
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Высокая пропускная способность Live Imaging микроколоний для измерения неоднородности в росте и экспрессии генов
В этой статье
Резюме
Фенотипы роста дрожжей точно измеряются с помощью высокой параллельной покадровой визуализации обездвиженные клетки, растущие в микроколонии. Одновременно можно контролировать стрессоустойчивость, экспрессию белка и локализацию белка, создавая интегрированные наборы данных для изучения того, как экологические и генетические различия, а также неоднородность генной экспрессии среди изогенных клеток, модулируют рост.
Аннотация
Точные измерения неоднородности между штаммом и внутри штамма в ставках роста микробов имеют важное значение для понимания генетических и экологических факторов, вводимых в стрессоустойчивость, патогенность и другие ключевые компоненты фитнеса. Эта рукопись описывает микроскоп на основе анализа, который отслеживает примерно 105 Saccharomyces cerevisiae микроколонии за эксперимент. После автоматической покадровой визуализации дрожжей, обездвиженные в многоукрашенной пластине, темпы роста микроколонии легко анализируются с помощью пользовательского программного обеспечения для анализа изображений. Для каждой микроколонии также можно контролировать экспрессию и локализацию флуоресцентных белков и выживание острого стресса. Этот анализ позволяет точно оценить средние темпы роста штаммов, а также всестороннее измерение неоднородности роста, экспрессии генов и стрессоустойчивости в клональных популяциях.
Введение
Фенотипы роста вносят критический вклад в фитнес дрожжей. Естественный отбор может эффективно различать линии с темпами роста, отличающимися от обратного эффективного размера популяции, который может превышать 108 особей1. Кроме того, изменчивость темпов роста среди людей в популяции является эволюционно актуальным параметром, так как она может служить основой для стратегий выживания,таких как хеджирование ставок 2,3,4,5,6. Поэтому анализы, позволяющие проводить высокотоктяжелые измерения фенотипов роста и их распределения, имеют решающее значение для изучения микроорганизмов. Анализ роста микроколонии, описанный здесь, может генерировать индивидуальные измерения скорости роста на 105 микроколоний за эксперимент. Таким образом, этот анализ обеспечивает мощный протокол для изучения дрожжевой эволюционной генетики и геномики. Он поддается особенно хорошо для тестирования, как изменчивость в популяциях генетически идентичных одиночных клеток генерируется, поддерживается, и способствует популяции фитнес7,8,9,10.
Метод, описанный здесь (Рисунок 1) использует периодически захваченных, с низким увеличением яркое поле изображения клеток, растущих в жидких средствах массовой информации на 96- или 384-хорошо стеклянной нижней пластины для отслеживания роста в микроколонии. Клетки придерживаются лектина конканавалина А, который покрывает дно микроскопической пластины, и образуют двумерные колонии. Поскольку микроколонии растут в монослойном, область микроколонии сильно коррелирует с номером клетки7. Таким образом, точные оценки темпов роста микроколонии и времени задержки могут быть получены с помощью пользовательского программного обеспечения для анализа изображений, которое отслеживает скорость изменения площади каждой микроколонии. Кроме того, экспериментальная установка может контролировать изобилие и даже субклеточную локализацию флуоресцентно помеченных белков, выраженных в этих микроколониях. Вниз по течению обработки данных из этого микроколонии роста анализа может быть достигнуто путем пользовательского анализа или существующего программного обеспечения анализа изображений, таких как обработка изображений легко (PIE)11, алгоритм для надежного распознавания колонии области и высокой пропускной способности анализа роста с низким увеличением, яркие изображения, которая доступна через GitHub12.
Поскольку оценки темпов роста, полученные в результате анализа микроколонии роста, генерируются на основе большого числа одноколонных измерений, они чрезвычайно точны, при этом стандартные ошибки на несколько порядков меньше, чем сами оценки для эксперимента разумного размера. Таким образом, сила анализа для выявления различий в темпах роста между различными генотипами, обработками или условиями окружающей среды высока. Формат мультиэлементной пластины позволяет сравнивать множество различных комбинаций среды и генотипа в одном эксперименте. Если штаммы constitutively выражают различные флуоресцентные маркеры, они могут быть смешаны в том же хорошо и отличается последующим анализом изображения, который может увеличить мощность дальше, позволяя хорошо за колодец нормализации данных.

Рисунок 1: Схематическое представление протокола. Этот протокол следует за 2 главным образом шагами, которые подготовка экспериментальной плиты и подготовка клеток к изображению. Рандомизация пластин и рост клеток должны проводиться до и в течение дня эксперимента. Повторное смешивание клеток на каждом шагу во время разбавления необходимо в шагах до покрытия, и поэтому подготовка экспериментальной пластины сначала рекомендуется так, что она готова к покрытие сразу после завершения разбавления клеток. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
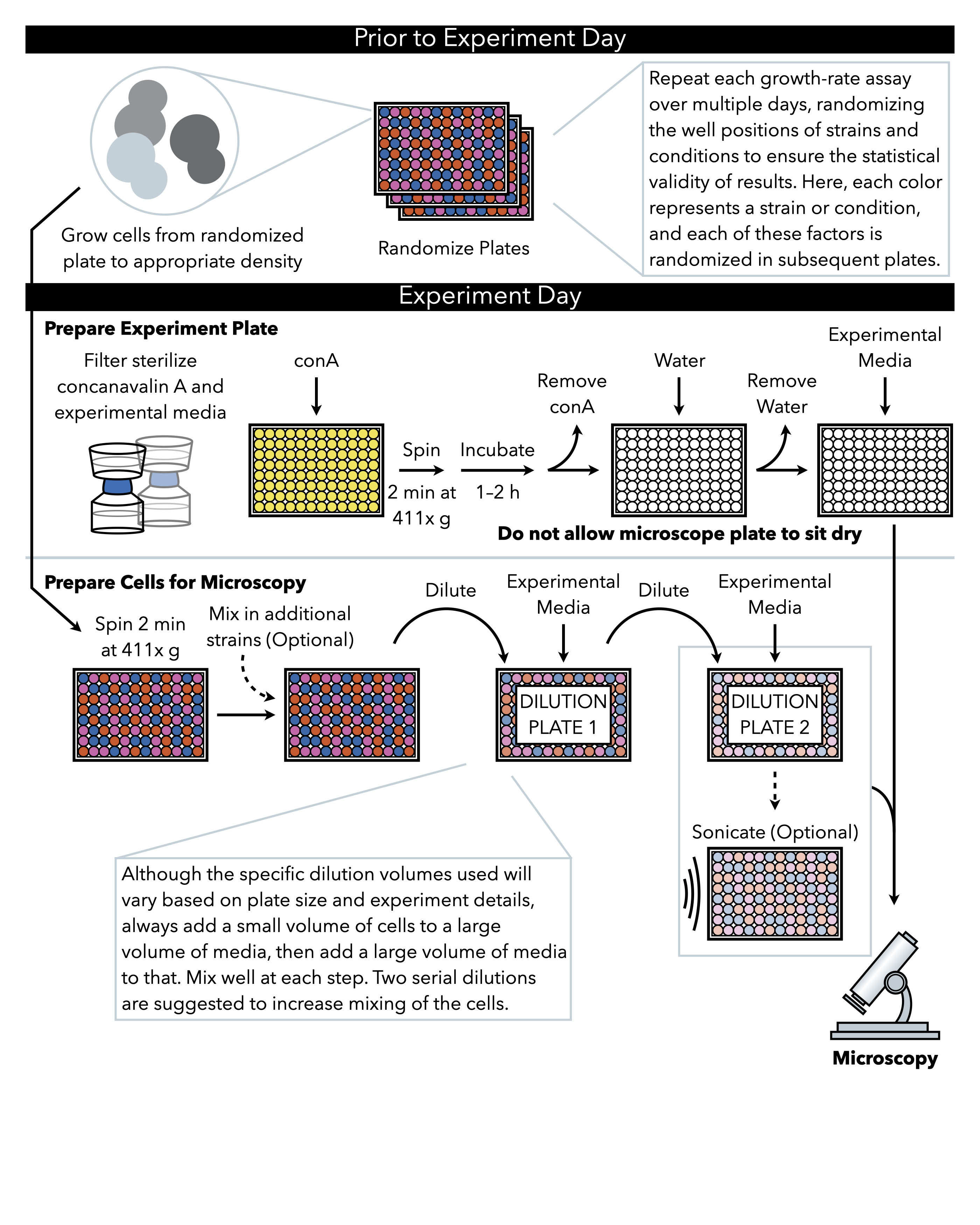{kind=link}
протокол
1. Подготовка рандомизированных пластин (до дня эксперимента)
- План штаммов и условий, которые будут проверены с анализом роста. В этот момент, случайным образом назначить штаммов и условий для любого хорошо.
ПРИМЕЧАНИЕ: При рассмотрении установки пластины, желательно включить более одного репликации на штамм и состояние роста на одной пластине для учета хорошо связанных шума в измерениях. Более подробную информацию можно посмотреть в обсуждении. - Вычислительно рандомизировать расположение каждого штамма и состояние окружающей среды для репликации пластин, которые будут работать в разные дни.
- Выращиваем все клетки, которые будут использоваться в эксперименте для насыщения в дрожжевой экстракт-пептон-декстроза (YEPD; 2% глюкозы) среды в шейкере при температуре 30 градусов по Цельсию (или любой другой соответствующей температуре).
- Создайте рандомизированные фондовые пластины либо вручную, либо с помощью робота для обработки жидкости. Добавьте 10 МКЛ назначенных насыщенных клеток к каждому колодец стерильной пластины культуры U-нижней ткани. Если несколько штаммов будут протестированы в одной колодец, не комбинировать их на данный момент; это сочетание будет сделано непосредственно перед разбавления клеток в день эксперимента, чтобы убедиться, что все штаммы находятся в правильной концентрации, когда покрытие в качестве микроколонии основателей клеток.
- Добавьте 10 йл 30% глицерола к каждому колодец каждой пластины. Пипетка вверх и вниз так, что клетки и глицерол становятся хорошо смешанными.
- Печать каждой пластины с крышкой фольги и заморозить вниз сразу при -70 градусов по Цельсию, пока не готов к использованию.
ПРИМЕЧАНИЕ: Важно создать все рандомизированные пластины в один и тот же день, и заморозить их, так что условия предварительного роста клеток в каждой пластине будут идентичны и не будет генерировать технические изменения в анализе скорости роста.
2. Предварительный рост дрожжей
ПРИМЕЧАНИЕ: Как правило, это начинается до дня эксперимента и сильно зависит от экспериментального вопроса. Подробнее об обсуждении.
- Удалите складе пластины (10 ЗЛ дрожжей, 10 глицерола на колодец) из -70 градусов по Цельсию морозильник и добавить 180 йл средств массовой информации, которые будут использоваться для эксперимента. Если эксперимент будет проводиться с использованием питательных веществ, ограничивающих средства массовой информации, не предварительно выращивать дрожжи для насыщения в питательных ограничивающих средств массовой информации, как споруляция дрожжей может произойти. Вместо этого, предварительно расти в не ограничивающих средств массовой информации.
- Выращиваем дрожжи при встряхивании при 30 градусах Цельсия. Подумайте, следует ли запустить анализ, начиная с ячеек в фазе журнала или в стационарной фазе, чтобы определить, если разбавления клеток несколько раз до эксперимента будет необходимо. Если дрожжи штаммов или условий в анализе, как ожидается, имеют значительно разные темпы роста, то два дня до роста период будет необходимо для того, чтобы все различные условия для достижения стационарной фазы.
3. Настройка микроскопа
- Микроскопная подготовка пластины
- Убедитесь, что микроскоп инкубатор и нагрева микроскоп камеры до желаемой температуры роста для экспериментальных условий. Для стандартных экспериментов с использованием saccharomyces cerevisiae клетки, инкубатор должен нагревать камеру микроскопа до 30 градусов по Цельсию, чтобы гарантировать, что условия роста для клеток будет правильным во время анализа скорости роста.
- Дезинфицировать рабочую скамью, пипетки и другие инструменты с 70% этанола. Получить микроскоп пластины и поместить его на скамейке на вершине ворса и статических свободной салфетки.
ПРИМЕЧАНИЕ: Никогда не прикасайтесь к нижней части микроскопа пластины, даже в перчатках, и всегда установить микроскоп пластины вниз на верхней части ворса и статических свободных протрите в любое время он касается любой поверхности. Это предотвращает пятна или царапины от препятствует измерению темпов роста после того, как эксперимент в настоящее время изображены. - Оттепель 5 мл 5x раствора конканавалина А, разбавить до 1x водой, и фильтр стерилизовать через шприц оснащен 0,2-мкм фильтр.
- Фильтр стерилизовать все другие жидкости, которые будут использоваться в анализе с фильтром 0,2 мкм, в том числе экспериментальных средств массовой информации, для удаления любых кристаллов или мусора, которые могут материализоваться в растворах. Наличие кристаллов приведет к снижению качества изображений микроскопии.
- Pipet 200 йл раствора конканавалина А в каждом колодец микроскопа пластины.
- Центрифуга пластины в течение 2 мин при 411 х тяжести(г) с ворсинки и статические свободные протрите под пластиной, чтобы обеспечить, чтобы раствор конканавалина равномерно охватывает дно каждой хорошо и что Есть нет пузырьков воздуха.
- Накройте тарелку крышкой и дайте ей посидеть 1-2 ч. Точное время, в течение которое находится пластина, является гибким, но важно быть последовательным между различными пробегами эксперимента.
- Удалите весь раствор конканавалина А из пластины либо путем всасывания, либо путем принудительного выгрузки его в раковину или сосуд. Будьте осторожны, чтобы не коснуться стеклянной части пластины. Это приемлемо, если в скважинах остаются некоторые капли раствора конканавалина А.
- Вымойте колодцы микроскопа пластины, добавив 400 йл стерильной воды. Удалите воду, как это было сделано с конканавалином А на предыдущем этапе. Не позволяйте пластине сидеть сухой.
- Немедленно добавьте в тарелку экспериментальные средства роста 185 йл. К этой пластине будет добавлено 15 МКЛ правильно разбавленных клеток.
- Разбавление дрожжевых клеток
ПРИМЕЧАНИЕ: В приведенных ниже шагах описывается разбавление дрожжей из насыщенной культуры(примерно 10 8 клеток/мл) в 400 раз для достижения концентрации 250 000 клеток/мл, 15 из которых будут разбавлены в 400 мкл в стеклянной нижней пластине, что даст окончательное число примерно 4000 клеток на колодец в пластине 96-колодец. При использовании пластины 384 хорошо окончательное количество клеток на колодец должно быть около 700 и разбавления должны быть скорректированы соответствующим образом. Это соотношение должно быть скорректировано для ячеек, собранных в фазе журнала, растущих в более богатых или бедных средствах предварительного роста, или из различных штаммов. Окончательная плотность ячеек на колодец должна быть уменьшена при запуске анализа скорости роста на периоды времени более 10 ч.- Настройка двух пластин культуры 96-колодец для серийных разбавлений: этикетка, как пластина 1 и 2, и добавить 90 йл экспериментальных средств роста (т.е. средства массовой информации, что дрожжи будут расти в на микроскоп) для каждой серийной пластины разбавления.
ПРИМЕЧАНИЕ: Независимо от того, что окончательное разбавление используется, по крайней мере два серийных разбавления клеток рекомендуется, в каждом из которых небольшой объем дрожжей pipetted в больший объем экспериментальных средств массовой информации, а затем большой объем экспериментальных средств массовой информации смешивается в энергично с пипетки (как в шагах 3.2.5 и 3.2.6 ниже). - Извлеките пластину клеток из предварительного роста и центрифуги пластины в течение 2 мин при 411 x g.
ПРИМЕЧАНИЕ: Очень важно не перекрестно загрязнять различные скважины в пластине. Цель этого шага центрифугации перед удалением покрытия фольги из пластин заключается в обеспечении того, чтобы заполненные дрожжами капли из одной скважины не вылетали из фольги и в конечном итоге в других скважинах. Будьте осторожны, чтобы никогда не наклонять или агитировать пластины, чтобы избежать дрожжей вступать в контакт с фольгой покрытия после центрифугации. - Тщательно очистить фольгу и повторного перерасхода клеток энергично трубопроводных клеток с пипетки установить примерно половину общего объема в пластине при перемещении трубы вокруг хорошо перемешать. Убедитесь, что все ячейки были повторно использованы со дна скважин.
- Если будет использоваться несколько штаммов в отдельных скважинах, штаммы должны быть смешаны в это время в соотношении, необходимом для эксперимента. Если эталонный штамм будет использоваться для генерации измерений скорости роста, соотношение ссылки на тестовый штамм должно быть 1:1.
- Пипетка 10 йл дрожжей из средств роста в разбавленной пластины 1. Добавьте 100 МКЛ экспериментальных средств роста к каждой из них к окончательному объему 200 МКЛ на колодец. Пипет вверх и вниз энергично.
- Пипет 10 МКЛ дрожжей из пластины 1 в тарелку 2. Добавьте 100 МКЛ экспериментальных средств роста к каждой хорошо, и трубы вверх и вниз энергично смешивать.
ПРИМЕЧАНИЕ: Эти шаги разбавления имеют решающее значение, чтобы помочь отдельным кластерам дрожжей, которые застряли вместе в конце стадии предварительного роста и обеспечить, чтобы примерно равное количество дрожжевых клеток в конечном итоге в каждой хорошо. Наличие последовательного количества дрожжей в каждой хорошо помогает удалить экспериментальный шум и предубеждения в измерениях темпов роста (см. результаты).
- Настройка двух пластин культуры 96-колодец для серийных разбавлений: этикетка, как пластина 1 и 2, и добавить 90 йл экспериментальных средств роста (т.е. средства массовой информации, что дрожжи будут расти в на микроскоп) для каждой серийной пластины разбавления.
- Соникация
ПРИМЕЧАНИЕ: Sonication является необязательным,и только должны быть выполнены для дрожжей штаммов, которые имеют высокую склонность придерживаться друг к другу (например, некоторые дикие штаммы). Для лабораторных штаммов, sonication, как правило, не является необходимым и может быть пропущен, продолжая шаг 3.4.- Дезинфицировать 96-контактный sonicator голову с 70% этанола, поместив его в 96-хорошо пластины заполнены 70% этанола и сухой с ворсинкой и статических свободной салфетки.
- Установите программу sonication которая достаточно сильна для того чтобы сломать врозь flocculated клетки дрожжа, но не убивает клетки или причиняет повышенные реакции усилия. Возможно, потребуется некоторое тестирование для определения наилучшей звуковой программы для данного эксперимента. Программа sonication, используемая в этом эксперименте: амплитуда 10, время процесса 10 с, пульс-на 1 с, пульс-офф 1 с. Эта точная программа, вероятно, не применима ко всем sonicators поэтому тестирование предлагается до дня эксперимента.
- Смешайте дрожжи в серийной пластине разбавления 2 еще раз, трубя вверх и вниз энергично пять раз.
- Поместите разбавленную пластину 2 на платформу и закрелите ее соникаторными булавками в клеточной подвеске, но не касаясь нижней части пластины. Запустите программу sonication, используя соответствующую защиту уха.
- После того, как программа запускается, очистить sonicator голову с 70% этанола, а затем с водой, а затем сразу же приступить к микроскопу подготовки пластины так, что клетки не flocculate снова.
- Подготовка плиты для микроскопа:
- Pipet 15 МКЛ дрожжей из серийного разбавления пластины 2 в микроскоп пластины объемом 200 йл. Добавить 200 йл экспериментальных средств роста к каждой хорошо до конечного объема 400 йл на колодец, и трубы вверх и вниз энергично смешивать.
- Обложка пластины с дышащий мембраны. Важно хорошо запечатать пластину этой мембраной, например, с помощью резинового ролика.
- Чтобы прилипнуть дрожжевые клетки к конканавалину А на стеклянной поверхности, центрифуга пластины с ворсинки и статические свободные протрите под ним в течение 2 мин при 411 х г.
- На микроскоп, протрите верхнюю и нижнюю часть пластины с ворса и статические свободные салфетки, и удар сжатого воздуха на пластину, чтобы избавиться от мусора.
- Поместите пластину на микроскоп, убедившись, что она является уровневой и что А1 хорошо находится в левом верхнем углу.
4. Измерения скорости роста замедленной микроскопии
ПРИМЕЧАНИЕ: Во время тайм-промежуток микроскопии следующие функции управляются компьютером: x, y, и z положение, ставни, и флуоресценции фильтров. Аппаратная система автофокусировки является оптимальной для предотвращения дрейфа фокусного самолета во время замедленной визуализации. Кроме того, можно использовать цикл автоматической фокусировки на основе программного обеспечения. Для поддержания влажности в микроскопии камеры, рекомендуется держать стакан с очищенной водой в камере в течение всего эксперимента.
- Создайте список позиций (x,y) для изображения, так что каждый микроскоп пластины хорошо полностью изображен. Избегайте перекрывающихся изображений, чтобы ни одна ячейка не анализировалась несколько раз.
- Изображение в ярком поле с diascopic освещение (DIA) при увеличении 15x. Установите экспозицию до 5 мс.
- Увеличьте изображение в цифровом виде, чтобы клетки были хорошо видны. Используйте фокусировки ручки для определения идеального фокуса для эксперимента в четырех колодцах на углу пластины и в одном колодцы в центре пластины. Сосредоточьтесь таким образом, чтобы получить максимальный контраст клеток.
- Установите позицию z (или позицию автофокуса) для эксперимента, чтобы быть средним положением z/autofocus, идентифицированным для каждой из этих скважин. Если микроскопная пластина хорошо сделана и стекло дно не имеет дефектов, идеальное положение фокусировки должно быть аналогичным для каждого хорошо.
ПРИМЕЧАНИЕ: При анализе изображений с обработкой изображений легко (PIE) анализизображений трубопровода 11,12, это полезно для клеток, чтобы быть немного не в фокусе на микроскоп так, что есть темный обод за пределами клетки и светлого цвета интерьера, который помогает в точном распознавании колонии и размер оценки. - При использовании флуоресцентных штаммов определите каналы и экспозиции изображения, гарантируя, что пиксели не будут переэкспонированы. При установке времени экспозиции для флуоресцентных каналов, выключите режим "живого захвата" на микроскопе, чтобы избежать подвергая клетки флуоресценции возбуждения в течение длительных периодов времени, так как это может как photobleach клеток и вызвать стресс.
- Настройка приобретения последовательности времени для захвата изображений с желаемым временным интервалом в течение желаемого периода времени.
- Запустите эксперимент.
Результаты
Новизна этого протокола заключается в том, что темпы роста могут быть рассчитаны для отдельных клеток в популяции, отслеживая их рост в микроколонии через промежуток времени изображения(рисунок 2A). Поскольку микроколонии растут в течение многих часов в планарной манер?...
Обсуждение
Описанный здесь протокол представляет ею универсальный анализ, позволяющий одновременно контролировать рост клеток и экспрессию генов на уровне отдельных микроколоний. Сочетание этих двух условий дает уникальную биологическую информацию. Например, предыдущая работа использовала э?...
Раскрытие информации
Авторов нечего раскрывать.
Благодарности
Мы благодарим Наоми Зив, Сашу Леви и Шуанг Ли за их вклад в разработку этого протокола, Дэвида Грешема за совместное оборудование и Мариссу Нолл за помощь в производстве видео. Эта работа была поддержана Национальными институтами здравоохранения грант R35GM118170.
Материалы
| Name | Company | Catalog Number | Comments |
| General Materials | |||
| 500 mL Bottletop Filter .22 µm PES Sterilizing, Low Protein Binding, w/45mm Neck | Fisher | CLS431154 | used to filter the media |
| BD Falcon*Tissue Culture Plates, microtest u-bottom | Fisher | 08-772-54 | 96-well culture tubes used to freeze cells, pre-grow cells, and dilutions |
| BD Syringes without Needle, 50 mL | Fisher | 13-689-8 | Used to filter the Concanavalin A |
| Costar Sterile Disposable Reagent Reservoirs | Fisher | 07-200-127 | reagent reservoirs used to pipette solutions with multichannel pipette |
| Costar Thermowell Aluminum Sealing Tape | Fisher | 07-200-684 | 96-well plate seal for pre-growth and freezing |
| lint and static free Kimwipes | Fisher | 06-666A | lint and static free wipes to keep microscope plate bottom free of debris and scratches |
| Nalgene Syringe Filters | ThermoFisher Scientific | 199-2020 | 0.2 μm pore size, 25 mm diameter; used to filter concanavalin A solution |
| Media Components | |||
| Minimal chemically defined media (MD; 2% glucose) | alternative microscopy media used for yeast pre-growth and growth during microscopy | ||
| Synthetic Complete Media (SC; 2% glucose) | microscopy media used for yeast pre-growth and growth during microscopy | ||
| Yeast extract-peptone-dextrose (YEPD; 2% glucose) medium | cell growth prior to freezing down randomized plates | ||
| Microscopy Materials | |||
| Breathe-Easy sealing membrane | Millipore Sigma | Z380059-1PAK | breathable membranes used to seal plate during microscopy experiment. At this stage breathable membranes are recommended because they prevent condensation in the wells and allow for better microscopy images |
| Brooks 96-well flat clear glass bottom microscope plate | Dot Scientific | MGB096-1-2-LG-L | microscope plate |
| Concanavalin A from canavalia ensiformis (Jack Bean), lyophilized powder | Millipore Sigma | 45-C2010-1G | Make 5x concanavalin A solution and freeze 5ml of 5x concanavalin A in 50 mL conical tubes at -80 °C |
| Strains Used | |||
| MAH.5, MAH.96, MAH.52, MAH.66, MAH.11, MAH.58, MAH.135, MAH.15, MAH.44, MAH.132 | Haploid mutation accumulation strains in a laboratory background, described in Hall and Joseph 2010 | ||
| EP026.2A-2C | Progeny of the ancestral Hall and Joseph 2010 mutation accumulation strain, transformed with YFR054cΔ::Scw11P::GFP | ||
| Equipment | |||
| Misonix Sonicator S-4000 with 96-pin attachment | Sonicator https://www.labx.com/item/misonix-inc-s-4000-sonicator/4771281 | ||
| Nikon Eclipse Ti-E with Perfect Focus System | Inverted microscope with automated stage and autofocus system |
Ссылки
- Geiler-Samerotte, K. A., Hashimoto, T., Dion, M. F., Budnik, B. A., Airoldi, E. M., Drummond, D. A. Quantifying condition-dependent intracellular protein levels enables high-precision fitness estimates. PloS one. 8 (9), 75320 (2013).
- Kussell, E., Leibler, S. Phenotypic diversity, population growth, and information in fluctuating environments. Science. 309 (5743), 2075-2078 (2005).
- Thattai, M., van Oudenaarden, A. Stochastic gene expression in fluctuating environments. Genetics. 167 (1), 523-530 (2004).
- King, O. D., Masel, J. The evolution of bet-hedging adaptations to rare scenarios. Theoretical population biology. 72 (4), 560-575 (2007).
- Acar, M., Mettetal, J. T., van Oudenaarden, A. Stochastic switching as a survival strategy in fluctuating environments. Nature genetics. 40 (4), 471-475 (2008).
- Avery, S. V. Microbial cell individuality and the underlying sources of heterogeneity. Nature reviews. Microbiology. 4 (8), 577-587 (2006).
- Levy, S. F., Ziv, N., Siegal, M. L. Bet hedging in yeast by heterogeneous, age-correlated expression of a stress protectant. PLoS biology. 10 (5), 1001325 (2012).
- van Dijk, D., et al. Slow-growing cells within isogenic populations have increased RNA polymerase error rates and DNA damage. Nature communications. 6, 7972 (2015).
- Ziv, N., Shuster, B. M., Siegal, M. L., Gresham, D. Resolving the Complex Genetic Basis of Phenotypic Variation and Variability of Cellular Growth. Genetics. 206 (3), 1645-1657 (2017).
- Li, S., Giardina, D. M., Siegal, M. L. Control of nongenetic heterogeneity in growth rate and stress tolerance of Saccharomyces cerevisiae by cyclic AMP-regulated transcription factors. PLoS genetics. 14 (11), 1007744 (2018).
- Plavskin, Y., Li, S., Ziv, N., Levy, S. F., Siegal, M. L. Robust colony recognition for high-throughput growth analysis from suboptimal low-magnification brightfield micrographs. bioRxiv. , (2018).
- Ziv, N., Siegal, M. L., Gresham, D. Genetic and nongenetic determinants of cell growth variation assessed by high-throughput microscopy. Molecular biology and evolution. 30 (12), 2568-2578 (2013).
- Hall, D. W., Joseph, S. B. A high frequency of beneficial mutations across multiple fitness components in Saccharomyces cerevisiae. Genetics. 185 (4), 1397-1409 (2010).
- Saleemuddin, M., Husain, Q. Concanavalin A: a useful ligand for glycoenzyme immobilization--a review. Enzyme and microbial technology. 13 (4), 290-295 (1991).
- Geiler-Samerotte, K. A., Bauer, C. R., Li, S., Ziv, N., Gresham, D., Siegal, M. L. The details in the distributions: why and how to study phenotypic variability. Current opinion in biotechnology. 24 (4), 752-759 (2013).
- Nakagawa, S., Schielzeth, H. Repeatability for Gaussian and non-Gaussian data: a practical guide for biologists. Biological reviews of the Cambridge Philosophical Society. 85 (4), 935-956 (2010).
- Bolker, J. A. Exemplary and surrogate models: two modes of representation in biology. Perspectives in biology and medicine. 52 (4), 485-499 (2009).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены