
Method Article
Coloração e Imagem de Alta Resolução de Modelos Organoides e Esferoides Tridimensionais
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
Aqui, fornecemos protocolos detalhados, robustos e complementares para realizar imagens de coloração e resolução subcelular de modelos de cultura celular tridimensional fixa que variam de 100 μm a vários milímetros, permitindo assim a visualização de sua morfologia, composição do tipo celular e interações.
Resumo
Modelos de cultura celular tridimensionais (3D) in vitro, como organoides e esferoides, são ferramentas valiosas para muitas aplicações, incluindo desenvolvimento e modelagem de doenças, descoberta de medicamentos e medicina regenerativa. Para explorar plenamente esses modelos, é crucial estudá-los nos níveis celular e subcelular. No entanto, caracterizar esses modelos de cultura de células 3D in vitro pode ser tecnicamente desafiador e requer conhecimentos específicos para realizar análises eficazes. Aqui, este artigo fornece protocolos detalhados, robustos e complementares para realizar imagens de coloração e resolução subcelular de modelos de cultura de células 3D fixas in vitro que variam de 100 μm a vários milímetros. Esses protocolos são aplicáveis a uma ampla variedade de organoides e esferoides que diferem em suas condições de célula de origem, morfologia e cultura. Desde a colheita da estrutura 3D até a análise da imagem, esses protocolos podem ser concluídos dentro de 4 a 5 dias. Resumidamente, as estruturas 3D são coletadas, fixadas e podem então ser processadas por meio de incorporação de parafina e coloração histológica/imuno-histoquímica, ou diretamente imunomarcadas e preparadas para limpeza óptica e reconstrução 3D (profundidade de 200 μm) por microscopia confocal.
Introdução
Nas últimas décadas, os avanços na biologia de células-tronco e nas tecnologias de cultura 3D in vitro anunciaram uma revolução na biologia e na medicina. Modelos celulares de maior complexidade em 3D tornaram-se muito populares, pois permitem que as células cresçam e interajam com uma estrutura extracelular circundante, recapitulando de perto aspectos dos tecidos vivos, incluindo sua arquitetura, organização celular e interações, ou mesmo características de difusão. Como tal, os modelos de cultura de células 3D podem fornecer insights únicos sobre o comportamento das células em tecidos em desenvolvimento ou doentes in vitro. Organoides e esferoides são estruturas 3D multicelulares, variando de vários micrômetros a milímetros, e são as estruturas 3D in vitro mais proeminentes. Ambos podem ser cultivados dentro de um andaime de suporte, incluindo (i) hidrogéis derivados de animais (extrato de membrana basal, colágeno), plantas (alginato / agarose), ou sintetizados a partir de produtos químicos, ou (ii) matrizes inertes contendo poros para promover a proliferação e o crescimento celular.
Organoides e esferoides também podem se desenvolver sem a presença de um andaime de suporte, confiando nas células para se auto-montar em aglomerados. Isso depende de diferentes técnicas, como o uso de materiais não adesivos para inibir a fixação celular, a tensão superficial e a força gravitacional (por exemplo, técnicas de queda suspensa) ou a rotação circular constante dos vasos (por exemplo, cultura do rotador). Em todos os casos, essas técnicas facilitam as interações célula-célula e célula-matriz para superar as limitações da cultura tradicional de células monocamadas1. Os termos "organoides" e "esferoides" foram usados de forma intercambiável no passado, mas existem diferenças fundamentais entre esses dois modelos de cultura de células 3D. Os organoides são aglomerados celulares 3D in vitro derivados de células-tronco pluripotentes ou células-tronco específicas de tecidos, nos quais as células se auto-organizam espontaneamente em progenitores e tipos celulares diferenciados e que recapitulam pelo menos algumas funções do órgão de interesse2. Os esferoides compreendem uma gama mais ampla de estruturas 3D multicelulares formadas sob condições não aderentes e podem surgir de uma grande diversidade de tipos celulares, como linhagens celulares imortalizadas ou células primárias3. Assim, inerente às suas origens intrínsecas de células-tronco, os organoides têm uma maior propensão à auto-montagem, viabilidade e estabilidade do que os esferoides.
No entanto, em essência, esses dois modelos são estruturas 3D compostas de múltiplas células, e as técnicas desenvolvidas para estudá-los são, portanto, muito semelhantes. Por exemplo, abordagens de imagem poderosas no nível de resolução de célula única são necessárias para sondar a complexidade celular de organoides e esferoides. Aqui, resumindo a experiência deste grupo e a de líderes no campo dos organoides4, este artigo descreve procedimentos detalhados para realizar colorações bidimensionais (2D) e 3D de montagem completa, imagens e análises da composição celular e subcelular e organização espacial de organoides e esferoides variando de 100 μm a vários milímetros. De fato, este procedimento apresenta dois tipos diferentes e complementares de coloração e aquisição de imagens para analisar uma grande variedade de tamanhos e tipos de modelos de cultura de células 3D in vitro. O uso de um (análise 3D de montagem inteira) ou outro (análise de seção 2D) dependerá do modelo estudado e das respostas buscadas. A análise 3D de montagem completa por microscopia confocal pode, por exemplo, ser aplicada para visualizar células em cultura 3D de até 200 μm de profundidade, independentemente do tamanho total da estrutura 3D, enquanto a análise de seções 2D fornece insights sobre amostras de qualquer tamanho, embora no nível 2D. Este procedimento tem sido aplicado com sucesso em uma variedade de organoides4,5 e esferoides derivados de células humanas e murinas, provenientes de diferentes camadas germinativas embrionárias. A visão geral do procedimento é mostrada na Figura 1. Os principais estágios, as relações entre eles, os passos decisivos e o tempo esperado são indicados.

Figura 1: Visão geral esquemática do procedimento. Os modelos de cultura de células 3D in vitro são coletados e fixados, em seguida, preparados para coloração de montagem inteira 3D (opção a) ou incorporados em parafina para seccionamento e coloração 2D (opção b). Para experimentos de coloração 3D de montagem completa, as estruturas 3D fixas são imunomarcadas após a etapa de fixação. Uma etapa opcional de limpeza óptica pode ser realizada para melhorar a qualidade da imagem e a profundidade da microscopia óptica, reduzindo o espalhamento da luz durante o processamento da imagem. As imagens são capturadas em um microscópio confocal invertido ou em um sistema confocal de alto conteúdo e analisadas usando o software apropriado. Para a incorporação de parafina, as estruturas 3D são processadas diretamente (opção b.1 para grandes estruturas ≥ 400 μm) ou incluídas em um gel (b.2; pequenas estruturas ≤ 400 μm) para desidratação e incorporação de parafina. Os blocos de parafina são então cortados e corados (coloração histológica ou imunoquímica). As imagens de seções 2D são obtidas em um scanner de lâmina digital ou um microscópio vertical e analisadas em uma plataforma de análise de imagens usando análise quantitativa digital rápida. Por favor, clique aqui para ver uma versão maior desta figura.
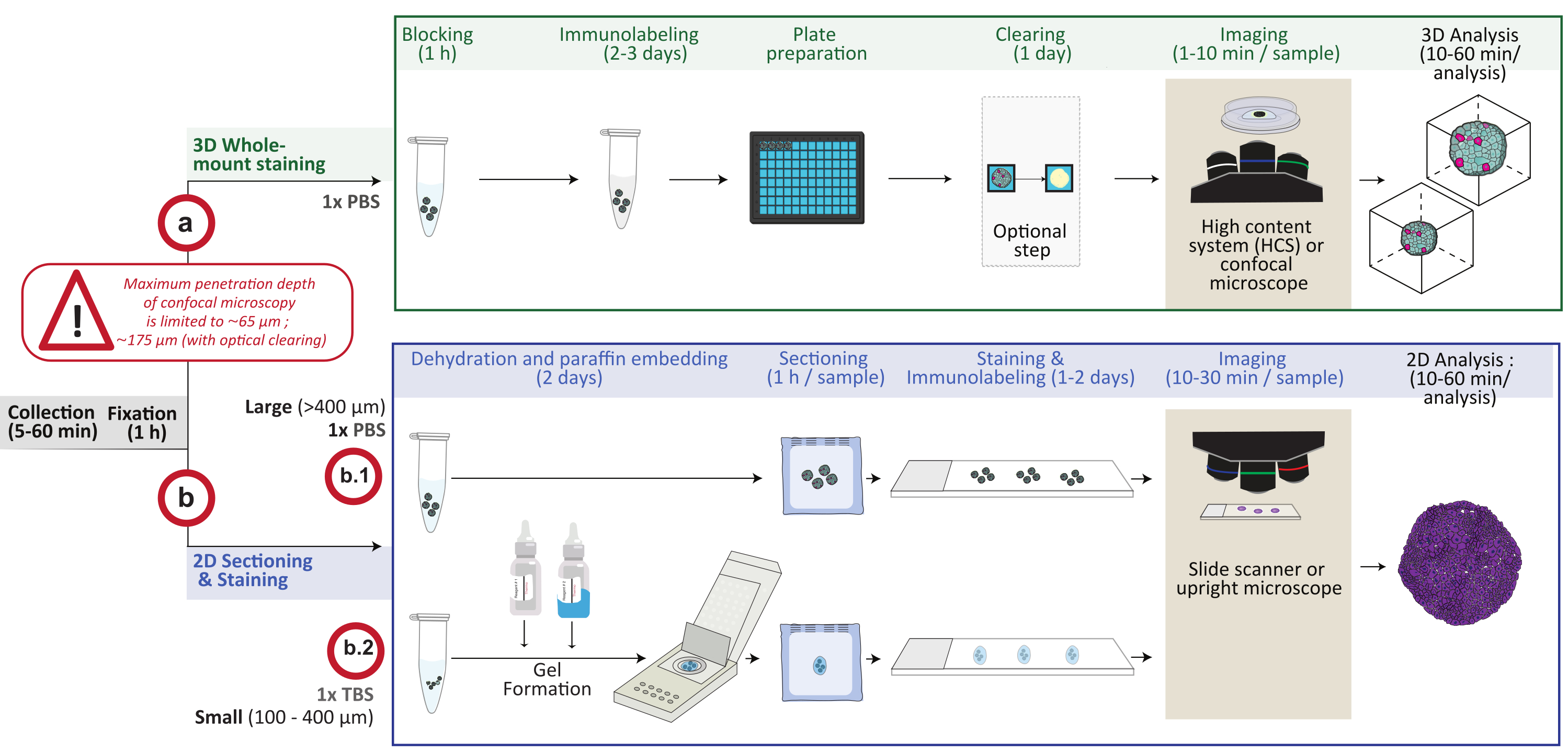{kind=link}
Protocolo
NOTA: Uma perda de ≤25% do número inicial de estruturas 3D deve ser esperada durante as etapas que envolvem mudanças de reagentes e lavagem no procedimento a seguir. Planeje usar um número final de pelo menos dez estruturas 3D, com um tamanho variando de 100 a 500 μm, por condição testada para realizar análises de imagem qualitativas e quantitativas. Se necessário, para estruturas maiores, corte as extremidades das pontas da pipeta de 1 mL para evitar quebrar as estruturas. Para todas as etapas, se a sedimentação da estrutura 3D for muito longa, as células podem ser giradas suavemente a 50 × g por 5 min à temperatura ambiente (RT). Dependendo da questão investigada, as vantagens/desvantagens de tal etapa de fiação devem ser consideradas, pois a centrifugação pode comprometer a forma das estruturas 3D. Evite fiar a >100 × g.
1. Coleta e fixação de modelos de cultura de células 3D
NOTA: Tenha cuidado para não aspirar as estruturas 3D, que serão apenas frouxamente presas à parede do tubo.
- Colheita de modelos de cultura de células 3D incorporados em uma matriz
NOTA: Esta seção descreve a recuperação de estruturas 3D cultivadas em gotas de um extrato de membrana basal do sarcoma murino de Engelbreth-Holm-Swarm (BME), mas pode ser adaptada a outras matrizes. Veja a discussão para pontos cruciais sobre ECM.- Remova o meio de cultura dos poços sem interromper a matriz 3D. Sobrecubra o interior e o exterior de uma ponta de pipeta de 1 ml com proteína (chamada de ponta pré-revestida de 1 ml a seguir) mergulhando todo o comprimento da ponta em albumina sérica bovina a 0,1% (BSA) em solução salina tamponada com fosfato (PBS) (chamada solução de 0,1% PBS-BSA a partir de agora) e pipetando 1 mL desta solução para cima e para baixo duas vezes.
NOTA: Este pré-revestimento impedirá que as células grudem na ponta e minimizará qualquer perda. - Sobrecubra o interior de um tubo de centrífuga (15 mL) com proteína (chamada de tubo de centrífuga pré-revestido doravante) enchendo repetidamente com solução PBS-BSA a 0,1% e esvaziando o tubo.
NOTA: Isso impedirá que as células grudem no tubo e minimizará qualquer perda. - Usando a ponta pré-revestida de 1 mL, ressuspense cuidadosamente as estruturas 3D do poço usando 1 mL de PBS 1x gelado e transfira suavemente a suspensão contendo as estruturas 3D para o tubo de centrífuga pré-revestido.
- Adicione suavemente 13 mL de PBS gelado 1x e permita que as estruturas 3D se sedimentem no gelo por pelo menos 10 min.
NOTA: Se necessário, girar durante 5 min a 50 × g a 4 °C. Evite girar >100 × g, pois isso comprometerá a forma das estruturas 3D. - Remova o sobrenadante. Usando uma ponta pré-revestida de 1 mL, ressuscite suavemente as estruturas 3D em 1 mL de PBS 1x gelado. Repetir os passos 1.1.4 a 1.1.5 para obter um pellet homogéneo sem qualquer resíduo de matriz 3D.
NOTA: A remoção eficiente da matriz é influenciada pelo tipo de matriz, pelo número e pelo tamanho das estruturas 3D e requer otimização para diferentes condições de cultura. Para estruturas 3D cultivadas em BME, a recuperação da remoção da matriz normalmente leva de 45 a 60 minutos. - Usando uma ponta pré-revestida de 1 mL, transfira a suspensão PBS 1x 1x 1 mL contendo as estruturas 3D para um tubo de centrífuga pré-revestido de 1,5 mL e prossiga com a seção 1.3.
- Remova o meio de cultura dos poços sem interromper a matriz 3D. Sobrecubra o interior e o exterior de uma ponta de pipeta de 1 ml com proteína (chamada de ponta pré-revestida de 1 ml a seguir) mergulhando todo o comprimento da ponta em albumina sérica bovina a 0,1% (BSA) em solução salina tamponada com fosfato (PBS) (chamada solução de 0,1% PBS-BSA a partir de agora) e pipetando 1 mL desta solução para cima e para baixo duas vezes.
- Colheita de modelos flutuantes de cultura de células 3D
- Usando uma ponta pré-revestida de 1 mL, colete e transfira cuidadosamente as estruturas 3D para um tubo de centrífuga pré-revestido de 1,5 mL. Deixe as estruturas 3D sedimentarem, ou girem por 5 min a 50 × g em RT.
- Remova o sobrenadante. Usando uma ponta pré-revestida de 1 mL, ressuspeite as estruturas 3D em 1 mL de 1x PBS. Prossiga com a secção 1.3.
- Fixação de modelos de cultura de células 3D
- Permita que as estruturas 3D se sedimentem. Remova cuidadosamente o sobrenadante; sob um exaustor, ressuscite suavemente as estruturas 3D em 1 mL de formalina usando uma ponta pré-revestida de 1 mL.
NOTA: A formalina contém formaldeído, que é perigoso. Manipule o produto químico em um exaustor químico. Use luvas de borracha e óculos de segurança. - Incubar as estruturas 3D por 30 min no RT.
NOTA: Uma etapa de fixação de 30 minutos com formalina é necessária para a imunocoloração de uma ampla gama de estruturas 3D (variando em tamanho, forma e origem). No entanto, em geral, tempos de fixação mais longos (>3 h) são mais adequados para preservar a fluorescência das proteínas repórteres. - Deixe as estruturas 3D sedimentarem ou girem por 5 min a 50 × g no RT. Remova suavemente a formalina e substitua-a por 1 mL de 1x PBS. Repita esta etapa de lavagem em 1x PBS duas vezes. Conservar as amostras a 4 °C e proceder aos pontos 2 ou 3.
NOTA: O protocolo pode ser pausado aqui e as células podem ser mantidas a 4 °C para armazenamento a longo prazo (>1 ano).
- Permita que as estruturas 3D se sedimentem. Remova cuidadosamente o sobrenadante; sob um exaustor, ressuscite suavemente as estruturas 3D em 1 mL de formalina usando uma ponta pré-revestida de 1 mL.
2.3D coloração de montagem inteira, imagens e análise de modelos de cultura de células 3D
NOTA: Como os organoides estão frouxamente presos à parede do tubo, manuseie-os suavemente, pois todas as mudanças de reagentes seguintes podem causar perda de amostra. Antes de começar, certifique-se da disponibilidade dos controles corretos para coloração. Controles positivos e negativos podem ser células, nas quais a proteína de interesse é conhecida por ser superexpressa ou ausente, respectivamente. Incubar amostras sem o anticorpo primário para determinar se o sinal observado é devido à ligação não específica do anticorpo secundário. Como algumas células tendem a exibir altos níveis de autofluorescência, use controles desprovidos de anticorpos secundários para determinar se a fluorescência observada é proveniente da autofluorescência de fundo. A imunomarcação e a visualização do repórter fluorescente podem ser combinadas.
- Coloração de montagem inteira 3D
- Preparar a solução de bloqueio de permeabilização (PB) suplementando 1x PBS com 0,1%-1% de um surfactante não iônico (ver Tabela de Materiais), 1% de dimetilsulfóxido, 1% de BSA e 1% de soro de burro (ou do animal em que os anticorpos secundários foram criados).
NOTA: Otimize cuidadosamente a concentração do surfactante não iônico dependendo da localização do alvo: membrana (0-0,5%), citoplasma (0,5-1%) e núcleo (1%). Esta solução pode ser armazenada a 4°C por até 1 mês. A BSA geralmente funciona bem para a etapa de bloqueio, mas em caso de alto ruído de fundo, realize um teste empírico para obter os melhores resultados possíveis para uma determinada combinação de anticorpos. - Transfira os organoides do tubo de centrífuga de 1,5 mL para um tubo de 0,5 mL usando uma ponta pré-revestida de 1 mL. Deixe o sedimento organoide, remova suavemente o PBS 1x e substitua-o por 0,5 mL de solução de PB. Incubar os organoides com agitação horizontal suave (30-50 rpm) por 1 h no RT.
- Deixe o sedimento organoide, remova suavemente a solução de PB e lave duas vezes em 1 mL PBS-BSA 0,1% por 3 min.
NOTA: Esperar por 3 min permite que as estruturas sedimentem no fundo do tubo. - Remova suavemente o PBS-BSA 0,1% e adicione 250 μL de anticorpo primário diluído na concentração apropriada em solução PB:1x PBS (1:10). Para preparar 10 mL de PB:1x PBS (1:10) solução, diluir 1 mL de PB solução em 9 mL de PBS 1x. Incubar durante 2-3 dias com agitação horizontal suave (30-50 rpm) a 4 °C.
NOTA: Um tempo de incubação de anticorpos apropriado é crucial para uma penetração adequada de anticorpos, pois as estruturas 3D às vezes podem atingir tamanhos grandes. - Deixe o sedimento organoide e remova suavemente a solução primária de anticorpos. Lave 5x em PBS-BSA 0,1% por 3 min por lavagem e depois 2x em 1 mL PBS-BSA 0,1% por 15 min por lavagem com agitação horizontal suave.
- Adicionar 250 μL de anticorpo secundário diluído a 1:250 em solução de PB:1x PBS (1:10). Incubar durante 24 h a 4°C com agitação horizontal suave (30-50 rpm). Para esta etapa, proteja as amostras da luz.
- Adicionar 250 μL de Hoechst 33342 (solução-mãe de 20 μM) diluído a 1:1000 em solução PB:1x PBS (1:10) e incubar durante mais 2 h a 4 °C com uma agitação horizontal suave (30-50 rpm).
- Deixar sedimentar os organoides e remover suavemente a solução que contém o anticorpo secundário + Hoechst 33342. Lave os organoides 5x em 1 mL de 1x PBS por 3 min por lavagem e depois 2x em 1 mL de 1x PBS por 15 min por lavagem com agitação horizontal suave (30-50 rpm).
NOTA: É crucial lavar extensivamente as amostras para evitar ruído de fundo ou perda de sinal. - Armazenar as amostras em PBS a 4 °C até à aquisição da imagem. Prossiga com a secção 2.2.
NOTA: O protocolo pode ser pausado aqui, e as amostras podem ser armazenadas a 4 °C por vários meses, protegidas da luz.
- Preparar a solução de bloqueio de permeabilização (PB) suplementando 1x PBS com 0,1%-1% de um surfactante não iônico (ver Tabela de Materiais), 1% de dimetilsulfóxido, 1% de BSA e 1% de soro de burro (ou do animal em que os anticorpos secundários foram criados).
- Preparação da amostra para imagens confocais
- Usando uma ponta pré-revestida de 1 mL, transfira cuidadosamente os organoides para 50 μL do PBS 1x por poço em uma microplaca de poliestireno preto de 96 poços. Prossiga com a etapa 2.2.3 ou a seção 2.3.
NOTA: Nesta fase, a amostra pode ser protegida da luz e armazenada a 4 °C durante muitas semanas. - Clareira
NOTA: A etapa de limpeza é opcional e pode ser usada para imunomarcar organoides ou para detectar fluorescência endógena. A limpeza pode causar encolhimento da estrutura 3D, mas não altera a morfologia geral, exceto para organoides esféricos monocamadas com grandes lúmens4. Para esses organoides císticos, pule a etapa de limpeza e realize imagens de tecidos profundos6.- Preparar uma solução de depuração de glicerol-frutose a 2,5 M contendo 50% v/v de glicerol, 11% v/v de água destilada e 45% p/v de frutose, misturando num agitador magnético pelo menos durante a noite até que a solução esteja completamente solubilizada e homogénea. Conservar a 4 °C no escuro até 1 mês.
- Remova o máximo de 1x PBS possível sem tocar nos organoides. Adicionar 200 μL da solução de limpeza utilizando uma ponta de pipeta de 1 ml após a remoção da extremidade e ressuspender suavemente para evitar a formação de bolhas. Incubar no RT durante, pelo menos, 12 h e prosseguir com a secção 3.
NOTA: Como a solução de limpeza é viscosa, pequenos volumes são difíceis de manusear. Para facilitar o manuseio, verifique se a solução está em RT e pipeta lentamente. Para uma limpeza ideal, deixe a amostra sedimentar na solução de limpeza por pelo menos 24 horas antes da imagem. Se as estruturas 3D estiverem flutuando no momento da aquisição, execute um giro opcional por 10 minutos a <100 × g no RT, ou permita mais tempo (um a vários dias) para deixá-las sedimentar. O protocolo pode ser pausado nesta etapa antes de prosseguir para a imagem se estiver protegido da luz e armazenado a 4 °C (por semanas) ou -20 °C (por meses).
- Usando uma ponta pré-revestida de 1 mL, transfira cuidadosamente os organoides para 50 μL do PBS 1x por poço em uma microplaca de poliestireno preto de 96 poços. Prossiga com a etapa 2.2.3 ou a seção 2.3.
- Aquisição e análise de imagens
NOTA: A tecnologia de seccionamento de imagens será necessária para criar imagens em estruturas 3D.- Use microscópios confocais e favoreça objetivos de imersão com maior abertura numérica (NA) em comparação com o ar. Escolha os objetivos de ampliação (10x, 20x, 40x) de acordo com o tamanho das estruturas 3D, reconstrução da imagem (costura) e soluções usadas para a análise.
- Ao selecionar o modo de aquisição, leve em consideração a profundidade de foco do objetivo usado para definir a etapa para o empilhamento Z; permitem uma renderização 3D ideal.
NOTA: As soluções de análise de imagem variam e a análise precisará ser ajustada ao software usado. Por exemplo, esse protocolo de análise foi estabelecido em um software de análise de alto conteúdo (consulte Tabela de Materiais e Figura Suplementar 1 para detalhes) e fornece dados sobre segmentação de objetos, cálculo de propriedades e seleção de população de células dentro de um objeto reconstruído em 3D.
3. Seccionamento, coloração, imagem e análise 2D de modelos de cultura de células 3D
NOTA: Os modelos de cultura de células 3D variam em tamanho. Prossiga com as secções 3.1 ou 3.2 para uma incorporação eficiente de parafina (Figura 2). Aguarde tempo suficiente para a sedimentação da estrutura 3D antes de qualquer lavagem e troca de reagentes. Tenha cuidado para não aspirar os organoides que estarão flutuando no fundo do tubo. Para a incorporação de parafina, consulte a Figura 2 para obter orientação.
- Incorporação de parafina de grandes modelos de cultura de células 3D (Ø ≥ 400 μm)
- No dia anterior à incorporação, pré-aqueça dois frascos de 150 mL cheios de parafina (banhos de parafina), um pequeno molde de incorporação de metal por amostra e pinça fina a 65 °C.
- Usando uma ponta pré-revestida de 1 mL, transfira cuidadosamente os organoides em 1x PBS para um tubo de vidro de fundo plano com uma tampa de garrafa revestida de politetrafluoroetileno. Deixe o sedimento organoide, remova cuidadosamente o PBS 1x e substitua-o por etanol a 70%. Incubar por pelo menos 30 min.
- Deixe os organoides sedimentarem e remova cuidadosamente o etanol a 70%. Substitua-o por 1 mL de solução de eosina Y pronta para uso. Mexa o tubo e manche por pelo menos 30 min. Retire cuidadosamente a solução de eosina e desidrate os organoides em três lavagens sucessivas com 1 mL de etanol a 100% por ~30 min cada.
NOTA: O etanol, um líquido inflamável e volátil, causa irritação ocular e do trato respiratório grave. Manipule-o em um capuz de fumaça e use óculos de proteção. - Retire cuidadosamente o etanol a 100% e, sob um exaustor químico, limpe os organoides em 3 lavagens sucessivas com 1 mL de xileno por ~30 min cada.
NOTA: O xileno é um líquido tóxico inflamável cujos vapores podem causar irritação. Manipule-o em um exaustor. Evite o contato direto com a pele e use luvas de borracha e óculos de proteção. - Sob um capô químico, prepare um de tecido microgêmeo branco colocando um pedaço de almofada de biópsia (previamente embebido em xileno) dentro de um dos compartimentos do. Transfira cuidadosamente as estruturas 3D usando uma pipeta Pasteur de plástico pré-revestida de 2 mL para a almofada de biópsia. Cubra-os com outra almofada de biópsia embebida em xileno para evitar que os organoides se movam e feche o.
- Se várias amostras forem processadas, coloque o em um banho de xileno para aguardar o processamento adicional. Uma vez que todas as amostras sejam transferidas para, colocar as num banho de parafina pré-aquecido durante 30 minutos a 65 °C. Transfira as para um banho de parafina pré-aquecido fresco durante a noite.
- Após a impregnação da parafina, pegue um molde de incorporação pré-aquecido e adicione a parafina aquecida a ele. Coloque a almofada de biópsia contendo as estruturas 3D no molde e agite-a suavemente até que todos os organoides caiam para o fundo do molde. Coloque com muito cuidado as estruturas 3D no centro do molde usando pinças finas pré-aquecidas. Prossiga com a secção 3.3.
NOTA: Tenha cuidado para não perturbar as estruturas 3D com a pinça; empurre, mas não os belisque.
- Incorporação de parafina de pequenos modelos de cultura de células 3D (Ø ≤ 400 μm)
- No dia anterior à incorporação, pré-aqueça dois frascos de 150 mL cheios de parafina (banhos de parafina), um pequeno molde de incorporação de metal por amostra e pinça fina a 65 °C.
- Remova cuidadosamente o PBS 1x da suspensão organoide. Realize suavemente 3 lavagens em 1 mL de 1x solução salina tamponada com Tris (TBS). Remova o máximo de 1x TBS possível sem tocar nos organoides.
NOTA: Tenha cuidado para não aspirar a amostra. Se necessário, execute um spin de 5 min a 50 x g no RT. Os vestígios remanescentes de fosfato interferirão nas etapas seguintes, impedindo notavelmente a polimerização do gel. Portanto, não use soluções PBS durante qualquer etapa de processamento. Para esta etapa, um kit comercial, contendo, Reagente #1 (fluido transparente) e Reagente #2 (fluido colorido), foi usado para facilitar o procedimento embutido em parafina sem potencialmente perder pequenos fragmentos (ver Tabela de Materiais). Siga as instruções do kit. As são pré-montadas com papéis de apoio e inserções de cartão já no lugar. - Adicione 2 gotas de Reagente #2 no tubo e misture suavemente batendo no tubo. Adicione 2 gotas de Reagente #1 e misture novamente tocando para fazer o gel solidificar. Usando a pinça fina, retire o gel do tubo e coloque-o no poço do.
- Sob o exaustor, desidratar a amostra colocando o em banhos sucessivos da seguinte forma (usar os frascos de 150 mL e usar etanol fresco ou xileno para cada banho): etanol 70%, 30 min; etanol 96%, 30 min; etanol 100%, três lavagens, 30 min cada; xileno, três lavagens, 30 min cada.
- Coloque as num banho de parafina pré-aquecido durante 30 minutos a 65 °C e transfira-as para um banho de parafina pré-aquecido fresco durante a noite. Após a impregnação da parafina, pegue um molde de incorporação pré-aquecido e adicione parafina aquecida nele. Abra o, desaloje cuidadosamente o gel com pinça fina e coloque o gel que contém as estruturas 3D no centro do molde de incorporação. Prossiga com a secção 3.3.
- Etapas comuns para a incorporação de parafina
- Transfira suavemente o molde para uma área fria para deixar a parafina solidificar em uma camada fina, o que manterá as estruturas 3D na posição apropriada. Adicione um de tecido em cima do molde e adicione parafina quente para cobrir este de plástico. Remova o molde assim que estiver completamente solidificado e prossiga com a secção 3.4.
NOTA: Os blocos de parafina podem ser armazenados à temperatura ambiente durante anos.
- Transfira suavemente o molde para uma área fria para deixar a parafina solidificar em uma camada fina, o que manterá as estruturas 3D na posição apropriada. Adicione um de tecido em cima do molde e adicione parafina quente para cobrir este de plástico. Remova o molde assim que estiver completamente solidificado e prossiga com a secção 3.4.

Figura 2: Visão geral do procedimento de incorporação de parafina de grandes e pequenos modelos de cultura de células 3D in vitro.
(A) Procedimento padrão para a incorporação de parafina. Após a fixação e desidratação, as estruturas 3D são coradas com eosina para facilitar sua visualização (superior e inferior esquerda). As estruturas 3D são cuidadosamente colocadas na almofada de biópsia (azul) no usando um pipeta Pasteur de 2 mL (meio). Após a impregnação da parafina, as estruturas 3D são suavemente jogadas na parafina líquida usando fórceps e suavemente agitadas na almofada de biópsia. Pequenas estruturas 3D são perdidas durante esta etapa, pois não podem ser liberadas da almofada (canto inferior direito: falha na incorporação). Somente grandes estruturas 3D serão incorporadas (canto superior direito: incorporação bem-sucedida). As pontas de seta apontam para culturas 3D. (B) Alternativa ao protocolo padrão de incorporação de parafina. Depois de ter fixado pequenas estruturas 3D, um kit comercial é usado para manter as células em um gel e facilitar sua transferência para o molde após a impregnação da parafina (à direita: incorporação bem-sucedida). Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
- Corte e coloração de blocos
- Corte cortes de 4 μm usando um micrótomo padrão e execute técnicas histológicas e imuno-histoquímicas padrão. Prossiga com a secção 3.5.
NOTA: Slides específicos (ver Tabela de Materiais) foram utilizados para uma melhor adesão das seções. As lâminas podem ser armazenadas à temperatura ambiente ou a 4 °C durante anos.
- Corte cortes de 4 μm usando um micrótomo padrão e execute técnicas histológicas e imuno-histoquímicas padrão. Prossiga com a secção 3.5.
- Aquisição e análise de imagens
- Realize imagens usando um scanner de lâmina digital ou microscópio vertical e analise dados usando uma plataforma para análise quantitativa digital rápida que relata dados de expressão morfológica e multiplexada célula por célula em seções inteiras de estrutura 3D (consulte a Figura 2 Suplementar para obter detalhes).
Observação : O objetivo de 20x é usado rotineiramente por esse grupo.
- Realize imagens usando um scanner de lâmina digital ou microscópio vertical e analise dados usando uma plataforma para análise quantitativa digital rápida que relata dados de expressão morfológica e multiplexada célula por célula em seções inteiras de estrutura 3D (consulte a Figura 2 Suplementar para obter detalhes).
Resultados
Este protocolo fornece uma visão geral das etapas críticas para a coloração 2D e 3D de montagem completa, bem como análises de imagem e quantitativas de modelos de cultura de células 3D (Figura 3 e Figura 4). É aplicável a uma ampla gama de modelos de cultura de células 3D - de esferoides a organoides de diferentes espécies hospedeiras ou tecidos - e permite a aquisição de informações precisas e quantitativas sobre arquitetura, organização celular e interações em níveis celulares e subcelulares (Figura 3 e Figura 4). Os laboratórios podem precisar otimizar as técnicas histológicas e imuno-histoquímicas 2D e as concentrações de anticorpos de acordo com suas próprias necessidades.
Ambos os métodos produzem informações biológicas valiosas. A coloração 3D de montagem completa e a microscopia confocal fornecem informações visuais sobre a composição celular e a posição espacial com um campo de profundidade de até 200 μm (Figura 3B). No entanto, o seccionamento 2D é conveniente para estruturas 3D maiores revelarem características morfológicas celulares detalhadas em toda a seção de estruturas 3D que, de outra forma, podem ser difíceis de observar in situ devido ao espalhamento de luz que compromete a resolução em amostras maiores. Além disso, ambas as técnicas podem fornecer dados quantitativos. De fato, a resolução obtida permite a aplicação de algoritmos de segmentação celular e subcelular para a quantificação do número de células e a detecção da presença de vários marcadores celulares em diferentes subtipos celulares (Figura 3F e Figura 4). Em resumo, as técnicas de imagem aqui descritas são reprodutíveis, simples e complementares e representam ferramentas valiosas para o estudo da heterogeneidade celular.

Figura 3: Resultados representativos para montagem inteira 3D, imagens e análises de seções ópticas 3D e 2D. (A) Imagens confocais de esferoides de glioma humano (h) de alto grau cultivadas por uma semana e marcadas com Hoechst (azul), Olig2 (amarelo) e Actin (vermelho) (20x objetiva de água). Para todas as imagens adquiridas, as configurações do microscópio foram estabelecidas usando um controle positivo (acima) e, em seguida, o controle negativo foi visualizado usando configurações idênticas para controlar a falta de fluorescência na ausência de anticorpos primários (abaixo). (B) Representação ortogonal 3D de toda a montagem da coloração Ki67 realizada em (h) glioma esferoide de alto grau cultivado por uma semana (limpeza de glicerol-frutose; 20x objetiva de água, confocal). (C) Imagens confocais de (h) esferoide de glioma de alto grau cultivadas durante uma semana e marcadas com Hoechst (azul), Olig2 (amarelo) e Faloidina-488 (verde) (limpeza de glicerol-frutose; 20x objetivo de água). (D) Imagens confocais de esferoides humanos (h) rabdomiossarcoma (superior) e de células da crista neural do rato (m) (em baixo) cultivados durante uma semana e marcados com Hoechst (azul), Actina (vermelho) e Ki67 (verde), respetivamente (depuração de glicerol-frutose; 20x objetivo seco). (E) Imagens confocais de (h) esferoide de glioma de alto grau cultivadas durante uma semana e marcadas com Hoechst (azul) e Ki67 (verde) (limpeza de glicerol-frutose; 40x objetiva de água) (canto superior esquerdo). As imagens segmentadas no canal Hoechst e nas regiões nucleares Ki67-positivas (+) no canal verde foram geradas utilizando software de análise de alto teor (ver Figura 1 Suplementar e Tabela de materiais) (abaixo). A saída dada é a porcentagem de núcleos Ki67+ por estrutura 3D segmentada (canto superior direito). Barra de escala = 100 μm. Clique aqui para ver uma versão maior desta figura.
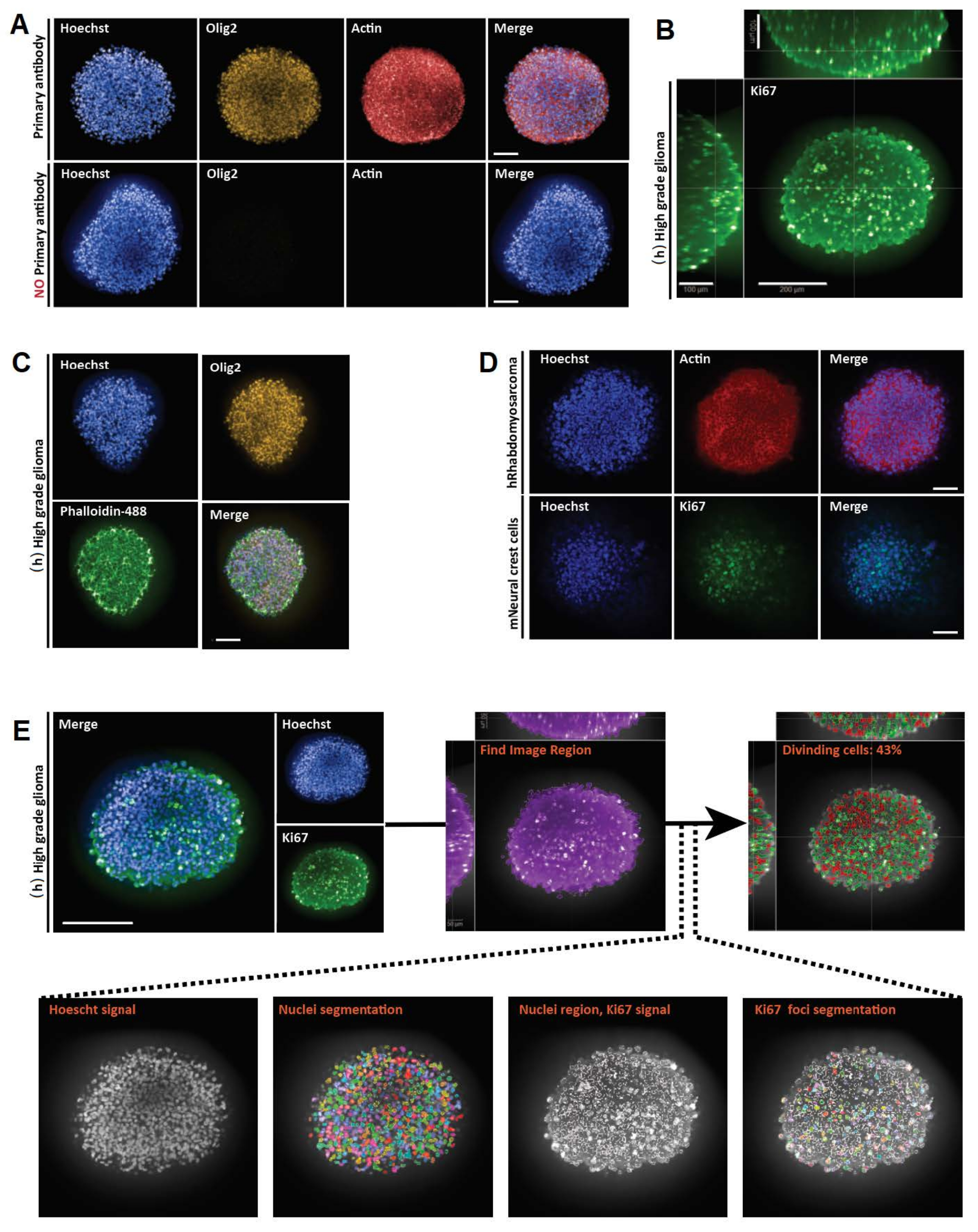{kind=link}

Figura 4: Resultados representativos para imagens e análises de seções ópticas 2D. (A, D) Imagens de seção 2D de um modelo de célula 3D (esferoides de rabdomiossarcoma humano cultivados por um mês) obtidas com um scanner de lâmina digital e analisadas em uma plataforma para análise quantitativa digital rápida. (A) Coloração H&E e detecção de células de acordo com o seu tamanho. Barra de escala = 500 μm. (B) O histograma mostra a porcentagem de células < 100 μm 2 e > 100 μm2 detectadas usando software para análise quantitativa digital rápida (esquerda: Halo) ou contagem manual (direita: MC). (C) Coloração Ki67 e detecção de células de acordo com a intensidade do seu sinal de 3,3'-diaminobenzidina (DAB). Negativo (azul), fracamente positivo (amarelo), positivo (vermelho). Barra de escala = 500 μm. (D) O histograma mostra a porcentagem de células Ki67-negativas, fracamente positivas e positivas. Abreviaturas: H&E = hematoxilina e eosina; MC = contagem manual. Por favor, clique aqui para ver uma versão maior desta figura.
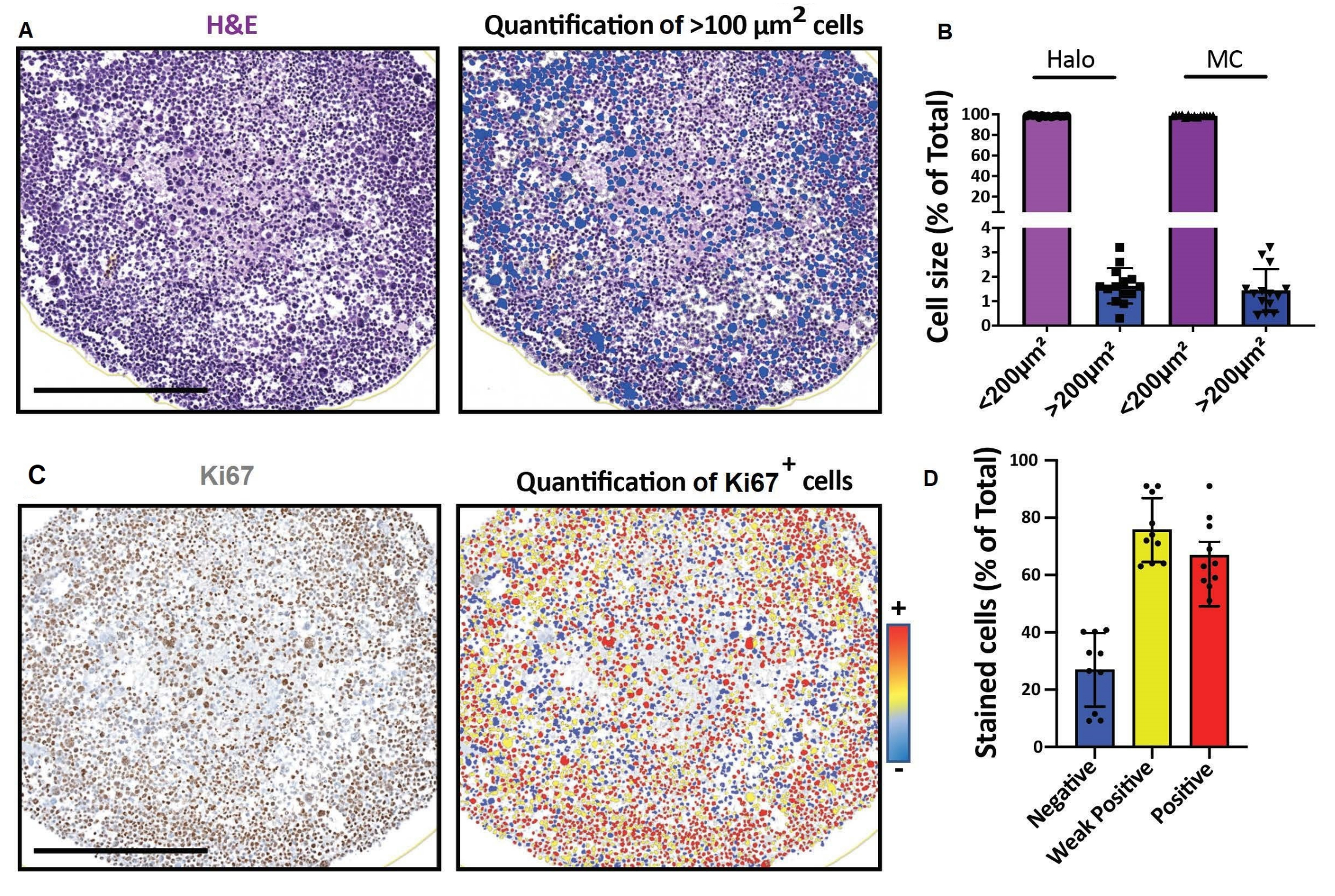{kind=link}
Figura 1 Suplementar: Visão geral das etapas no software de análise de imagens. As análises são baseadas na associação de blocos de construção. Cada bloco de construção corresponde a uma função - segmentação, cálculo, associação, definição de saída - e oferece vários algoritmos e seleções de variáveis para corresponder à amostra biológica que está sendo visualizada. O software fornece vários protocolos de análise RMS (Ready Made Solution) que podem ser facilmente usados e modificados. Os protocolos integrados de análise de imagem podem ser salvos, aplicados a diferentes conjuntos de dados e compartilhados entre usuários. Resumidamente, o protocolo de análise implica segmentação sequencial de objetos: esferoide, núcleos e, finalmente, bolsas Ki67 (A488). Em seguida, a intensidade média dos bolsões Ki67 é calculada para discriminar ainda mais os eventos positivos. Finalmente, os núcleos que abrangem bolsas positivas Ki67 são selecionados positivamente. Clique aqui para baixar este arquivo.
Figura 2 Suplementar: Visão geral das etapas do procedimento do software de análise quantitativa. Passo 1. Carregue os arquivos usando a guia Estudos. Os arquivos serão abertos na seção Ações de imagem. Passo 2. Abra a guia Anotações e clique em Ações de camada para criar uma nova camada ao redor da estrutura usando a ferramenta de círculo da barra de ferramentas. Para estruturas não circulares, a ferramenta caneta pode ser usada. Passo 3. A barra de ferramentas pode ser usada para projetar anotações e visualizar a quantificação com a  ferramenta. Passo 4. Abra a guia Análise e selecione as melhores condições para a análise da amostra (vários ensaios podem ser necessários aqui). Passo 4.1. Use a seção Seleção de manchas para configurar a condição de coloração. No caso de várias manchas, elas podem ser adicionadas e renomeadas, e a cor virtual pode ser modificada. A detecção de localização pode ser especificada - coloração nuclear ou citoplasmática. Passo 4.2. Use a seção Detecção de Célula para configurar a detecção de célula. Esta seção será a mais importante para a análise. A seção Limiar de Contraste Nuclear permitirá a detecção de todos os núcleos. Atenção deve ser dada no caso de haver vários tamanhos de população, o software pode detectar várias células em vez de uma grande única. As seções de tamanho nuclear e agressividade da segmentação nuclear podem ser usadas para quantificar as faixas populacionais de tamanho celular. Passo 5. Descrição sobre como executar a análise de amostra. Siga as etapas mostradas na figura. A seção Camada de Anotação executará a configuração somente neste slide. A quantificação pode ser visualizada usando a ferramenta. Repita os passos 4.1-5 até que a quantificação adequada seja alcançada. Passos 6-6.1. Essas etapas permitem que você desenhe uma figura usando o software. Passo 7. Gráficos de quantificação obtidos via software podem ser salvos. Passo 8. Os dados podem ser exportados. Clique aqui para baixar este arquivo.
ferramenta. Passo 4. Abra a guia Análise e selecione as melhores condições para a análise da amostra (vários ensaios podem ser necessários aqui). Passo 4.1. Use a seção Seleção de manchas para configurar a condição de coloração. No caso de várias manchas, elas podem ser adicionadas e renomeadas, e a cor virtual pode ser modificada. A detecção de localização pode ser especificada - coloração nuclear ou citoplasmática. Passo 4.2. Use a seção Detecção de Célula para configurar a detecção de célula. Esta seção será a mais importante para a análise. A seção Limiar de Contraste Nuclear permitirá a detecção de todos os núcleos. Atenção deve ser dada no caso de haver vários tamanhos de população, o software pode detectar várias células em vez de uma grande única. As seções de tamanho nuclear e agressividade da segmentação nuclear podem ser usadas para quantificar as faixas populacionais de tamanho celular. Passo 5. Descrição sobre como executar a análise de amostra. Siga as etapas mostradas na figura. A seção Camada de Anotação executará a configuração somente neste slide. A quantificação pode ser visualizada usando a ferramenta. Repita os passos 4.1-5 até que a quantificação adequada seja alcançada. Passos 6-6.1. Essas etapas permitem que você desenhe uma figura usando o software. Passo 7. Gráficos de quantificação obtidos via software podem ser salvos. Passo 8. Os dados podem ser exportados. Clique aqui para baixar este arquivo.
Discussão
A cultura de células é uma ferramenta indispensável para descobrir mecanismos biológicos fundamentais envolvidos no desenvolvimento de tecidos e órgãos, função, regeneração e ruptura e doença. Embora a cultura de células 2D monocamadas tenha predominado, pesquisas recentes mudaram para culturas que geram estruturas 3D mais reflexivas das respostas celulares in vivo, devido notavelmente à organização espacial adicional e aos contatos célula-célula que influenciam a expressão gênica e o comportamento celular e, portanto, poderiam fornecer dados mais preditivos7. No entanto, muitos desafios permanecem, incluindo a necessidade de técnicas de coloração e imagem fáceis de usar para visualização microscópica detalhada e avaliação de estruturas 3D complexas nos níveis celular e subcelular. Nesse contexto, protocolos detalhados, robustos e complementares foram fornecidos para realizar a coloração e imagens de resolução celular e subcelular de modelos de cultura celular 3D fixa in vitro que variam de 100 μm a vários milímetros de tamanho.
Este procedimento apresenta duas estratégias diferentes para lidar com uma grande variedade de tamanhos e tipos de modelos de cultura de células 3D in vitro. A escolha de um (análise de montagem inteira 3D) ou outro (análise de seccionamento 2D) dependerá do modelo utilizado e do problema investigado. A análise 3D de montagem completa por microscopia confocal permite a visualização de células com um campo de profundidade de até 200 μm, independentemente do tamanho total da estrutura 3D, enquanto o seccionamento 2D é aplicável a amostras de qualquer tamanho, mas a visualização permanece dimensional 2D. Abaixo estão algumas sugestões para solução de problemas e considerações técnicas.
A perda de estruturas 3D durante o fluxo de trabalho é a desvantagem mais comum. Eles podem permanecer aderentes às pontas e tubos, e é por isso que as pontas e tubos de pré-revestimento com solução PBS-BSA 0,1% são fundamentais. Além disso, é crucial deixar as estruturas 3D sedimentarem entre as mudanças de reagentes e realizar toda a pipetagem com muito cuidado. Como mencionado no procedimento, para todas as etapas, se a sedimentação da estrutura 3D for muito longa, as células podem ser giradas suavemente a 50 × g por 5 min no RT. Dependendo do objetivo do estudo, as vantagens/desvantagens de tal etapa de fiação devem ser consideradas, pois a centrifugação pode comprometer a forma das estruturas 3D. Além disso, deve-se tomar cuidado para preservar essa morfologia durante a etapa de fixação, pois os organoides císticos tendem a entrar em colapso. A fixação de estruturas com menos de 400 μm de tamanho deve evitar mudanças estruturais.
Para uma imunomarcação ideal, a recuperação de organoides de suas matrizes 3D é um passo crucial. A matriz 3D pode impedir a penetração adequada de anticorpos ou levar a uma alta coloração de fundo devido à ligação não específica à matriz. A remoção da ECM pode alterar a morfologia dos segmentos externos dos organoides (notavelmente no caso de pequenas saliências celulares que se estendem das estruturas 3D estudadas) e dificultar parcialmente as análises. Para tais estruturas 3D, a matriz pode ser mantida durante todo o procedimento; no entanto, as condições de cultura devem ser cuidadosamente adaptadas para cultivar células em uma quantidade mínima de matriz, a fim de evitar a penetração insuficiente de soluções e anticorpos e evitar sucessivas etapas de lavagem destinadas a reduzir o ruído de fundo excessivo 6,8.
A etapa de limpeza óptica descrita neste protocolo na seção de coloração de montagem inteira 3D é pertinente para a imagem de estruturas 3D de até 150-200 μm de profundidade, em vez de 50-80 μm sem limpeza. Em comparação com outras metodologias de compensação que muitas vezes requerem várias semanas e utilizam agentes de compensação tóxicos, uma etapa de compensação rápida e segura publicada anteriormente foi utilizada neste protocolo 4,9. Além disso, essa etapa de limpeza é reversível e novos anticorpos podem ser adicionados à coloração inicial sem perda de resolução ou brilho4. No entanto, dependendo do modelo de cultura de células 3D estudado, uma profundidade de 150-200 μm pode não ser suficiente para a imagem da estrutura 3D de forma informativa, e esse protocolo de limpeza pode causar alterações na morfologia geral de organoides esféricos, monocamadas, com grandes lúmens4. Os usuários devem projetar cuidadosamente seu experimento e, se necessário, otimizar o tempo da etapa de permeabilização/bloqueio (para permitir a penetração de anticorpos e solução), a etapa de limpeza (para penetrar mais profundamente em 200 μm, os espécimes devem ser totalmente limpos) e a aquisição de imagens. As duas tecnologias mais prevalentes disponíveis nas instalações centrais seriam a folha de luz e a microscopia confocal. Os usuários precisarão escolher cuidadosamente uma tecnologia com base no tamanho de suas estruturas 3D e sua questão biológica10. No entanto, em comparação com a microscopia confocal, a resolução da microscopia de folha de luz obtida para essas estruturas profundas permanece subótima para a obtenção de resolução subcelular.
Aqui, um processo detalhado e robusto foi relatado que é dedicado à incorporação de parafina de amostras únicas. Curiosamente, Gabriel et al. desenvolveram recentemente um protocolo que incorpora culturas de células 3D em parafina com um rendimento aumentado. Eles usaram um molde de polidimetilsiloxano (PDMS) para confinar 96 estruturas 3D em um padrão de microarray em um bloco, fornecendo novas perspectivas para estudos em modelos de tumores 3D abrangendo mais grupos, pontos de tempo, condições de tratamento e replicações11. No entanto, este método requer extensas habilidades e máquinas, nomeadamente para a fabricação do pré-molde usado para criar moldes PDMS.
Em resumo, este artigo descreve duas abordagens diferentes, complementares e adaptáveis, permitindo a aquisição de informações precisas e quantitativas sobre a composição arquitetônica e celular de modelos celulares 3D. Ambos os parâmetros são cruciais para o estudo de processos biológicos, como a heterogeneidade celular intratumoral e seu papel na resistência aos tratamentos.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Este trabalho foi apoiado pelo Prêmio de Inovação Robert J. Arceci #604303 da St Baldrick.
Materiais
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Biopsy pad Q Path blue | VWR | 720-2254 | |
| Cassettes macrostar III Blc couv. Char. x1500 | VWR | 720-2233 | |
| Cassette microtwin white | VWR | 720-2183 | |
| Chemical hood | Erlab | FI82 5585-06 | |
| Filter tips 1000 µL | Star lab Tip-One | S1122-1730 | |
| Fine forceps | Pyramid innovation | R35002-E | |
| Flat-bottom glass tubes with PTFE lined 2 mL | Fisher Scientific | 11784259 | Excellent for environmental samples, pharmaceuticals and diagnostic reagents. PTFE is designed for the ultimate in product safety. PTFE provides totally inert inner seal and surface facing the sample or product. |
| Glass bottom dish plate 35 mm | Ibedi | 2018003 | |
| Horizontal agitation | N-BIOTEK | NB-205 | |
| Incubator prewarmed to 65 °C | Memmert Incubator | LAB129 | |
| Inox molds 15x15 | VWR | 720-1918 | |
| Microscope Slides Matsunami TOMO-11/90 | Roche diagnostics | 8082286001 | these slides are used for a better adhesion of sections |
| Microtome | Microm Microtech France | HM340E | |
| Panoramic scan II | 3dhistech | 2397612 | |
| Paraffin embedding equipment | Leica | EG1150C | |
| Plastic pipette Pasteur 2 mL | VWR | 612-1681 | |
| Q Path flacon 150mL cape blanc x250 | VWR | 216-1308 | Good for environmental samples, pharmaceuticals and diagnostic reagents. Polypropylene (PP) are rigid, solid, provide excellent stress crack and impact resistance and have a good oil and alcohol barrier and chemical resistance. PE-lined cap is stress crack resistant and offers excellent sealing characteristics. |
| Set of micropipettors (p200, p1000) | Thermo Scientific | 11877351 (20-200) 11887351(p1000) | |
| OPERA PHENIX | PerkinElmer | HH14000000 | |
| SP5 inverted confocal microscope | Leica | LSM780 | |
| Tissue cassette | VWR | 720-0228 | |
| Zeiss Axiomager microscope | Leica | SIP 60549 | |
| Reagent | |||
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7030-100G | |
| Cytoblock (kit) | Thermofisher Scientific | 10066588 | |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | 57648266 | CAUTION: toxic and flammable. Vapors may cause irritation. Manipulate in a fume hood. Avoid direct contact with skin. Wear rubber gloves, protective eye goggles. |
| Eosin aqueous 1% | Sigma-Aldrich | HT110316 | |
| Ethanol 96% | VWR | 83804.360 | CAUTION: Causes severe eye irritation. Flammable liquid and vapor. Causes respiratory tract irritation. Manipulate in a fume hood. Wear protective eye goggles. |
| Ethanol 100% | VWR | 20821.365 | CAUTION: Causes severe eye irritation. Flammable liquid and vapor. Causes respiratory tract irritation. Manipulate in a fume hood. Wear protective eye goggles. |
| Formalin 4% | Microm Microtech France | F/40877-36 | CAUTION: Formalin contains formaldehyde which is hazardous. Manipulate in a fume hood. Avoid direct contact with skin. Wear rubber gloves and protective eye goggles. |
| Fructose | Sigma-Aldrich | F0127 | |
| Gill hematoxylin type II | Microm Microtech France | F/CP813 | |
| Glycerol | Sigma-Aldrich | G5516 | 500 mL |
| Hoechst 33342 | Life Technologies | H3570 | CAUTION: Suspected of causing genetic defects. Avoid direct contact with skin. Wear rubber gloves and protective eye goggles. |
| Normal donkey serum | Sigma-Aldrich | D9663 | 10 mL |
| Paraffin Wax tek III | Sakura | 4511 | |
| Phosphate Buffer Saline (PBS) 1 X | Gibco | 14190-094 | |
| Tris-Buffered Saline (TBS) 10X | Microm Microtech France | F/00801 | 100 mL |
| Triton X-100 | Sigma-Aldrich | T8532 | CAUTION: Triton X100 is hazardous. Avoid contact with skin and eyes. |
| Xylene | Sigma-Aldrich | 534056 | CAUTION: Xylene is toxic and flammable. Vapors may cause irritation. Manipulate in a fume hood. Avoid direct contact with skin. Wear rubber gloves, protective eye goggles. |
| Solutions | |||
| Clearing solution | Glycerol-Fructose clearing solution is 60% (vol/w) glycerol and 2.5 M fructose. To prepare 10 mL of this solution, mix 6 mL of glycerol and 4.5 g of fructose. Complete to 10 mL with dH2O. Use a magnetic stirrer overnight. Refractive index = 1.4688 at room temperature (RT: 19–23 °C). Store at 4 °C in dark for up to 1 month. | ||
| PBS-BSA 0,1% solution | To prepare 0,1% (vol/wt) PBS-BSA 0,1% solution, dissolve 500 mg of BSA in 50 mL of PBS-1X (store at 4°C for up to 2 weeks). And dilute 1mL of this solution into 9mL of PBS-1X. This solution can be used to precoat the tip and centrifugation tube. | ||
| Permeabilisation-blocking solution (PB solution) | The PBSDT blocking solution is PBS-1X supplemented with 0.1% – 1% Tritonx-100 (depending on the protein localization membrane/nucleus), 1% DMSO, 1% BSA and 1% donkey serum (or from the animal in which the secondary antibodies were raised). This solution can be stored at 4°C for up to 1 month. | ||
| PB:PBS-1X (1:10) solution | PB:PBS-1X (1:10) solution is a 10 time diluted PB solution. To prepare 10 mL of this solution dilute 1 mL of PB solution in 9 mL of PBS-1X. | ||
| Software | |||
| Halo software | Indicalabs | NM 87114 | |
| Harmony software | PerkinElmer | HH17000010 |
Referências
- Ryu, N. E., Lee, S. H., Park, H. Spheroid culture system methods and applications for mesenchymal stem cells. Cells. 8 (12), 1-13 (2019).
- Bartfeld, S., Clevers, H. Stem cell-derived organoids and their application for medical research and patient treatment. Journal of Molecular Medicine. 95 (7), 729-738 (2017).
- Cui, X., Hartanto, Y., Zhang, H. Advances in multicellular spheroids formation. Journal of the Royal Society, Interface. 14 (127), (2017).
- Dekkers, J. F., et al. High-resolution 3D imaging of fixed and cleared organoids. Nature Protocols. 14, 1756-1771 (2019).
- Broutier, L., et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nature Protocols. 11 (9), 1724-1743 (2016).
- Rezanejad, H., Lock, J. H., Sullivan, B. A., Bonner-Weir, S. Generation of pancreatic ductal organoids and whole-mount immunostaining of intact organoids. Current Protocols in Cell Biology. 83 (1), 82 (2019).
- Edmondson, R., Broglie, J. J., Adcock, A. F., Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay and Drug Development Technologies. 12 (4), 207-218 (2014).
- McCray, T., Richards, Z., Marsili, J., Prins, G. S., Nonn, L. Handling and assessment of human primary prostate organoid culture. Journal of Visualized Experiments: JoVE. (143), e59051 (2019).
- Ueda, H. R., et al. Tissue clearing and its applications in neuroscience. Nature Reviews: Neuroscience. 21 (2), 61-79 (2020).
- Lazzari, G., et al. Light sheet fluorescence microscopy versus confocal microscopy: in quest of a suitable tool to assess drug and nanomedicine penetration into multicellular tumor spheroids. European Journal of Pharmaceutics and Biopharmaceutics. 142, 195-203 (2019).
- Gabriel, J., Brennan, D., Elisseeff, J. H., Beachley, V. Microarray embedding/sectioning for parallel analysis of 3D cell spheroids. Scientific Reports. 9, 16287 (2019).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados