
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Imagerie en direct à haut débit des microcolonies pour mesurer l’hétérogénéité de la croissance et de l’expression des gènes
Dans cet article
Résumé
Les phénotypes de croissance de levure sont précisément mesurés par l’imagerie time-lapse très parallèle des cellules immobilisées se développent dans les microcolonies. Simultanément, la tolérance au stress, l’expression des protéines et la localisation des protéines peuvent être surveillées, générant des ensembles de données intégrés pour étudier comment les différences environnementales et génétiques, ainsi que l’hétérogénéité de l’expression génique entre les cellules isogéniques, modulent la croissance.
Résumé
Des mesures précises de l’hétérogénéité entre les souches et à l’intérieur de la souche dans les taux de croissance microbienne sont essentielles pour comprendre les apports génétiques et environnementaux dans la tolérance au stress, la pathogénie et d’autres composantes clés de la condition physique. Ce manuscrit décrit un test au microscope qui suit environ 105microcoloniesSaccharomyces cerevisiae par expérience. Après l’imagerie automatisée en accéléré de levure immobilisée dans une plaque multiwell, les taux de croissance de la microcolonie sont facilement analysés à l’aide d’un logiciel d’analyse d’image personnalisé. Pour chaque microcolonie, l’expression et la localisation des protéines fluorescentes et la survie du stress aigu peuvent également être surveillées. Cet essai permet une estimation précise des taux de croissance moyens des souches, ainsi qu’une mesure complète de l’hétérogénéité de la croissance, de l’expression des gènes et de la tolérance au stress au sein des populations clonales.
Introduction
Les phénotypes de croissance contribuent de façon critique à la condition physique de levure. La sélection naturelle peut faire une distinction efficace entre les lignées dont les taux de croissance diffèrent par l’inverse de la taille effective de la population, qui peut dépasser 108 individus1. En outre, la variabilité des taux de croissance entre les individus au sein d’une population est un paramètre d’évolution pertinent, car il peut servir de base à des stratégies de survie telles que lacouverture des paris 2,3,4,5,6. Par conséquent, les analyses qui permettent des mesures très précises des phénotypes de croissance et de leurs distributions sont essentielles pour l’étude des micro-organismes. L’analyse de croissance de la microcolonie décrite ici peut générer des mesures individuelles du taux de croissance pour ~105 microcolonies par expérience. Cet essai fournit donc un protocole puissant pour étudier la génétique évolutive de levure et la génomique. Il se prête particulièrement bien à tester comment la variabilité au sein des populations de cellules individuelles génétiquement identiques est générée, maintenue et contribue à la condition physique de la population7,8,9,10.
La méthode décrite ici (figure 1) utilise périodiquement capturées, images de faible grossissement brightfield des cellules qui poussent dans les médias liquides sur une plaque de fond de verre de 96 ou 384 puits pour suivre la croissance en microcolonies. Les cellules adhèrent à la concanavalin A de lectine, qui recouvre le fond de la plaque de microscope, et forment des colonies bidimensionnelles. Puisque les microcolonies se développent dans un monocouche, la zone de microcolonie est fortement corrélée avec le numéro de cellule7. Par conséquent, des estimations précises du taux de croissance de la microcolonie et du temps de décalage peuvent être générées avec un logiciel personnalisé d’analyse d’image qui suit le taux de changement de la zone de chaque microcolonie. En outre, la configuration expérimentale peut surveiller les abondances et même les localisations subcellulaires des protéines étiquetées fluorescentes exprimées dans ces microcolonies. Le traitement en aval des données de cet essai de croissance de microcolonie peut être réalisé par analyse personnalisée ou par des logiciels existants d’analyse d’image, tels que Processing Images Easily (PIE)11, un algorithme pour la reconnaissance robuste de la zone de colonie et l’analyse de croissance à haut débit à partir de faible grossissement, images brightfield, qui est disponible via GitHub12.
Étant donné que les estimations du taux de croissance dérivées de l’analyse de microcolonie-croissance sont générées à partir d’un grand nombre de mesures d’une seule colonie, elles sont extrêmement précises, avec des erreurs standard plusieurs ordres de grandeur plus petits que les estimations elles-mêmes pour une expérience de taille raisonnable. Par conséquent, la puissance de l’analyse pour détecter les différences de taux de croissance entre les différents génotypes, traitements ou conditions environnementales est élevée. Le format multiwell-plaque permet de comparer de nombreuses combinaisons d’environnement et de génotypes différentes en une seule expérience. Si les souches expriment constitutivement différents marqueurs fluorescents, elles peuvent être mélangées dans le même puits et distinguées par une analyse d’image ultérieure, ce qui pourrait augmenter encore la puissance en permettant une normalisation des données bien par puits.

Figure 1: Représentation schématique du protocole. Ce protocole suit deux étapes principales, qui sont la préparation de la plaque expérimentale et la préparation des cellules à l’image. La randomisation des plaques et la croissance des cellules doivent être effectuées avant et jusqu’au jour de l’expérience. Le mélange répété des cellules à chaque étape pendant la dilution est impératif dans les étapes jusqu’au placage, et donc la préparation de la plaque expérimentale d’abord est recommandée afin qu’elle soit prête pour le placage immédiatement après l’achèvement de la dilution cellulaire. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
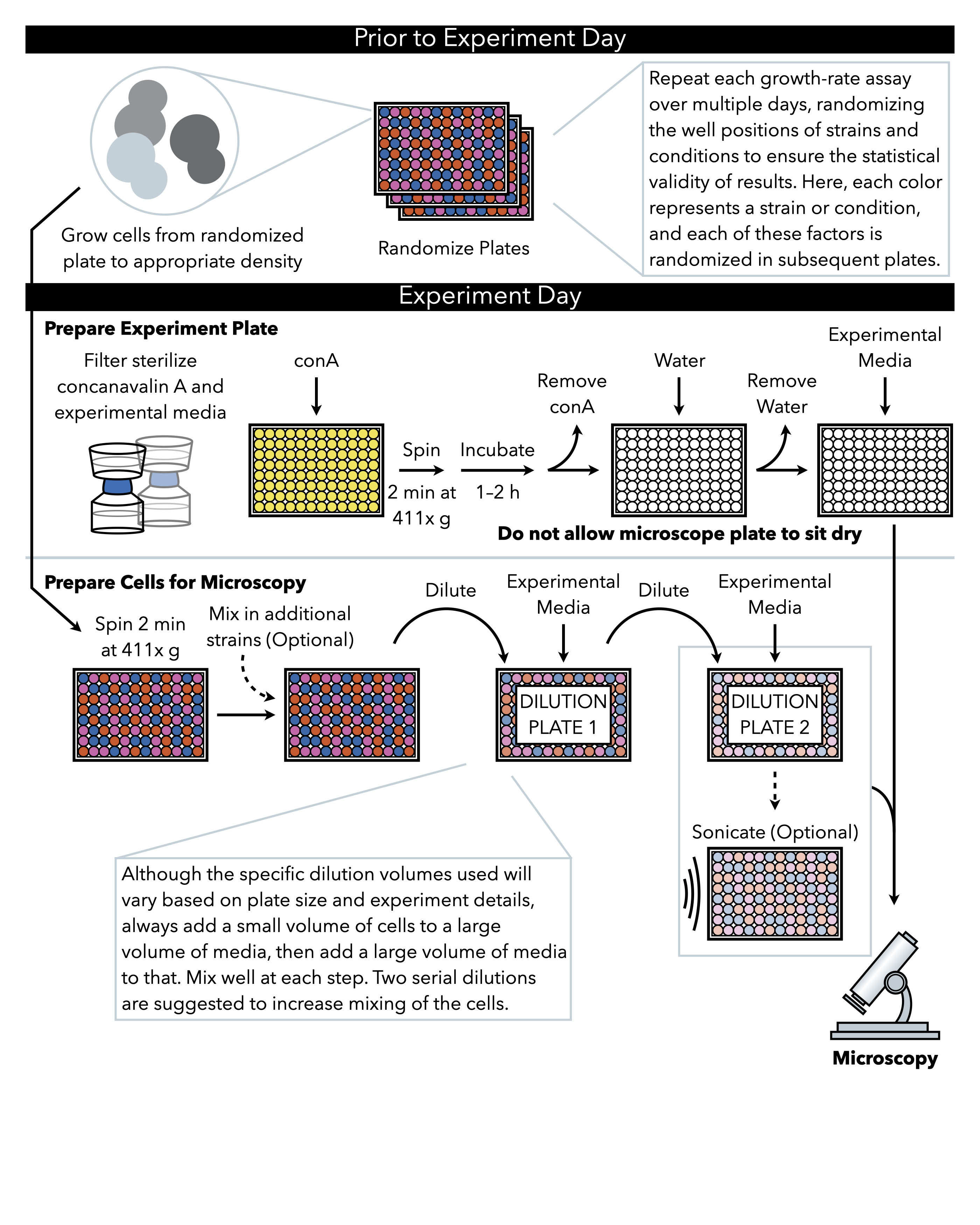{kind=link}
Protocole
1. Préparation de plaques randomisées (avant le jour de l’expérience)
- Planifiez les souches et les conditions à tester avec l’analyse de croissance. À ce stade, assigner au hasard des souches et des conditions à n’importe quel puits.
REMARQUE : Lors de l’examen de la configuration de la plaque, il est conseillé d’inclure plus d’une réplique par souche et état de croissance sur une seule plaque pour tenir compte du bruit bien apparenté dans les mesures. Voir Discussion pour plus de détails. - Calculez aléatoirement l’emplacement de chaque souche et l’état de l’environnement pour les répliques de plaques qui seront exécutés à des jours différents.
- Cultiver toutes les cellules qui seront utilisées dans l’expérience à saturation dans la levure extrait-peptone-dextrose (YEPD; 2% de glucose) milieu dans un shaker à 30 °C (ou toute autre température appropriée).
- Créez les plaques de stock randomisées manuellement ou avec un robot de manipulation liquide. Ajouter 10 μL des cellules saturées désignées à chaque puits d’une plaque stérile de culture tissulaire à fond U. Si plusieurs souches seront testées dans un seul puits, ne les combinez pas à ce stade; cette combinaison se fera juste avant les dilutions cellulaires le jour de l’expérience pour s’assurer que toutes les souches sont aux concentrations correctes lorsqu’elles sont plaquées cellules fondatrices de microcolonie.
- Ajouter 10 μL de 30% de glycérol à chaque puits de chaque assiette. Pipet de haut en bas de sorte que les cellules et le glycérol deviennent bien mélangés.
- Sceller chaque plaque avec un couvercle de papier d’aluminium et congeler immédiatement à -70 °C jusqu’à ce qu’elle soit prête à l’emploi.
REMARQUE : Il est important de créer toutes les plaques randomisées le même jour et de les congeler, de sorte que les conditions de pré-croissance des cellules de chaque plaque auront été identiques et ne généreront pas de variation technique dans l’analyse du taux de croissance.
2. Pré-croissance de la levure
REMARQUE : En règle générale, cela commence avant le jour de l’expérience et dépend fortement de la question expérimentale. Voir Discussion pour plus de détails.
- Retirer une plaque de bouillon (levure de 10 μL, 10 μL de glycérol par puits) du congélateur de -70 °C et ajouter 180 μL du média à utiliser pour l’expérience. Si l’expérience sera menée à l’aide de supports limitant les nutriments, ne pas pré-cultiver la levure à saturation dans le média limitant les nutriments que la sporulation de la levure peut se produire. Au lieu de cela, pré-croissance dans les médias non limitatifs.
- Faire pousser la levure en secouant à 30 °C. Examinez s’il faut faire fonctionner l’essai en commençant par les cellules en phase de journal ou en phase stationnaire pour déterminer si la dilution des cellules plusieurs fois avant l’expérience sera nécessaire. Si l’on s’attend à ce que les souches ou les conditions de levure dans l’essai aient des taux de croissance sensiblement différents, alors une période de pré-croissance de deux jours sera nécessaire pour que toutes les différentes conditions atteignent la phase stationnaire.
3. Configuration du microscope
- Préparation de plaque de microscope
- Assurez-vous que l’incubateur de microscope est en place et chauffez la chambre de microscope à la température de croissance désirée pour les conditions expérimentales. Pour les expériences standard utilisant les cellules de Saccharomyces cerevisiae, l’incubateur doit chauffer la chambre de microscope à 30 °C pour s’assurer que les conditions de croissance des cellules seront correctes pendant l’analyse du taux de croissance.
- Assainir l’établi, les pipettes et autres outils avec 70 % d’éthanol. Récupérez une plaque de microscope et placez-la sur le banc au-dessus d’un lingette sans peluche et statique.
REMARQUE : Ne touchez jamais le fond de la plaque de microscope, même avec des gants, et placez toujours la plaque de microscope sur le dessus d’un lingette sans peluche et statique chaque fois qu’elle touche n’importe quelle surface. Cela empêche les bavures ou les rayures d’empêcher les mesures du taux de croissance une fois que l’expérience est imitée. - Décongeler 5 mL de solution 5x concanavalin A, diluer à 1x avec de l’eau, et filtrer stériliser à travers une seringue équipée d’un filtre de 0,2-μm.
- Filtre stériliser tous les autres liquides qui seront utilisés dans l’analyse avec un filtre de 0,2 μm, y compris les médias expérimentaux, pour enlever les cristaux ou les débris qui peuvent avoir matérialisé dans les solutions. La présence de cristaux réduirait la qualité des images de microscopie.
- Pipet 200 μL de concanavalin Une solution dans chaque puits de la plaque de microscope.
- Centrifugeuse de la plaque pendant 2 min à 411 x gravité (g) avec un lingette sans peluche et statique sous la plaque, pour s’assurer que la solution concanavalin A couvre uniformément le fond de chaque puits et qu’il n’y a pas de bulles d’air.
- Couvrir la plaque avec son couvercle et laisser reposer pendant 1-2 h. L’heure précise à l’emplacement de la plaque est flexible, mais il est important d’être cohérent entre les différentes séries de l’expérience.
- Retirez toutes les concanavalin Une solution de la plaque soit par aspiration, soit en la déchargeant de force dans l’évier ou dans un réceptacle. Veillez à ne pas toucher la partie vitrée de la plaque. Il est acceptable que certaines gouttes de concanavalin une solution restent dans les puits.
- Lavez les puits de plaque de microscope en ajoutant 400 μL d’eau stérile. Retirez l’eau comme cela a été fait avec la concanavalin A dans l’étape précédente. Ne laissez pas la plaque reposer au sec.
- Ajouter immédiatement 185 μL de supports de croissance expérimentale dans la plaque. 15 μL de cellules correctement diluées seront ajoutés à cette plaque.
- Dilution des cellules de levure
REMARQUE : Les étapes ci-dessous décrivent une dilution de levure provenant d’une culture saturée(environ 10 8 cellules/mL) 400 fois pour atteindre une concentration de 250 000 cellules/mL, dont 15 μL seront diluées en 400 μL dans la plaque de fond de verre, donnant un nombre final d’environ 4 000 cellules par puits dans une plaque de 96 puits. Si vous utilisez une plaque de 384 puits, le nombre final de cellules par puits devrait être d’environ 700 et les dilutions devraient être ajustées en conséquence. Ce ratio doit être ajusté pour les cellules collectées en phase de notation, en croissance dans des supports de pré-croissance plus riches ou plus pauvres, ou à partir de différentes souches. La densité finale des cellules par puits doit être réduite lors de l’exécution du taux de croissance d’essai pour les périodes de temps de plus de 10 h.- Mettre en place deux plaques de culture de 96 puits pour les dilutions en série : étiqueter comme plaque 1 et 2, et ajouter 90 μL de supports de croissance expérimentaux (c.-à-d. les supports dans qui la levure poussera au microscope) à chaque plaque de dilution en série.
REMARQUE : Quelle que soit la dilution finale utilisée, au moins deux dilutions périodiques des cellules sont recommandées, dans chacune d’elles un petit volume de levure est canalisé dans un plus grand volume de médias expérimentaux, puis un grand volume de médias expérimentaux est mélangé vigoureusement avec un pipet (comme dans les étapes 3.2.5 et 3.2.6 ci-dessous). - Récupérer la plaque des cellules de la pré-croissance et centrifugeuse de la plaque pendant 2 min à 411 x g.
REMARQUE : Il est très important de ne pas contaminer différents puits dans la plaque. Le but de cette étape de centrifugation avant d’enlever le revêtement de papier d’aluminium des plaques est de s’assurer que les gouttelettes remplies de levure d’un puits ne s’envolent pas du papier d’aluminium et finissent dans d’autres puits. Veillez à ne jamais incliner ou agiter les plaques pour éviter que la levure entre en contact avec le revêtement de papier d’aluminium après centrifugation. - Peler délicatement le papier d’aluminium et resuspendre les cellules en pipetant vigoureusement les cellules à l’aide d’une pipette réglée à environ la moitié du volume total de la plaque tout en déplaçant le pipet autour du puits pour mélanger. Vérifiez que toutes les cellules ont été réutilisées à partir du fond des puits.
- Si plusieurs souches dans les puits individuels seront utilisées, les souches devraient être mélangées à ce moment-là au rapport nécessaire à l’expérience. Si une souche de référence sera utilisée pour générer des mesures du taux de croissance, le rapport de référence à la souche d’essai doit être de 1:1.
- Pipet 10 μL de levure du média de croissance dans la plaque de dilution 1. Ajouter 100 μL de supports de croissance expérimentaux à chaque puits à un volume final de 200 μL par puits. Pipet de haut en bas vigoureusement.
- Pipet 10 μL de levure de la plaque 1 dans la plaque 2. Ajouter 100 μL de supports de croissance expérimentaux à chaque puits, et pipet de haut en bas vigoureusement à mélanger.
REMARQUE : Ces étapes de dilution sont essentielles pour aider à séparer les grappes de levure qui sont collées ensemble à la fin du stade de pré-croissance et pour s’assurer qu’un nombre à peu près égal de cellules de levure se retrouvent dans chaque puits. Le fait d’avoir un nombre constant de levures dans chaque puits aide à éliminer le bruit expérimental et les biais dans les mesures du taux de croissance (voir Résultats représentatifs).
- Mettre en place deux plaques de culture de 96 puits pour les dilutions en série : étiqueter comme plaque 1 et 2, et ajouter 90 μL de supports de croissance expérimentaux (c.-à-d. les supports dans qui la levure poussera au microscope) à chaque plaque de dilution en série.
- Sonication
REMARQUE : La sonication est facultativeet ne doit être effectuée que pour les souches de levure qui ont une forte propension à adhérer les unes aux autres (p. ex., certaines souches sauvages). Pour les souches de laboratoire, la sonication n’est généralement pas nécessaire et peut être ignorée en procédant à l’étape 3.4.- Assainir une tête de sonicateur de 96 broches avec 70% d’éthanol en le plaçant dans une plaque de 96 puits remplie à 70% d’éthanol et sèche avec un lingette sans peluche et statique.
- Définissez un programme de sonication suffisamment solide pour briser les cellules de levure flocculated, mais ne tue pas les cellules ou ne provoque pas de réponses élevées au stress. Certains tests peuvent être nécessaires pour identifier le meilleur programme de sonication pour une expérience donnée. Le programme de sonication utilisé dans cette expérience est: amplitude = 10, temps de traitement = 10 s, pulse-on = 1 s, pulse-off = 1 s. Ce programme exact n’est probablement pas applicable à tous les sonicateurs, de sorte que les tests sont suggérés avant le jour de l’expérience.
- Mélanger la levure dans la plaque de dilution en série 2 une fois de plus en pipetting de haut en bas vigoureusement cinq fois.
- Placez la plaque de dilution 2 sur la plate-forme et fixez-la avec les broches du sonicateur dans la suspension cellulaire, mais ne touchez pas le fond de la plaque. Exécutez le programme de sonication à l’aide d’une protection adéquate de l’oreille.
- Après le programme fonctionne, nettoyer la tête du sonicateur avec 70% d’éthanol, puis avec de l’eau, puis procéder immédiatement à la préparation des plaques de microscope afin que les cellules ne flocculate à nouveau.
- Préparer la plaque pour microscope:
- Pipet 15 μL de levure de la plaque de dilution en série 2 dans la plaque de microscope à un volume de 200 μL. Ajouter 200 μL de supports de croissance expérimentale à chaque puits à un volume final de 400 μL par puits, et pipet de haut en bas vigoureusement pour mélanger.
- Couvrir la plaque d’une membrane respirante. Il est important de bien sceller la plaque avec cette membrane, par exemple à l’aide d’un rouleau de caoutchouc.
- Pour adhérer les cellules de levure à la concanavalin A sur la surface de verre, centrifugez la plaque avec un lint- et statique-libre essuyer en dessous pendant 2 min à 411 x g.
- Au microscope, essuyez le haut et le bas de la plaque à l’aide d’un lingette sans peluche et statique, et soufflez l’air comprimé sur la plaque pour vous débarrasser des débris.
- Placez la plaque sur le microscope, en vous assurant qu’elle est de niveau et que le puits A1 se trouve dans le coin supérieur gauche.
4. Mesures du taux de croissance de la microscopie en accéléré
REMARQUE : Pendant la microscopie en accéléré, les caractéristiques suivantes sont contrôlées par ordinateur : position x, y et z, volets et filtres de fluorescence. Un système de mise au point automatique basé sur le matériel est optimal pour prévenir la dérive des avions focaux pendant l’imagerie en accéléré. Alternativement, une boucle de mise au point automatique basée sur un logiciel peut être utilisée. Pour maintenir l’humidité dans la chambre de microscope, il est conseillé de garder un bécher avec de l’eau purifiée dans la chambre pendant toute la durée de l’expérience.
- Créez une liste de positions (x,y) à l’image, de sorte que chaque puits microscope-plaque est entièrement image. Évitez les images qui se chevauchent afin qu’aucune cellule ne soit analysée plusieurs fois.
- Image dans le champ lumineux avec l’illumination diascopique (DIA) à un grossissement de 15x. Réglez l’exposition à ~5 ms.
- Zoomez sur l’image numériquement afin que les cellules soient clairement visibles. Utilisez les boutons de mise au point pour identifier la mise au point idéale pour l’expérience dans les quatre puits sur le coin de la plaque et dans un puits au centre de la plaque. Concentrez-vous de manière à obtenir un contraste maximal des cellules.
- Définissez la position z (ou position d’autofocus) pour que l’expérience soit une moyenne des positions z/autofocus identifiées pour chacun de ces puits. Si la plaque de microscope est bien faite et que le fond en verre n’a pas de défauts, les positions de mise au point idéales devraient être similaires pour chaque puits.
REMARQUE: Lors de l’analyse des images avec le traitement des images facilement (PIE) pipeline d’analyse d’image11,12, il est utile pour les cellules d’être légèrement hors de discussion sur le microscope de sorte qu’il ya une jante sombre à l’extérieur de la cellule et un intérieur de couleur claire, ce qui aide à la reconnaissance précise des colonies et des estimations de taille. - Si vous utilisez des souches fluorescentes, identifiez les canaux et les expositions à l’image, en vous assurant qu’aucun pixel n’est surexposé. Lors de la fixation du temps d’exposition pour les canaux fluorescents, éteignez le mode « capture en direct » sur le microscope pour éviter d’exposer les cellules à l’excitation de fluorescence pendant de longues périodes de temps, car cela peut à la fois photobleach les cellules et causer du stress.
- Configurer l’acquisition de la séquence de temps pour capturer des images à l’intervalle de temps désiré pour la durée désirée.
- Exécutez l’expérience.
Résultats
La nouveauté de ce protocole est que le taux de croissance peut être calculé pour les cellules individuelles au sein d’une population en suivant leur croissance en microcolonies par imagerie en accéléré (figure 2A). Étant donné que les microcolonies poussent pendant de nombreuses heures de façon planaire en raison de la présence de concanavalin A, leurs zones peuvent être suivies tout au long de l’expérience, et un ajustement linéaire au changement dans le journal naturel de...
Discussion
Le protocole décrit ici est un test polyvalent qui permet de surveiller simultanément la croissance cellulaire et l’expression des gènes au niveau des microcolonies individuelles. La combinaison de ces deux modalités donne des idées biologiques uniques. Par exemple, des travaux antérieurs ont utilisé cet essai pour montrer une corrélation négative entre l’expression du gène TSL1 et le taux de croissance de la microcolonie dans les cellules isogéniques de type sauvage en mesurant les
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous remercions Naomi Ziv, Sasha Levy et Shuang Li pour leur contribution au développement de ce protocole, David Gresham pour l’équipement partagé, et Marissa Knoll pour leur aide à la production vidéo. Ces travaux ont été soutenus par la subvention R35GM118170 des National Institutes of Health.
matériels
| Name | Company | Catalog Number | Comments |
| General Materials | |||
| 500 mL Bottletop Filter .22 µm PES Sterilizing, Low Protein Binding, w/45mm Neck | Fisher | CLS431154 | used to filter the media |
| BD Falcon*Tissue Culture Plates, microtest u-bottom | Fisher | 08-772-54 | 96-well culture tubes used to freeze cells, pre-grow cells, and dilutions |
| BD Syringes without Needle, 50 mL | Fisher | 13-689-8 | Used to filter the Concanavalin A |
| Costar Sterile Disposable Reagent Reservoirs | Fisher | 07-200-127 | reagent reservoirs used to pipette solutions with multichannel pipette |
| Costar Thermowell Aluminum Sealing Tape | Fisher | 07-200-684 | 96-well plate seal for pre-growth and freezing |
| lint and static free Kimwipes | Fisher | 06-666A | lint and static free wipes to keep microscope plate bottom free of debris and scratches |
| Nalgene Syringe Filters | ThermoFisher Scientific | 199-2020 | 0.2 μm pore size, 25 mm diameter; used to filter concanavalin A solution |
| Media Components | |||
| Minimal chemically defined media (MD; 2% glucose) | alternative microscopy media used for yeast pre-growth and growth during microscopy | ||
| Synthetic Complete Media (SC; 2% glucose) | microscopy media used for yeast pre-growth and growth during microscopy | ||
| Yeast extract-peptone-dextrose (YEPD; 2% glucose) medium | cell growth prior to freezing down randomized plates | ||
| Microscopy Materials | |||
| Breathe-Easy sealing membrane | Millipore Sigma | Z380059-1PAK | breathable membranes used to seal plate during microscopy experiment. At this stage breathable membranes are recommended because they prevent condensation in the wells and allow for better microscopy images |
| Brooks 96-well flat clear glass bottom microscope plate | Dot Scientific | MGB096-1-2-LG-L | microscope plate |
| Concanavalin A from canavalia ensiformis (Jack Bean), lyophilized powder | Millipore Sigma | 45-C2010-1G | Make 5x concanavalin A solution and freeze 5ml of 5x concanavalin A in 50 mL conical tubes at -80 °C |
| Strains Used | |||
| MAH.5, MAH.96, MAH.52, MAH.66, MAH.11, MAH.58, MAH.135, MAH.15, MAH.44, MAH.132 | Haploid mutation accumulation strains in a laboratory background, described in Hall and Joseph 2010 | ||
| EP026.2A-2C | Progeny of the ancestral Hall and Joseph 2010 mutation accumulation strain, transformed with YFR054cΔ::Scw11P::GFP | ||
| Equipment | |||
| Misonix Sonicator S-4000 with 96-pin attachment | Sonicator https://www.labx.com/item/misonix-inc-s-4000-sonicator/4771281 | ||
| Nikon Eclipse Ti-E with Perfect Focus System | Inverted microscope with automated stage and autofocus system |
Références
- Geiler-Samerotte, K. A., Hashimoto, T., Dion, M. F., Budnik, B. A., Airoldi, E. M., Drummond, D. A. Quantifying condition-dependent intracellular protein levels enables high-precision fitness estimates. PloS one. 8 (9), 75320 (2013).
- Kussell, E., Leibler, S. Phenotypic diversity, population growth, and information in fluctuating environments. Science. 309 (5743), 2075-2078 (2005).
- Thattai, M., van Oudenaarden, A. Stochastic gene expression in fluctuating environments. Genetics. 167 (1), 523-530 (2004).
- King, O. D., Masel, J. The evolution of bet-hedging adaptations to rare scenarios. Theoretical population biology. 72 (4), 560-575 (2007).
- Acar, M., Mettetal, J. T., van Oudenaarden, A. Stochastic switching as a survival strategy in fluctuating environments. Nature genetics. 40 (4), 471-475 (2008).
- Avery, S. V. Microbial cell individuality and the underlying sources of heterogeneity. Nature reviews. Microbiology. 4 (8), 577-587 (2006).
- Levy, S. F., Ziv, N., Siegal, M. L. Bet hedging in yeast by heterogeneous, age-correlated expression of a stress protectant. PLoS biology. 10 (5), 1001325 (2012).
- van Dijk, D., et al. Slow-growing cells within isogenic populations have increased RNA polymerase error rates and DNA damage. Nature communications. 6, 7972 (2015).
- Ziv, N., Shuster, B. M., Siegal, M. L., Gresham, D. Resolving the Complex Genetic Basis of Phenotypic Variation and Variability of Cellular Growth. Genetics. 206 (3), 1645-1657 (2017).
- Li, S., Giardina, D. M., Siegal, M. L. Control of nongenetic heterogeneity in growth rate and stress tolerance of Saccharomyces cerevisiae by cyclic AMP-regulated transcription factors. PLoS genetics. 14 (11), 1007744 (2018).
- Plavskin, Y., Li, S., Ziv, N., Levy, S. F., Siegal, M. L. Robust colony recognition for high-throughput growth analysis from suboptimal low-magnification brightfield micrographs. bioRxiv. , (2018).
- Ziv, N., Siegal, M. L., Gresham, D. Genetic and nongenetic determinants of cell growth variation assessed by high-throughput microscopy. Molecular biology and evolution. 30 (12), 2568-2578 (2013).
- Hall, D. W., Joseph, S. B. A high frequency of beneficial mutations across multiple fitness components in Saccharomyces cerevisiae. Genetics. 185 (4), 1397-1409 (2010).
- Saleemuddin, M., Husain, Q. Concanavalin A: a useful ligand for glycoenzyme immobilization--a review. Enzyme and microbial technology. 13 (4), 290-295 (1991).
- Geiler-Samerotte, K. A., Bauer, C. R., Li, S., Ziv, N., Gresham, D., Siegal, M. L. The details in the distributions: why and how to study phenotypic variability. Current opinion in biotechnology. 24 (4), 752-759 (2013).
- Nakagawa, S., Schielzeth, H. Repeatability for Gaussian and non-Gaussian data: a practical guide for biologists. Biological reviews of the Cambridge Philosophical Society. 85 (4), 935-956 (2010).
- Bolker, J. A. Exemplary and surrogate models: two modes of representation in biology. Perspectives in biology and medicine. 52 (4), 485-499 (2009).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.