von mindestens zehn 3D-Strukturen mit einer Größe von 100 bis 500 μm pro getestetem Zustand ein, um qualitative und quantitative Bildanalysen durchzuführen. Schneiden Sie bei größeren Strukturen bei Bedarf die Enden der 1-ml-Pipettenspitzen ab, um ein Brechen der Strukturen zu vermeiden. Wenn die 3D-Struktursedimentation zu lang ist, können die Zellen bei 50 × g für 5 min bei Raumtemperatur (RT) schonend gedreht werden. Je nach Fragestellung sollten Vor- und Nachteile eines solchen Spinnschrittes abgewogen werden, da die Zentrifugation die Form der 3D-Strukturen beeinträchtigen kann. Vermeiden Sie das Schleudern bei >100 × g.
1. Sammlung und Fixierung von 3D-Zellkulturmodellen
HINWEIS: Achten Sie darauf, die 3D-Strukturen, die nur lose an der Rohrwand befestigt werden, nicht abzusaugen.
- Ernte von 3D-Zellkulturmodellen, die in eine Matrix eingebettet sind
HINWEIS: Dieser Abschnitt beschreibt die Gewinnung von 3D-Strukturen, die in Tropfen eines Basalmembranextrakts aus dem murinen Engelbreth-Holm-Schwarm-Sarkom (BME) gezüchtet wurden, kann aber an andere Matrices angepasst werden. In der Diskussion finden Sie die wichtigsten Punkte in Bezug auf ECM.- Entfernen Sie das Nährmedium aus den Vertiefungen, ohne die 3D-Matrix zu stören. Die Innen- und Außenseite einer 1-ml-Pipettenspitze wird mit Protein (im Folgenden als vorbeschichtete 1-ml-Spitze bezeichnet) vorbeschichtet, indem Sie die gesamte Länge der Spitze in 0,1%iges Rinderserumalbumin (BSA) in phosphatgepufferte Kochsalzlösung (PBS) (im Folgenden PBS-BSA 0,1% ige Lösung genannt) tauchen und 1 ml dieser Lösung zweimal auf und ab pipettieren.
HINWEIS: Diese Vorbeschichtung verhindert, dass die Zellen an der Spitze haften bleiben und minimiert Verluste. - Überschichten Sie die Innenseite eines Zentrifugenröhrchens (15 ml) mit Protein (im Folgenden vorbeschichtetes Zentrifugenröhrchen genannt), indem Sie wiederholt mit 0,1%iger PBS-BSA-Lösung füllen und das Röhrchen entleeren.
HINWEIS: Dadurch wird verhindert, dass die Zellen an der Röhre haften bleiben und Verluste minimiert werden. - Resuspendieren Sie mit der vorbeschichteten 1-ml-Spitze die 3D-Strukturen des Bohrlochs vorsichtig mit 1 mL eiskaltem 1x PBS und übertragen Sie die Suspension mit den 3D-Strukturen vorsichtig in das vorbeschichtete Zentrifugenröhrchen.
- Geben Sie vorsichtig 13 ml eiskaltes 1x PBS hinzu und lassen Sie die 3D-Strukturen mindestens 10 Minuten lang auf Eis absetzen.
HINWEIS: Bei Bedarf 5 min bei 50 × g bei 4 °C schleudern. Vermeiden Sie Drehungen >100 × g, da dies die Form der 3D-Strukturen beeinträchtigt. - Entfernen Sie den Überstand. Mit einer vorbeschichteten 1-ml-Spitze die 3D-Strukturen vorsichtig in 1 ml eiskaltem 1x PBS resuspendieren. Wiederholen Sie die Schritte 1.1.4 bis 1.1.5, um ein homogenes Pellet ohne 3D-Matrixrückstände zu erhalten.
HINWEIS: Eine effiziente Matrixentfernung wird durch die Art der Matrix, die Anzahl und Größe der 3D-Strukturen beeinflusst und erfordert eine Optimierung für unterschiedliche Kulturbedingungen. Bei 3D-Strukturen, die in BME gezüchtet wurden, dauert die Wiederherstellung aus der Matrixentfernung in der Regel 45-60 Minuten. - Die 1 mL 1x PBS-Suspension, die die 3D-Strukturen enthält, wird mit einer vorbeschichteten 1-ml-Spitze in ein vorbeschichtetes 1,5-ml-Zentrifugenröhrchen überführt und mit Abschnitt 1.3 fortgefahren.
- Entfernen Sie das Nährmedium aus den Vertiefungen, ohne die 3D-Matrix zu stören. Die Innen- und Außenseite einer 1-ml-Pipettenspitze wird mit Protein (im Folgenden als vorbeschichtete 1-ml-Spitze bezeichnet) vorbeschichtet, indem Sie die gesamte Länge der Spitze in 0,1%iges Rinderserumalbumin (BSA) in phosphatgepufferte Kochsalzlösung (PBS) (im Folgenden PBS-BSA 0,1% ige Lösung genannt) tauchen und 1 ml dieser Lösung zweimal auf und ab pipettieren.
- Gewinnung von schwimmenden 3D-Zellkulturmodellen
- Sammeln Sie die 3D-Strukturen vorsichtig mit einer vorbeschichteten 1-ml-Spitze und übertragen Sie sie in ein vorbeschichtetes 1,5-ml-Zentrifugenröhrchen. Lassen Sie die 3D-Strukturen sedimentieren oder schleudern Sie für 5 min bei 50 × g bei RT.
- Entfernen Sie den Überstand. Resuspendieren Sie die 3D-Strukturen mit einer vorbeschichteten 1-ml-Spitze in 1 mL 1x PBS. Fahren Sie mit Abschnitt 1.3 fort.
- Fixierung von 3D-Zellkulturmodellen
- Lassen Sie die 3D-Strukturen sedimentieren. Entfernen Sie vorsichtig den Überstand; Unter einem Abzug die 3D-Strukturen in 1 ml Formalin mit einer vorbeschichteten 1-ml-Spitze vorsichtig resuspendieren.
HINWEIS: Formalin enthält Formaldehyd, das gefährlich ist. Manipulieren Sie die Chemikalie in einer chemischen Haube. Tragen Sie Gummihandschuhe und Schutzbrillen. - Inkubieren Sie die 3D-Strukturen für 30 Minuten bei RT.
HINWEIS: Ein 30-minütiger Fixierungsschritt mit Formalin ist für die Immunfärbung einer Vielzahl von 3D-Strukturen (unterschiedlich in Größe, Form und Herkunft) erforderlich. Im Allgemeinen sind jedoch längere Fixationszeiten (>3 h) besser geeignet, um die Fluoreszenz von Reporterproteinen zu erhalten. - Lassen Sie die 3D-Strukturen sedimentieren oder schleudern Sie für 5 min bei 50 × g bei RT. Entfernen Sie das Formalin vorsichtig und ersetzen Sie es durch 1 ml 1x PBS. Wiederholen Sie diesen Waschschritt in 1x PBS zweimal. Lagern Sie die Proben bei 4 °C und fahren Sie mit Abschnitt 2 oder Abschnitt 3 fort.
HINWEIS: Das Protokoll kann hier pausiert werden, und die Zellen können bei 4 °C für die Langzeitlagerung (>1 Jahr) gehalten werden.
- Lassen Sie die 3D-Strukturen sedimentieren. Entfernen Sie vorsichtig den Überstand; Unter einem Abzug die 3D-Strukturen in 1 ml Formalin mit einer vorbeschichteten 1-ml-Spitze vorsichtig resuspendieren.
2.3D Färbung, Bildgebung und Analyse von 3D-Zellkulturmodellen
HINWEIS: Da die Organoide lose an der Röhrchenwand befestigt sind, sollten Sie sie vorsichtig behandeln, da alle folgenden Reagenzienwechsel zu Probenverlust führen können. Stellen Sie vor Beginn sicher, dass die richtigen Kontrollen für die Färbung verfügbar sind. Positiv- und Negativkontrollen können Zellen sein, von denen bekannt ist, dass das interessierende Protein entweder überexprimiert bzw. nicht vorhanden ist. Inkubieren Sie Proben ohne den primären Antikörper, um festzustellen, ob das beobachtete Signal auf eine unspezifische Bindung des sekundären Antikörpers zurückzuführen ist. Da einige Zellen dazu neigen, hohe Autofluoreszenzwerte aufzuzeigen, verwenden Sie Kontrollen ohne sekundäre Antikörper, um festzustellen, ob die beobachtete Fluoreszenz von der Hintergrundautofluoreszenz herrührt. Immunmarkierung und Visualisierung von Fluoreszenzreportern können kombiniert werden.
- 3D-Färbung der gesamten Montierung
- Bereiten Sie die Permeabilisierungsblocklösung (PB) vor, indem Sie 1x PBS mit 0,1%-1% eines nichtionischen Tensids (siehe Materialtabelle), 1% Dimethylsulfoxid, 1% BSA und 1% Eselserum (oder aus dem Tier, in dem die sekundären Antikörper erhöht wurden) ergänzen.
HINWEIS: Optimieren Sie sorgfältig die Konzentration des nichtionischen Tensids in Abhängigkeit von der Lokalisation des Ziels: Membran (0-0,5%), Zytoplasma (0,5-1%) und Kern (1%). Diese Lösung kann bis zu 1 Monat bei 4 °C gelagert werden. BSA funktioniert in der Regel gut für den Blockierungsschritt, aber bei hohem Hintergrundrauschen führen Sie einen empirischen Test durch, um die bestmöglichen Ergebnisse für eine bestimmte Kombination von Antikörpern zu erhalten. - Übertragen Sie die Organoide aus dem 1,5-ml-Zentrifugenröhrchen in ein 0,5-ml-Röhrchen mit einer vorbeschichteten 1-ml-Spitze. Lassen Sie die Organoide sedimentieren, entfernen Sie vorsichtig das 1x PBS und ersetzen Sie es durch 0,5 ml PB-Lösung. Inkubieren Sie die Organoide unter sanftem horizontalem Rühren (30-50 U/min) für 1 h bei RT.
- Lassen Sie die Organoide sedimentieren, entfernen Sie vorsichtig die PB-Lösung und waschen Sie zweimal in 1 mL PBS-BSA 0,1% für 3 min.
HINWEIS: Wenn Sie 3 Minuten warten, können sich die Strukturen am Boden der Röhre absetzen. - Entfernen Sie vorsichtig das PBS-BSA 0,1% und fügen Sie 250 μl primären Antikörper hinzu, der in der entsprechenden Konzentration in PB:1x PBS-Lösung (1:10) verdünnt ist. Zur Herstellung von 10 mL PB:1x PBS-Lösung (1:10) wird 1 ml PB-Lösung in 9 mL 1x PBS verdünnt. 2-3 Tage unter leichtem horizontalem Rühren (30-50 U/min) bei 4 °C inkubieren.
HINWEIS: Eine geeignete Antikörper-Inkubationszeit ist entscheidend für eine geeignete Antikörperpenetration, da 3D-Strukturen manchmal große Größen erreichen können. - Lassen Sie die Organoide sedimentieren und entfernen Sie vorsichtig die primäre Antikörperlösung. Waschen Sie 5x in PBS-BSA 0,1% für 3 min pro Waschgang und dann 2x in 1 mL PBS-BSA 0,1% für 15 min pro Waschgang unter sanftem horizontalem Rühren.
- Man fügt 250 μL Sekundärantikörper hinzu, verdünnt bei 1:250 in PB:1x PBS-Lösung (1:10). 24 Stunden bei 4°C unter leichtem horizontalem Rühren (30-50 U/min) inkubieren. Schützen Sie die Proben für diesen Schritt vor Licht.
- 250 μL Hoechst 33342 (20 μM Stammlösung) werden mit 1:1000 in PB:1x PBS-Lösung (1:10) verdünnt und weitere 2 h bei 4 °C unter leichtem horizontalem Rühren (30-50 U/min) inkubiert.
- Lassen Sie die Organoide sedimentieren und entfernen Sie vorsichtig die Lösung mit dem sekundären Antikörper + Hoechst 33342. Waschen Sie die Organoide 5x in 1 ml 1x PBS für 3 min pro Waschgang und dann 2x in 1 ml 1x PBS für 15 min pro Waschgang unter sanftem horizontalem Rühren (30-50 rpm).
HINWEIS: Es ist wichtig, die Proben ausgiebig zu waschen, um Hintergrundgeräusche oder Signalverlust zu vermeiden. - Lagern Sie die Proben bis zur Bildaufnahme bei 4 °C in PBS. Fahren Sie mit Abschnitt 2.2 fort.
HINWEIS: Hier kann das Protokoll pausiert werden, und die Proben können mehrere Monate vor Licht geschützt bei 4 °C gelagert werden.
- Bereiten Sie die Permeabilisierungsblocklösung (PB) vor, indem Sie 1x PBS mit 0,1%-1% eines nichtionischen Tensids (siehe Materialtabelle), 1% Dimethylsulfoxid, 1% BSA und 1% Eselserum (oder aus dem Tier, in dem die sekundären Antikörper erhöht wurden) ergänzen.
- Probenvorbereitung für die konfokale Bildgebung
- Übertragen Sie die Organoide vorsichtig mit einer vorbeschichteten 1-ml-Spitze in 50 μL des 1x PBS pro Vertiefung in eine 96-Well-Mikroplatte aus schwarzem Polystyrol. Fahren Sie mit Schritt 2.2.3 oder Abschnitt 2.3 fort.
HINWEIS: In diesem Stadium kann die Probe vor Licht geschützt und für viele Wochen bei 4 °C gelagert werden. - Lichtung
HINWEIS: Der Clearing-Schritt ist optional und kann entweder zur Immunmarkierung von Organoiden oder zum Nachweis endogener Fluoreszenz verwendet werden. Das Clearing kann zu einer Schrumpfung der 3D-Struktur führen, verändert jedoch nicht die allgemeine Morphologie, mit Ausnahme von sphärischen einschichtigen Organoiden mit großen Lumen4. Überspringen Sie bei diesen zystischen Organoiden den Clearing-Schritt und führen Sie eine Tiefengewebsbildgebungdurch 6.- 2,5 M Glycerin-Fructose-Reinigungslösung, die 50 % v/v Glycerin, 11 % v/v destilliertes Wasser und 45 % w/v Fructose enthält, wird mindestens über Nacht an einem Magnetrührer gemischt, bis die Lösung vollständig aufgelöst und homogen ist. Bis zu 1 Monat bei 4 °C im Dunkeln lagern.
- Entfernen Sie so viel 1x PBS wie möglich, ohne die Organoide zu berühren. Geben Sie 200 μl der Reinigungslösung mit einer 1-ml-Pipettenspitze hinzu, nachdem Sie das Ende entfernt haben, und resuspendieren Sie vorsichtig, um die Bildung von Blasen zu verhindern. Bei RT mindestens 12 h inkubieren und mit Abschnitt 3 fortfahren.
HINWEIS: Da die Clearinglösung zähflüssig ist, sind kleine Volumina schwer zu handhaben. Um die Handhabung zu erleichtern, stellen Sie sicher, dass sich die Lösung bei RT befindet, und pipettieren Sie langsam. Um eine optimale Klärung zu gewährleisten, lassen Sie die Probe vor der Bildgebung mindestens 24 Stunden in der Klärlösung sedimentieren. Wenn 3D-Strukturen zum Zeitpunkt der Aufnahme schweben, führen Sie einen optionalen Spin für 10 Minuten bei <100 × g bei RT durch oder lassen Sie mehr Zeit (ein bis mehrere Tage), um sie sedimentieren zu lassen. Das Protokoll kann bei diesem Schritt pausiert werden, bevor mit der Bildgebung fortgefahren wird, wenn es vor Licht geschützt und bei 4 °C (für Wochen) oder -20 °C (für Monate) gelagert wird.
- Übertragen Sie die Organoide vorsichtig mit einer vorbeschichteten 1-ml-Spitze in 50 μL des 1x PBS pro Vertiefung in eine 96-Well-Mikroplatte aus schwarzem Polystyrol. Fahren Sie mit Schritt 2.2.3 oder Abschnitt 2.3 fort.
- Bildaufnahme und -analyse
HINWEIS: Für die Abbildung von 3D-Strukturen ist eine Bildschnitttechnologie erforderlich.- Verwenden Sie konfokale Mikroskope und bevorzugen Sie Immersionsobjektive mit höherer numerischer Apertur (NA) im Vergleich zu Luft. Wählen Sie die Vergrößerungsobjektive (10x, 20x, 40x) entsprechend der Größe der 3D-Strukturen, der Bildrekonstruktion (Stitching) und der für die Analyse verwendeten Lösungen.
- Berücksichtigen Sie bei der Auswahl des Erfassungsmodus die Tiefenschärfe des Objektivs, das zur Definition des Schritts für das Z-Stacking verwendet wird. ermöglichen ein optimales 3D-Rendering.
HINWEIS: Die Bildanalyselösungen variieren und die Analyse muss an die verwendete Software angepasst werden. Zum Beispiel wurde dieses Analyseprotokoll auf einer High-Content-Analysesoftware erstellt (siehe Materialtabelle und ergänzende Abbildung 1 für Details) und liefert Daten zur Objektsegmentierung, zur Berechnung von Eigenschaften und zur Auswahl der Zellpopulation innerhalb eines 3D-rekonstruierten Objekts.
3. 2D-Schnitte, Färbung, Bildgebung und Analyse von 3D-Zellkulturmodellen
HINWEIS: 3D-Zellkulturmodelle variieren in der Größe. Fahren Sie mit Abschnitt 3.1 oder 3.2 fort, um eine effiziente Paraffineinbettung zu erfahren (Abbildung 2). Planen Sie ausreichend Zeit für die 3D-Struktursedimentation ein, bevor Sie sich waschen und das Reagenz wechseln. Achten Sie darauf, die Organoide, die am Boden der Röhre schwimmen, nicht abzusaugen. Informationen zur Einbettung von Paraffin finden Sie in Abbildung 2 .
- Paraffin-Einbettung von großen (Ø ≥ 400 μm) 3D-Zellkulturmodellen
- Am Tag vor dem Einbetten zwei mit Paraffin gefüllte 150-ml-Kolben (Paraffinbäder), eine kleine Metalleinbettform pro Probe und eine feine Pinzette auf 65 °C vorwärmen.
- Übertragen Sie die Organoide in 1x PBS vorsichtig mit einer vorbeschichteten 1-ml-Spitze in ein flaches Glasröhrchen mit einem mit Polytetrafluorethylen ausgekleideten Flaschenverschluss. Lassen Sie die Organoide sedimentieren, entfernen Sie vorsichtig das 1x PBS und ersetzen Sie es durch 70% Ethanol. Mindestens 30 Minuten inkubieren.
- Lassen Sie die Organoide sedimentieren und entfernen Sie vorsichtig das 70% ige Ethanol. Ersetzen Sie es durch 1 ml gebrauchsfertige Eosin-Y-Lösung. Streifen Sie die Tube und färben Sie sie mindestens 30 Minuten lang. Entfernen Sie vorsichtig die Eosinlösung und dehydrieren Sie die Organoide in drei aufeinanderfolgenden Wäschen mit 1 ml 100% igem Ethanol für jeweils ~ 30 Minuten.
HINWEIS: Ethanol, eine brennbare und flüchtige Flüssigkeit, verursacht schwere Reizungen der Augen und der Atemwege. Manipulieren Sie es in einem Abzug und tragen Sie eine schützende Augenbrille. - Entfernen Sie vorsichtig das 100% ige Ethanol und reinigen Sie die Organoide unter einer chemischen Haube in 3 aufeinanderfolgenden Wäschen mit 1 ml Xylol für jeweils ~ 30 Minuten.
HINWEIS: Xylol ist eine giftige, brennbare Flüssigkeit, deren Dämpfe Reizungen verursachen können. Manipulieren Sie es in einem Abzug. Vermeiden Sie direkten Kontakt mit der Haut und tragen Sie Gummihandschuhe und eine Schutzbrille. - Bereiten Sie unter einer chemischen Haube eine weiße Mikrozwillingsgewebekassette vor, indem Sie ein Stück Biopsiekissen (zuvor in Xylol getränkt) in eines der Fächer der Kassette legen. Übertragen Sie die 3D-Strukturen vorsichtig mit einer vorbeschichteten 2-ml-Pasteur-Kunststoffpipette auf das Biopsiekissen. Decken Sie sie mit einem anderen Biopsiekissen ab, das mit Xylol getränkt ist, um zu verhindern, dass sich die Organoide bewegen, und schließen Sie die Kassette.
- Wenn mehrere Proben verarbeitet werden, legen Sie die Kassette in ein Xylolbad, um auf die weitere Verarbeitung zu warten. Sobald alle Proben in Kassetten umgefüllt sind, legen Sie die Kassetten für 30 min bei 65 °C in ein vorgewärmtes Paraffinbad. Überführen Sie die Kassetten über Nacht in ein frisch vorgewärmtes Paraffinbad.
- Nehmen Sie nach der Paraffinimprägnierung eine vorgewärmte Einbettungsform und fügen Sie das erhitzte Paraffin hinzu. Legen Sie das Biopsiekissen mit den 3D-Strukturen in die Form und rühren Sie es vorsichtig, bis alle Organoide auf den Boden der Form fallen. Platzieren Sie die 3D-Strukturen mit einer vorgewärmten Feinzange sehr vorsichtig in der Mitte der Form. Fahren Sie mit Abschnitt 3.3 fort.
HINWEIS: Achten Sie darauf, die 3D-Strukturen nicht mit der Pinzette zu stören. Schieben, aber kneifen Sie sie nicht.
- Paraffin-Einbettung von kleinen (Ø ≤ 400 μm) 3D-Zellkulturmodellen
- Am Tag vor dem Einbetten zwei mit Paraffin gefüllte 150-ml-Kolben (Paraffinbäder), eine kleine Metalleinbettform pro Probe und eine feine Pinzette auf 65 °C vorwärmen.
- Entfernen Sie vorsichtig das 1x PBS aus der Organoid-Suspension. Führen Sie vorsichtig 3 Wäschen in 1 ml 1x Tris-gepufferter Kochsalzlösung (TBS) durch. Entfernen Sie so viel 1x TBS wie möglich, ohne die Organoide zu berühren.
HINWEIS: Achten Sie darauf, die Probe nicht abzusaugen. Falls erforderlich, führen Sie eine 5-minütige Drehung bei 50 x g bei RT durch. Verbleibende Spuren von Phosphat stören die folgenden Schritte und verhindern insbesondere die Gelpolymerisation. Verwenden Sie daher keine PBS-Lösungen während eines Verarbeitungsschritts. Für diesen Schritt wurde ein kommerzielles Kit verwendet, das Kassetten, Reagenz #1 (klare Flüssigkeit) und Reagenz #2 (farbige Flüssigkeit) enthielt, um das in Paraffin eingebettete Verfahren zu erleichtern, ohne möglicherweise winzige Fragmente zu verlieren (siehe Materialtabelle). Befolgen Sie die Anweisungen des Kits. Die Kassetten sind mit bereits vorhandenen Trägerpapieren und Kartoneinlagen vorkonfektioniert. - Geben Sie 2 Tropfen Reagenz #2 in das Röhrchen und mischen Sie vorsichtig, indem Sie auf das Röhrchen klopfen. Fügen Sie 2 Tropfen Reagenz #1 hinzu und mischen Sie erneut, indem Sie klopfen, um das Gel zu verfestigen. Entfernen Sie das Gel mit der feinen Pinzette aus der Tube und legen Sie es in die Vertiefung der Kassette.
- Unter dem Abzug wird die Probe entwässert, indem die Kassette wie folgt in aufeinanderfolgende Bäder gegeben wird (verwenden Sie die 150-ml-Kolben und verwenden Sie frisches Ethanol oder Xylol für jedes Bad): Ethanol 70%, 30 min; Ethanol 96%, 30 min; Ethanol 100%, drei Wäschen, je 30 min; Xylol, drei Wäschen, je 30 min.
- Stellen Sie die Kassetten für 30 min bei 65 °C in ein vorgewärmtes Paraffinbad und geben Sie sie über Nacht in ein frisch vorgewärmtes Paraffinbad. Nehmen Sie nach der Paraffinimprägnierung eine vorgewärmte Einbettungsform und fügen Sie erhitztes Paraffin hinzu. Öffnen Sie die Kassette, entfernen Sie das Gel vorsichtig mit einer feinen Pinzette und legen Sie das Gel mit den 3D-Strukturen auf die Mitte der Einbettform. Fahren Sie mit Abschnitt 3.3 fort.
- Allgemeine Schritte für die Einbettung von Paraffin
- Bringen Sie die Form vorsichtig in einen kalten Bereich, damit sich das Paraffin in einer dünnen Schicht verfestigt, wodurch die 3D-Strukturen in der richtigen Position gehalten werden. Fügen Sie eine Tissue-Kassette auf die Form und fügen Sie heißes Paraffin hinzu, um diese Kunststoffkassette zu bedecken. Entfernen Sie die Form, sobald sie vollständig erstarrt ist, und fahren Sie mit Abschnitt 3.4 fort.
HINWEIS: Paraffinblöcke können jahrelang bei Raumtemperatur gelagert werden.
- Bringen Sie die Form vorsichtig in einen kalten Bereich, damit sich das Paraffin in einer dünnen Schicht verfestigt, wodurch die 3D-Strukturen in der richtigen Position gehalten werden. Fügen Sie eine Tissue-Kassette auf die Form und fügen Sie heißes Paraffin hinzu, um diese Kunststoffkassette zu bedecken. Entfernen Sie die Form, sobald sie vollständig erstarrt ist, und fahren Sie mit Abschnitt 3.4 fort.

Abbildung 2: Überblick über das Verfahren zur Paraffineinbettung von großen und kleinen in vitro 3D-Zellkulturmodellen.
(A) Standardverfahren für die Einbettung von Paraffin. Nach der Fixierung und Dehydratisierung werden 3D-Strukturen mit Eosin gefärbt, um ihre Visualisierung zu erleichtern (oben und unten links). 3D-Strukturen werden vorsichtig mit einer 2-ml-Pasteur-Pipette (Mitte) auf das Biopsie-Pad (blau) in der Kassette gelegt. Nach der Paraffinimprägnierung werden die 3D-Strukturen mit einer Pinzette vorsichtig in das flüssige Paraffin getropft und im Biopsiepad sanft gerührt. Kleine 3D-Strukturen gehen bei diesem Schritt verloren, da sie sich nicht vom Pad lösen lassen (unten rechts: fehlgeschlagene Einbettung). Es werden nur große 3D-Strukturen eingebettet (oben rechts: erfolgreiche Einbettung). Pfeilspitzen zeigen auf 3D-Kulturen. (B) Alternative zum Standardprotokoll für die Einbettung von Paraffin. Nach der Fixierung kleiner 3D-Strukturen wird ein handelsübliches Kit verwendet, um Zellen in einem Gel zu erhalten und ihre Übertragung auf die Form nach der Paraffinimprägnierung zu erleichtern (rechts: erfolgreiche Einbettung). Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
- Blockschneiden und Färben
- Schneiden Sie 4-μm-Schnitte mit einem Standardmikrotom und führen Sie histologische und immunhistochemische Standardtechniken durch. Fahren Sie mit Abschnitt 3.5 fort.
HINWEIS: Für eine bessere Haftung der Querschnitte wurden spezielle Schienen (siehe Materialtabelle) verwendet. Die Objektträger können jahrelang bei Raumtemperatur oder bei 4 °C gelagert werden.
- Schneiden Sie 4-μm-Schnitte mit einem Standardmikrotom und führen Sie histologische und immunhistochemische Standardtechniken durch. Fahren Sie mit Abschnitt 3.5 fort.
- Bildaufnahme und -analyse
- Führen Sie die Bildgebung mit einem digitalen Objektträgerscanner oder einem aufrechten Mikroskop durch und analysieren Sie Daten mit einer Plattform für eine schnelle digitale quantitative Analyse, die morphologische und gemultiplexte Expressionsdaten auf Zellbasis über ganze 3D-Strukturabschnitte hinweg meldet (Details finden Sie in der ergänzenden Abbildung 2 ).
HINWEIS: Das 20x-Ziel wird von dieser Gruppe routinemäßig verwendet.
- Führen Sie die Bildgebung mit einem digitalen Objektträgerscanner oder einem aufrechten Mikroskop durch und analysieren Sie Daten mit einer Plattform für eine schnelle digitale quantitative Analyse, die morphologische und gemultiplexte Expressionsdaten auf Zellbasis über ganze 3D-Strukturabschnitte hinweg meldet (Details finden Sie in der ergänzenden Abbildung 2 ).
Ergebnisse
Dieses Protokoll bietet einen Überblick über die kritischen Schritte für die 2D- und 3D-Whole-Mount-Färbung sowie die Bildgebung und quantitative Analyse von 3D-Zellkulturmodellen (Abbildung 3 und Abbildung 4). Es ist auf eine breite Palette von 3D-Zellkulturmodellen anwendbar - von Sphäroiden bis hin zu Organoiden aus verschiedenen Wirtsspezies oder Geweben - und ermöglicht die Erfassung genauer und quantitativer Informationen über Architektur, Zellorganisation und Interaktionen auf zellulärer und subzellulärer Ebene (Abbildung 3 und Abbildung 4). Laboratorien müssen möglicherweise histologische und immunhistochemische 2D-Techniken und Antikörperkonzentrationen entsprechend ihren eigenen Bedürfnissen optimieren.
Beide Methoden liefern wertvolle biologische Informationen. 3D-Whole-Mount-Färbung und konfokale Mikroskopie liefern visuelle Informationen über die zelluläre Zusammensetzung und räumliche Position mit einem Tiefenfeld von bis zu 200 μm (Abbildung 3B). Die 2D-Schnitte sind jedoch für größere 3D-Strukturen geeignet, um detaillierte zelluläre morphologische Merkmale im gesamten Schnitt von 3D-Strukturen zu enthüllen, die aufgrund der Lichtstreuung, die die Auflösung in größeren Proben beeinträchtigt, in situ schwierig zu beobachten sind. Darüber hinaus können beide Techniken quantitative Daten liefern. In der Tat ermöglicht die erhaltene Auflösung die Anwendung von zellulären und subzellulären Segmentierungsalgorithmen zur Quantifizierung der Anzahl der Zellen und zum Nachweis des Vorhandenseins verschiedener Zellmarker in verschiedenen zellulären Subtypen (Abbildung 3F und Abbildung 4). Zusammenfassend lässt sich sagen, dass die hier beschriebenen bildgebenden Verfahren reproduzierbar, einfach und komplementär sind und wertvolle Werkzeuge zur Untersuchung der zellulären Heterogenität darstellen.

Abbildung 3: Repräsentative Ergebnisse für die 3D-Ganzmontage, Bildgebung und Analyse von optischen 3D- und 2D-Schnitten . (A) Konfokale Bilder von humanen (h) hochgradigen Gliom-Sphäroiden, die eine Woche lang kultiviert und mit Hoechst (blau), Olig2 (gelb) und Aktin (rot) (20x Wasserobjektiv) markiert wurden. Für alle aufgenommenen Bilder wurden die Mikroskopeinstellungen mit einer Positivkontrolle (oben) festgelegt, und dann wurde die Negativkontrolle mit identischen Einstellungen abgebildet, um das Fehlen von Fluoreszenz in Abwesenheit von primären Antikörpern zu kontrollieren (unten). (B) Orthogonale 3D-Whole-Mount-Darstellung der Ki67-Färbung, durchgeführt in (h) hochgradigem Gliom-Sphäroid, das eine Woche lang kultiviert wurde (Glycerin-Fructose-Clearing; 20x Wasserobjektiv, konfokal). (C) Konfokale Bilder von (h) hochgradigem Gliom-Sphäroid, das eine Woche lang kultiviert und mit Hoechst (blau), Olig2 (gelb) und Phalloidin-488 (grün) markiert wurde (Glycerin-Fructose-Clearing; 20x Wasserziel). (D) Konfokale Bilder von menschlichen (h) Rhabdomyosarkomen (oben) und Maus (m) Neuralleistenzellen (unten), die eine Woche lang kultiviert und mit Hoechst (blau), Aktin (rot) bzw. Ki67 (grün) markiert wurden (Glycerin-Fructose-Clearing; 20x trockenes Objektiv). (E) Konfokale Bilder von (h) hochgradigen Gliom-Sphäroiden, die eine Woche lang kultiviert und mit Hoechst (blau) und Ki67 (grün) markiert wurden (Glycerin-Fructose-Clearing; 40x Wasserobjektiv) (oben links). Segmentierte Bilder auf dem Hoechst-Kanal und Ki67-positiven (+) Kernregionen auf dem grünen Kanal wurden mit einer High-Content-Analyse-Software erzeugt (siehe ergänzende Abbildung 1 und Materialtabelle) (unten). Die Ausgabe ist der Prozentsatz der Ki67+ -Kerne pro segmentierter 3D-Struktur (oben rechts). Maßstabsbalken = 100 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
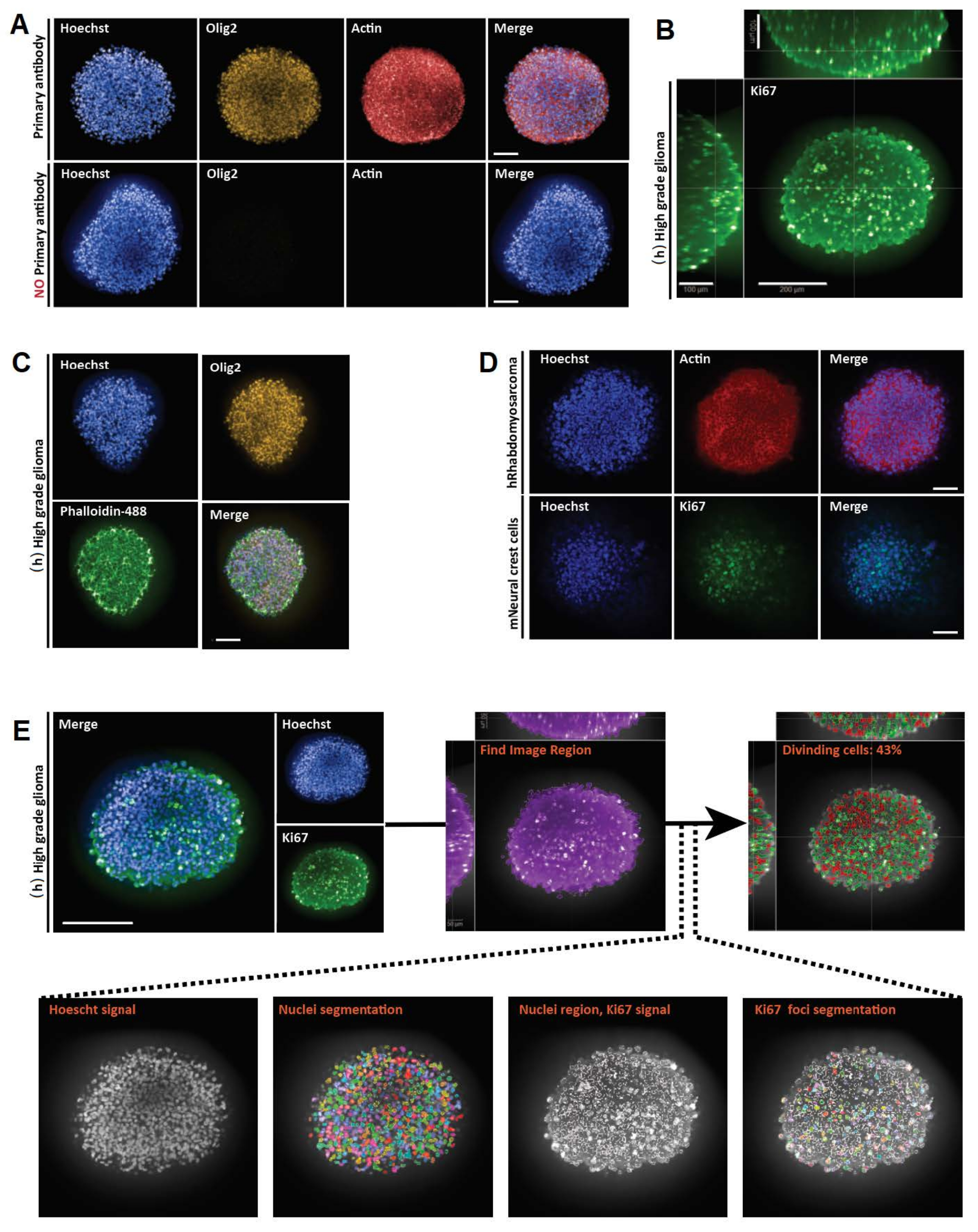{kind=link}

Abbildung 4: Repräsentative Ergebnisse für die Bildgebung und Analyse optischer 2D-Schnitte. (A, D) 2D-Schnittbilder eines 3D-Zellmodells (humane Rhabdomyosarkom-Sphäroide, die einen Monat lang kultiviert wurden), die mit einem digitalen Objektträgerscanner aufgenommen und auf einer Plattform für eine schnelle digitale quantitative Analyse analysiert wurden. (A) H&E-Färbung und Nachweis von Zellen entsprechend ihrer Größe. Maßstabsbalken = 500 μm. (B) Das Histogramm zeigt den Prozentsatz der Zellen < 100 μm 2 und > 100 μm2, die mit einer Software für schnelle digitale quantitative Analyse (links: Halo) oder manueller Zählung (rechts: MC) detektiert wurden. (C) Ki67-Färbung und Detektion von Zellen entsprechend der Intensität ihres 3,3'-Diaminobenzidin-Signals (DAB). Negativ (blau), schwach positiv (gelb), positiv (rot). Maßstabsbalken = 500 μm. (D) Das Histogramm zeigt den Prozentsatz der Ki67-negativen, schwach positiven und positiven Zellen. Abkürzungen: H&E = Hämatoxylin und Eosin; MC = manuelle Zählung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
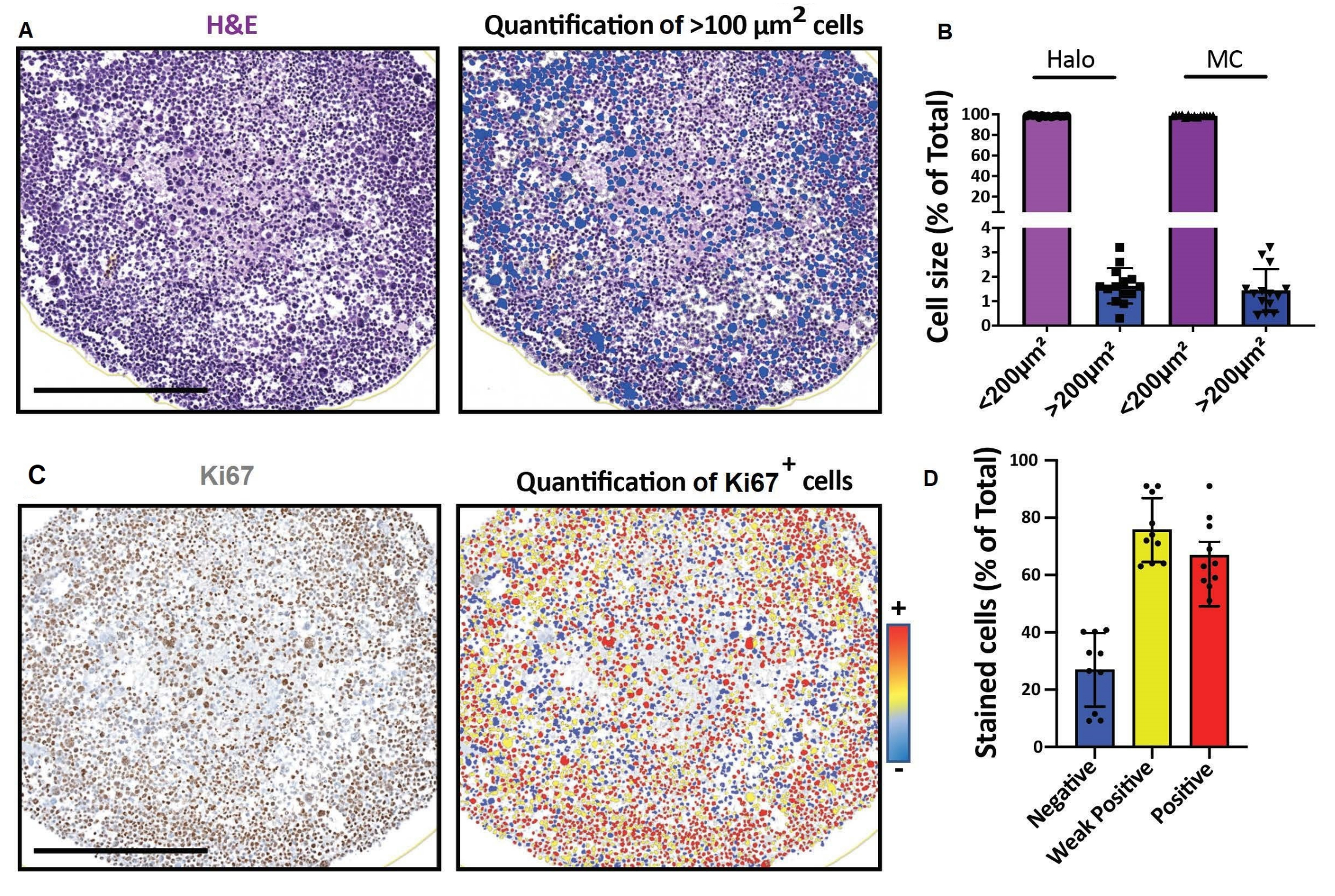{kind=link}
Ergänzende Abbildung 1: Übersicht über die Schritte in der Bildanalyse-Software. Analysen basieren auf der Assoziation von Bausteinen. Jeder Baustein entspricht einer Funktion - Segmentierung, Berechnung, Assoziation, Ausgabedefinition - und bietet mehrere Algorithmen und Variablenauswahlen, um der biologischen Probe zu entsprechen, die abgebildet wird. Die Software bietet mehrere RMS-Analyseprotokolle (Ready Made Solution), die einfach verwendet und geändert werden können. Integrierte Bildanalyseprotokolle können gespeichert, auf verschiedene Datensätze angewendet und von Benutzern gemeinsam genutzt werden. Kurz gesagt, das Analyseprotokoll impliziert eine sequentielle Objektsegmentierung: Sphäroide, Kerne und schließlich Ki67-Taschen (A488). Dann wird die mittlere Intensität der Ki67-Taschen berechnet, um die positiven Ereignisse weiter zu unterscheiden. Schließlich werden Kerne, die Ki67-positive Taschen umfassen, positiv ausgewählt. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung 2: Überblick über die Verfahrensschritte der quantitativen Analysesoftware. Schritt 1. Laden Sie die Dateien über die Registerkarte Studien hoch. Die Dateien werden im Abschnitt "Bildaktionen " geöffnet. Schritt 2. Öffnen Sie die Registerkarte Anmerkungen und klicken Sie dann auf Ebenenaktionen , um mit dem Kreiswerkzeug der Symbolleiste eine neue Ebene rund um die Struktur zu entwerfen. Für nicht kreisförmige Strukturen kann stattdessen das Stiftwerkzeug verwendet werden. Schritt 3. Die Symbolleiste kann verwendet werden, um Anmerkungen zu entwerfen und die Quantifizierung mit dem  Tool zu visualisieren. Schritt 4. Öffnen Sie die Registerkarte Analyse und wählen Sie die besten Bedingungen für die Analyse der Probe aus (hier können mehrere Versuche erforderlich sein). Schritt 4.1. Verwenden Sie den Abschnitt "Fleckenauswahl ", um die Färbebedingung festzulegen. Bei mehreren Flecken können diese hinzugefügt und umbenannt werden, und die virtuelle Farbe kann geändert werden. Die Lokalisationsdetektion kann spezifiziert werden - Kern- oder Zytoplasma-Färbung. Schritt 4.2. Verwenden Sie den Abschnitt Zellerkennung, um die Zellerkennung einzurichten. Dieser Abschnitt ist der wichtigste für die Analyse. Der Abschnitt "Kernkontrastschwelle " ermöglicht die Detektion aller Kerne. Es muss darauf geachtet werden, dass es mehrere Populationsgrößen gibt, die Software kann mehrere Zellen anstelle einer einzigen großen erkennen. Die Abschnitte " Kerngröße " und "Aggressivität der Kernsegmentierung " können verwendet werden, um Zellgrößenpopulationsbereiche zu quantifizieren. Schritt 5. Beschreibung zum Ausführen der Stichprobenanalyse. Befolgen Sie die in der Abbildung gezeigten Schritte. Im Abschnitt "Anmerkungsebene " wird die Einstellung nur auf dieser Folie ausgeführt. Die Quantifizierung kann mit dem Tool visualisiert werden. Wiederholen Sie die Schritte 4.1-5, bis eine geeignete Quantifizierung erreicht ist. Schritte 6-6.1. Diese Schritte ermöglichen es Ihnen, eine Figur mit der Software zu zeichnen. Schritt 7. Quantifizierungsgrafiken, die über Software erstellt wurden, können gespeichert werden. Schritt 8. Daten können exportiert werden. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Tool zu visualisieren. Schritt 4. Öffnen Sie die Registerkarte Analyse und wählen Sie die besten Bedingungen für die Analyse der Probe aus (hier können mehrere Versuche erforderlich sein). Schritt 4.1. Verwenden Sie den Abschnitt "Fleckenauswahl ", um die Färbebedingung festzulegen. Bei mehreren Flecken können diese hinzugefügt und umbenannt werden, und die virtuelle Farbe kann geändert werden. Die Lokalisationsdetektion kann spezifiziert werden - Kern- oder Zytoplasma-Färbung. Schritt 4.2. Verwenden Sie den Abschnitt Zellerkennung, um die Zellerkennung einzurichten. Dieser Abschnitt ist der wichtigste für die Analyse. Der Abschnitt "Kernkontrastschwelle " ermöglicht die Detektion aller Kerne. Es muss darauf geachtet werden, dass es mehrere Populationsgrößen gibt, die Software kann mehrere Zellen anstelle einer einzigen großen erkennen. Die Abschnitte " Kerngröße " und "Aggressivität der Kernsegmentierung " können verwendet werden, um Zellgrößenpopulationsbereiche zu quantifizieren. Schritt 5. Beschreibung zum Ausführen der Stichprobenanalyse. Befolgen Sie die in der Abbildung gezeigten Schritte. Im Abschnitt "Anmerkungsebene " wird die Einstellung nur auf dieser Folie ausgeführt. Die Quantifizierung kann mit dem Tool visualisiert werden. Wiederholen Sie die Schritte 4.1-5, bis eine geeignete Quantifizierung erreicht ist. Schritte 6-6.1. Diese Schritte ermöglichen es Ihnen, eine Figur mit der Software zu zeichnen. Schritt 7. Quantifizierungsgrafiken, die über Software erstellt wurden, können gespeichert werden. Schritt 8. Daten können exportiert werden. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Die Zellkultur ist ein unverzichtbares Werkzeug, um grundlegende biologische Mechanismen aufzudecken, die an der Entwicklung, Funktion, Regeneration und Störung von Geweben und Organen sowie an Krankheiten beteiligt sind. Obwohl die einschichtige 2D-Zellkultur vorherrschte, hat sich die neuere Forschung in Richtung Kulturen verlagert, die 3D-Strukturen erzeugen, die die zellulären Reaktionen in vivo besser widerspiegeln, insbesondere aufgrund zusätzlicher räumlicher Organisation und Zell-Zell-Kontakte, die die Genexpression und das zelluläre Verhalten beeinflussen und somit prädiktivere Daten liefern könnten7. Dennoch bleiben viele Herausforderungen bestehen, einschließlich des Bedarfs an benutzerfreundlichen Färbe- und Bildgebungstechniken für die detaillierte mikroskopische Visualisierung und Bewertung komplexer 3D-Strukturen auf zellulärer und subzellulärer Ebene. In diesem Zusammenhang wurden detaillierte, robuste und komplementäre Protokolle zur Verfügung gestellt, um Färbung und zelluläre und subzelluläre Bildgebung von fixierten In-vitro-3D-Zellkulturmodellen mit einer Größe von 100 μm bis zu mehreren Millimetern durchzuführen.
Dieses Verfahren stellt zwei verschiedene Strategien vor, um mit einer Vielzahl von Größen und Arten von In-vitro-3D-Zellkulturmodellen umzugehen. Die Wahl des einen (3D-Ganzmontageanalyse) oder des anderen (2D-Schnittanalyse) hängt vom verwendeten Modell und dem untersuchten Problem ab. Die 3D-Whole-Mount-Analyse mittels konfokaler Mikroskopie ermöglicht die Visualisierung von Zellen mit einem Tiefenfeld von bis zu 200 μm, unabhängig von der Gesamtgröße der 3D-Struktur, während die 2D-Schnitte für Proben jeder Größe anwendbar sind, die Visualisierung jedoch 2D-dimensional bleibt. Im Folgenden finden Sie einige Vorschläge zur Fehlerbehebung und technische Überlegungen.
Der Verlust von 3D-Strukturen während des Workflows ist der häufigste Nachteil. Sie können an den Spitzen und Rohren haften, weshalb die Vorbeschichtung von Spitzen und Rohren mit PBS-BSA 0,1% iger Lösung von entscheidender Bedeutung ist. Darüber hinaus ist es wichtig, die 3D-Strukturen zwischen den Reagenzienwechseln sedimentieren zu lassen und alle Pipettierungen sehr sorgfältig durchzuführen. Wie in der Prozedur erwähnt, können die Zellen bei einer zu langen 3D-Struktursedimentation bei 50 × g für 5 min bei RT vorsichtig gesponnen werden. Je nach Ziel der Studie sollten die Vor- und Nachteile eines solchen Spinnschritts berücksichtigt werden, da die Zentrifugation die Form der 3D-Strukturen beeinträchtigen kann. Darüber hinaus sollte darauf geachtet werden, dass diese Morphologie während des Fixierungsschritts erhalten bleibt, da zystische Organoide zum Kollaps neigen. Durch die Fixierung von Strukturen mit einer Größe von weniger als 400 μm sollen strukturelle Veränderungen verhindert werden.
Für eine optimale Immunmarkierung ist die Rückgewinnung von Organoiden aus ihren 3D-Matrizen ein entscheidender Schritt. Die 3D-Matrix kann eine ausreichende Penetration von Antikörpern behindern oder aufgrund der unspezifischen Bindung an die Matrix zu einer hohen Hintergrundfärbung führen. Die EZM-Entfernung kann die Morphologie der äußeren Segmente von Organoiden verändern (insbesondere bei kleinen Zellvorsprüngen, die sich von untersuchten 3D-Strukturen erstrecken) und die Analyse teilweise behindern. Bei solchen 3D-Strukturen kann die Matrix während des gesamten Verfahrens beibehalten werden. Die Kulturbedingungen sollten jedoch sorgfältig angepasst werden, um Zellen in einer minimalen Menge an Matrix zu züchten, um ein unzureichendes Eindringen von Lösungen und Antikörpern zu verhindern und aufeinanderfolgende Waschschritte zu vermeiden, die darauf abzielen, übermäßige Hintergrundgeräusche zu reduzieren 6,8.
Der in diesem Protokoll beschriebene optische Clearing-Schritt im Abschnitt 3D-Whole-Mount-Färbung ist für die Abbildung von 3D-Strukturen bis zu einer Tiefe von 150-200 μm anstelle von 50-80 μm ohne Clearing relevant. Im Vergleich zu anderen Clearing-Methoden, die oft mehrere Wochen in Anspruch nehmen und toxische Clearing-Mittel verwenden, wurde in diesem Protokoll 4,9 ein zuvor veröffentlichter schneller und sicherer Clearing-Schritt verwendet. Darüber hinaus ist dieser Clearing-Schritt reversibel, und neue Antikörper können der ersten Färbung ohne Verlust der Auflösung oder Helligkeit hinzugefügt werden4. Je nach untersuchtem 3D-Zellkulturmodell reicht jedoch eine Tiefe von 150-200 μm möglicherweise nicht aus, um die 3D-Struktur informativ abzubilden, und dieses Clearingprotokoll kann zu Veränderungen in der allgemeinen Morphologie von kugelförmigen, einschichtigen Organoiden mit großen Lumenführen 4. Anwender sollten ihr Experiment sorgfältig planen und, falls erforderlich, das Timing des Permeabilisierungs-/Blockierungsschritts (um das Eindringen von Antikörpern und Lösung zu ermöglichen), des Clearing-Schritts (um tiefer als 200 μm einzudringen, sollten die Proben vollständig freigegeben werden) und der Bildaufnahme optimieren. Die beiden am weitesten verbreiteten Technologien, die in Core Facilities verfügbar sind, sind die Lichtblatt- und die konfokale Mikroskopie. Benutzer müssen eine Technologie sorgfältig auswählen, die auf der Größe ihrer 3D-Strukturen und ihrer biologischen Fragebasiert 10. Im Vergleich zur konfokalen Mikroskopie bleibt die Auflösung der Lichtblattmikroskopie, die für solche tiefen Strukturen erhalten wird, jedoch suboptimal, um eine subzelluläre Auflösung zu erhalten.
Hier wurde über einen detaillierten und robusten Prozess berichtet, der der Paraffineinbettung einzelner Proben gewidmet ist. Interessanterweise haben Gabriel et al. kürzlich ein Protokoll entwickelt, das 3D-Zellkulturen in Paraffin mit erhöhtem Durchsatz einbettet. Sie verwendeten eine Polydimethylsiloxan (PDMS) -Form, um 96 3D-Strukturen in einem Microarray-Muster in einem Block einzuschließen, was neue Perspektiven für Studien zu 3D-Tumormodellen bietet, die mehr Gruppen, Zeitpunkte, Behandlungsbedingungen und Replikate umfassen11. Diese Methode erfordert jedoch umfangreiche Fähigkeiten und Maschinen, insbesondere für die Herstellung der Vorform, die zur Herstellung von PDMS-Formen verwendet wird.
Zusammenfassend beschreibt dieser Artikel zwei verschiedene, komplementäre und anpassungsfähige Ansätze, die es ermöglichen, genaue und quantitative Informationen über die architektonische und zelluläre Zusammensetzung von 3D-Zellmodellen zu erhalten. Beide Parameter sind entscheidend für die Untersuchung biologischer Prozesse wie der intratumoralen zellulären Heterogenität und ihrer Rolle bei der Resistenz gegen Behandlungen.
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
Diese Arbeit wurde durch den St Baldrick's Robert J. Arceci Innovation Award #604303 unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Biopsy pad Q Path blue | VWR | 720-2254 | |
| Cassettes macrostar III Blc couv. Char. x1500 | VWR | 720-2233 | |
| Cassette microtwin white | VWR | 720-2183 | |
| Chemical hood | Erlab | FI82 5585-06 | |
| Filter tips 1000 µL | Star lab Tip-One | S1122-1730 | |
| Fine forceps | Pyramid innovation | R35002-E | |
| Flat-bottom glass tubes with PTFE lined 2 mL | Fisher Scientific | 11784259 | Excellent for environmental samples, pharmaceuticals and diagnostic reagents. PTFE is designed for the ultimate in product safety. PTFE provides totally inert inner seal and surface facing the sample or product. |
| Glass bottom dish plate 35 mm | Ibedi | 2018003 | |
| Horizontal agitation | N-BIOTEK | NB-205 | |
| Incubator prewarmed to 65 °C | Memmert Incubator | LAB129 | |
| Inox molds 15x15 | VWR | 720-1918 | |
| Microscope Slides Matsunami TOMO-11/90 | Roche diagnostics | 8082286001 | these slides are used for a better adhesion of sections |
| Microtome | Microm Microtech France | HM340E | |
| Panoramic scan II | 3dhistech | 2397612 | |
| Paraffin embedding equipment | Leica | EG1150C | |
| Plastic pipette Pasteur 2 mL | VWR | 612-1681 | |
| Q Path flacon 150mL cape blanc x250 | VWR | 216-1308 | Good for environmental samples, pharmaceuticals and diagnostic reagents. Polypropylene (PP) are rigid, solid, provide excellent stress crack and impact resistance and have a good oil and alcohol barrier and chemical resistance. PE-lined cap is stress crack resistant and offers excellent sealing characteristics. |
| Set of micropipettors (p200, p1000) | Thermo Scientific | 11877351 (20-200) 11887351(p1000) | |
| OPERA PHENIX | PerkinElmer | HH14000000 | |
| SP5 inverted confocal microscope | Leica | LSM780 | |
| Tissue cassette | VWR | 720-0228 | |
| Zeiss Axiomager microscope | Leica | SIP 60549 | |
| Reagent | |||
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7030-100G | |
| Cytoblock (kit) | Thermofisher Scientific | 10066588 | |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | 57648266 | CAUTION: toxic and flammable. Vapors may cause irritation. Manipulate in a fume hood. Avoid direct contact with skin. Wear rubber gloves, protective eye goggles. |
| Eosin aqueous 1% | Sigma-Aldrich | HT110316 | |
| Ethanol 96% | VWR | 83804.360 | CAUTION: Causes severe eye irritation. Flammable liquid and vapor. Causes respiratory tract irritation. Manipulate in a fume hood. Wear protective eye goggles. |
| Ethanol 100% | VWR | 20821.365 | CAUTION: Causes severe eye irritation. Flammable liquid and vapor. Causes respiratory tract irritation. Manipulate in a fume hood. Wear protective eye goggles. |
| Formalin 4% | Microm Microtech France | F/40877-36 | CAUTION: Formalin contains formaldehyde which is hazardous. Manipulate in a fume hood. Avoid direct contact with skin. Wear rubber gloves and protective eye goggles. |
| Fructose | Sigma-Aldrich | F0127 | |
| Gill hematoxylin type II | Microm Microtech France | F/CP813 | |
| Glycerol | Sigma-Aldrich | G5516 | 500 mL |
| Hoechst 33342 | Life Technologies | H3570 | CAUTION: Suspected of causing genetic defects. Avoid direct contact with skin. Wear rubber gloves and protective eye goggles. |
| Normal donkey serum | Sigma-Aldrich | D9663 | 10 mL |
| Paraffin Wax tek III | Sakura | 4511 | |
| Phosphate Buffer Saline (PBS) 1 X | Gibco | 14190-094 | |
| Tris-Buffered Saline (TBS) 10X | Microm Microtech France | F/00801 | 100 mL |
| Triton X-100 | Sigma-Aldrich | T8532 | CAUTION: Triton X100 is hazardous. Avoid contact with skin and eyes. |
| Xylene | Sigma-Aldrich | 534056 | CAUTION: Xylene is toxic and flammable. Vapors may cause irritation. Manipulate in a fume hood. Avoid direct contact with skin. Wear rubber gloves, protective eye goggles. |
| Solutions | |||
| Clearing solution | Glycerol-Fructose clearing solution is 60% (vol/w) glycerol and 2.5 M fructose. To prepare 10 mL of this solution, mix 6 mL of glycerol and 4.5 g of fructose. Complete to 10 mL with dH2O. Use a magnetic stirrer overnight. Refractive index = 1.4688 at room temperature (RT: 19–23 °C). Store at 4 °C in dark for up to 1 month. | ||
| PBS-BSA 0,1% solution | To prepare 0,1% (vol/wt) PBS-BSA 0,1% solution, dissolve 500 mg of BSA in 50 mL of PBS-1X (store at 4°C for up to 2 weeks). And dilute 1mL of this solution into 9mL of PBS-1X. This solution can be used to precoat the tip and centrifugation tube. | ||
| Permeabilisation-blocking solution (PB solution) | The PBSDT blocking solution is PBS-1X supplemented with 0.1% – 1% Tritonx-100 (depending on the protein localization membrane/nucleus), 1% DMSO, 1% BSA and 1% donkey serum (or from the animal in which the secondary antibodies were raised). This solution can be stored at 4°C for up to 1 month. | ||
| PB:PBS-1X (1:10) solution | PB:PBS-1X (1:10) solution is a 10 time diluted PB solution. To prepare 10 mL of this solution dilute 1 mL of PB solution in 9 mL of PBS-1X. | ||
| Software | |||
| Halo software | Indicalabs | NM 87114 | |
| Harmony software | PerkinElmer | HH17000010 |
Referenzen
- Ryu, N. E., Lee, S. H., Park, H. Spheroid culture system methods and applications for mesenchymal stem cells. Cells. 8 (12), 1-13 (2019).
- Bartfeld, S., Clevers, H. Stem cell-derived organoids and their application for medical research and patient treatment. Journal of Molecular Medicine. 95 (7), 729-738 (2017).
- Cui, X., Hartanto, Y., Zhang, H. Advances in multicellular spheroids formation. Journal of the Royal Society, Interface. 14 (127), (2017).
- Dekkers, J. F., et al. High-resolution 3D imaging of fixed and cleared organoids. Nature Protocols. 14, 1756-1771 (2019).
- Broutier, L., et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nature Protocols. 11 (9), 1724-1743 (2016).
- Rezanejad, H., Lock, J. H., Sullivan, B. A., Bonner-Weir, S. Generation of pancreatic ductal organoids and whole-mount immunostaining of intact organoids. Current Protocols in Cell Biology. 83 (1), 82 (2019).
- Edmondson, R., Broglie, J. J., Adcock, A. F., Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay and Drug Development Technologies. 12 (4), 207-218 (2014).
- McCray, T., Richards, Z., Marsili, J., Prins, G. S., Nonn, L. Handling and assessment of human primary prostate organoid culture. Journal of Visualized Experiments: JoVE. (143), e59051 (2019).
- Ueda, H. R., et al. Tissue clearing and its applications in neuroscience. Nature Reviews: Neuroscience. 21 (2), 61-79 (2020).
- Lazzari, G., et al. Light sheet fluorescence microscopy versus confocal microscopy: in quest of a suitable tool to assess drug and nanomedicine penetration into multicellular tumor spheroids. European Journal of Pharmaceutics and Biopharmaceutics. 142, 195-203 (2019).
- Gabriel, J., Brennan, D., Elisseeff, J. H., Beachley, V. Microarray embedding/sectioning for parallel analysis of 3D cell spheroids. Scientific Reports. 9, 16287 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten