
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Expression bactérienne et purification de la métalloprotéinase-3 à matrice humaine par chromatographie d’affinité
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
La purification, la dialyse et l’activation de son étiquette sont utilisées pour augmenter les rendements de l’expression des protéines solubles du domaine catalytique de la métalloprotéinase-3 à matrice active chez les bactéries. Les fractions protéiques sont analysées via des gels SDS-PAGE.
Résumé
Les métalloprotéinases matricielles (MMP) appartiennent à la famille des protéases de metzincine avec des rôles centraux dans la dégradation et le remodelage de la matrice extracellulaire (ECM), ainsi que dans les interactions avec plusieurs facteurs de croissance et cytokines. La surexpression de MMP spécifiques est responsable de plusieurs maladies telles que le cancer, les maladies neurodégénératives et les maladies cardiovasculaires. Les MMP ont récemment été au centre de l’attention en tant que cibles pour développer des thérapies capables de traiter les maladies corrélées à la surexpression de la MMP.
Pour étudier le mécanisme MMP en solution, des méthodes d’expression et de purification des protéines recombinantes plus faciles et plus robustes sont nécessaires pour la production de MMP actifs et solubles. Cependant, le domaine catalytique de la plupart des MMP ne peut pas être exprimé chez Escherichia coli (E. coli) sous forme soluble en raison de l’absence de mécanisme post-traductionnel, alors que les systèmes d’expression des mammifères sont généralement coûteux et ont des rendements plus faibles. Les corps d’inclusion MMP doivent subir le processus fastidieux et laborieux de purification et de repliement approfondis, réduisant considérablement le rendement des MMP en conformation native. Cet article présente un protocole utilisant des cellules Rosetta2(DE3)pLysS (ci-après dénommées R2DP) pour produire le domaine catalytique de la métalloprotéinase matricielle-3 (MMP-3cd), qui contient une étiquette His-terminalE N suivie d’un pro-domaine (Hisx6-pro-MMP-3cd) pour une utilisation dans la purification d’affinité. Les cellules R2DP améliorent l’expression des protéines eucaryotes grâce à un plasmide résistant au chloramphénicol contenant des codons normalement rares dans les systèmes d’expression bactérienne. Par rapport à la lignée cellulaire traditionnelle de choix pour l’expression des protéines recombinantes, BL21 (DE3), la purification à l’aide de cette nouvelle souche a amélioré le rendement en Hisx6-pro-MMP-3cd purifié. Lors de l’activation et du dessalement, le domaine pro est clivé avec le N-terminal His-tag, fournissant un MMP-3cd actif pour une utilisation immédiate dans d’innombrables applications in vitro . Cette méthode ne nécessite pas d’équipement coûteux ou de protéines de fusion complexes et décrit la production rapide de MMP humains recombinants chez les bactéries.
Introduction
La plupart des protéines eucaryotes complexes subissent des modifications post-traductionnelles élaborées après leur expression, nécessitant un repliement des protéines hautement assisté et des cofacteurs pour être fonctionnels1. La production de grandes quantités de protéines humaines solubles dans un hôte bactérien reste un défi important en raison des coûts élevés et du manque de méthodes d’expression et de purification robustes, même pour des expériences de laboratoire à plus petite échelle2,3. Les MMP, des endopeptidases humaines de grand poids moléculaire, sont généralement exprimées sous forme de corps d’inclusion insolubles lorsqu’elles sont exprimées dans E. coli. L’extraction des MMP humains solubles conduit souvent à un processus de solubilisation et de repli laborieux et long4.
Les MMP jouent un rôle essentiel dans les processus physiologiques et pathogènes. Les MMP humains sont une famille de 23 endopeptidases de zinc, classées par structure et spécificité du substrat, et exprimées différemment en dépit d’un domaine catalytique hautement conservé5,6. Les MMP sont sécrétés sous forme de zymogènes inactifs, régulés par activation post-traductionnelle et leurs inhibiteurs endogènes, inhibiteurs tissulaires des métalloprotéinases (TIMPs)7,8,9,10. Bien qu’initialement reconnus pour leur rôle dans le renouvellement de l’ECM, les MMP ont également été impliqués dans le développement, la morphogenèse, la réparation des tissus et le remodelage8. La dérégulation des MMP a été notamment liée au cancer ainsi qu’aux maladies neurodégénératives, cardiovasculaires et fibrotiques, entre autres maladies5,7.
Le développement de méthodes robustes de production de MMP à grande échelle est essentiel pour assurer le succès des études futures des mécanismes MMP par le biais de tests biochimiques et cellulaires. Divers MMP ont déjà été exprimés dans des bactéries11, y compris des MMP marqués Hisx6, sans modifier l’activité MMP12,13,14,15. Cependant, ces méthodes comprennent des étapes longues et fastidieuses qui peuvent être difficiles à reproduire.
Les cellules de mammifères peuvent également être utilisées pour exprimer de nombreuses protéines humaines différentes tout en assurant les modifications post-traductionnelles appropriées16. Bien que le système d’expression des mammifères soit un choix idéal pour produire des protéines humaines recombinantes avec des modifications post-traductionnelles appropriées, les principaux inconvénients de cette méthode sont les faibles rendements initiaux, les milieux de croissance et les réactifs coûteux, les longs délais pour atteindre des lignes d’expression stables et le risque de contamination par d’autres espèces telles que les champignons ou les bactéries2,11 . De plus, la production de MMP dans les lignées cellulaires de mammifères produit des impuretés à partir de protéines cellulaires associées telles que les TIMPs ou les fibronectines11. Contrairement à la croissance cellulaire lente observée dans les cellules de mammifères, le système d’expression bactérienne offre une production de protéines à grande échelle dans un court laps de temps ainsi que des milieux plus simples et des besoins de croissance. Cependant, en raison de l’absence d’autres protéines cellulaires associées (c.-à-d. les TIMPs) dans les systèmes d’expression bactérienne, les MMP actifs à des concentrations plus élevées sont sujets à la dégradation par autoprotéolyse, ce qui entraîne un faible rendement en MMP17.
Cet article décrit une méthode détaillée pour l’expression bactérienne, la purification et l’activation de Hisx6-pro-MMP-3cd recombinant en utilisant E. coli comme hôte d’expression en raison de son prix abordable, de sa simplicité et de son succès dans la production de rendements plus élevés de MMP2,3,18. Étant donné qu’E. coli n’a pas la machinerie de repliement des protéines et le traitement post-traductionnel requis pour les MMP recombinants et d’autres protéines complexes, de nombreuses souches d’E. coli ont été conçues pour surmonter ces limitations, faisant d’E. coli un hôte plus approprié pour l’expression de MMP-3cd humain recombinant,19,20 . Par exemple, la souche R2DP utilisée dans cette étude améliore l’expression eucaryote en fournissant un plasmide résistant au chloramphénicol contenant des codons rarement utilisés chez E. coli.
Comme décrit dans ce protocole, après surexpression de corps d’inclusion relativement purs du vecteur pET-3a (Figure 1) dans les cellules R2DP, les protéines du domaine catalytique Hisx6-pro-MMP-3 (MMP-3cd) sont extraites et dénaturées4. Hisx6-pro-MMP-3cd3,19 a été purifié par chromatographie par étiquette d’affinité. Lors du repliement et de la dialyse, le pro-MMP-3cd (zymogène) a été activé par l’acétate de 4-aminophénylmercurique (APMA), et l’analyse SDS-PAGE est utilisée pour évaluer les rendements et la nécessité d’une purification supplémentaire5,21. Ce protocole décrit l’expression, la purification et l’activation de MMP-3cd soluble à titre d’exemple. Cependant, il peut également être utilisé comme guide pour l’expression d’autres MMP et protéases humaines ayant une expression similaire, et des mécanismes d’activation (Figure 2). Pour d’autres protéines autres que MMP-3cd, il est conseillé au lecteur de déterminer les compositions tampons optimales et les méthodes pour leur protéine cible avant d’essayer ce protocole.

Figure 1 : Carte plasmidique du plasmide pET-3a-Hisx6-pro-MMP-3cd. Le vecteur pET-3a comprend un gène de résistance à l’ampicilline. Une séquence de balises Hisx6 n-terminales est clonée dans le vecteur pET-3a, y compris pro-MMP-3cd, pour donner la construction pET-3a-Hisx6-pro-MMP-3cd sous contrôle du promoteur T7 entre les sites de restriction BamHI et NdeI. Veuillez cliquer ici pour voir une version agrandie de cette figure.
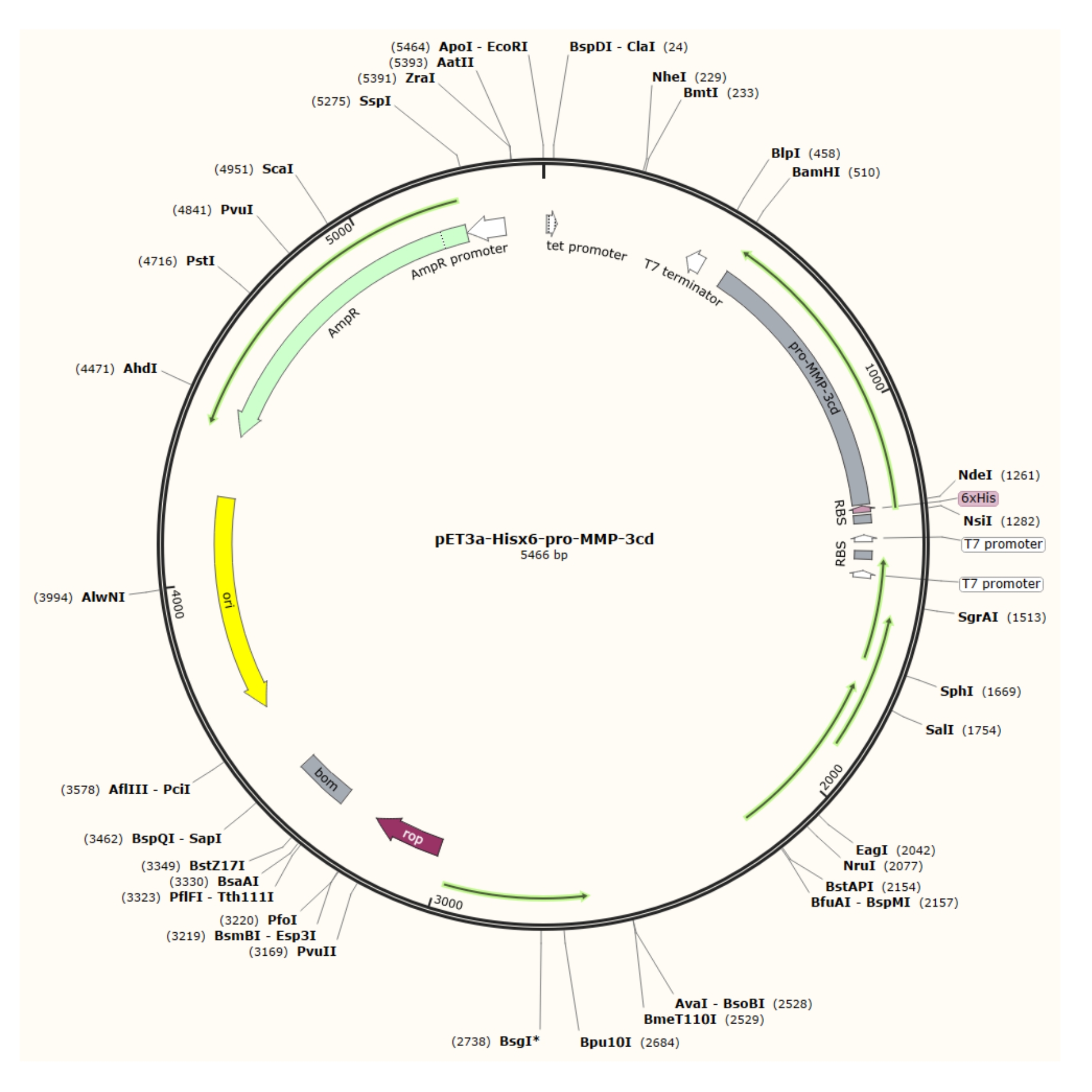{kind=link}

Figure 2 : Expression bactérienne du pro-MMP-3cd, purification, repliement et activation. 1.1 : Le plasmide pET-3a-Hisx6-pro-MMP-3cd a été transformé en cellules BL21(DE3) ou R2DP. 1.2 : L’expression de la protéine Pro-MMP-3cd a été induite à l’aide de l’IPTG. 1.3: La lyse chimique et la sonication sont utilisées pour extraire les protéines Hisx6-pro-MMP-3cd qui sont principalement insolubles et présentes dans les corps d’inclusion. L’urée a été utilisée pour dénaturer et solubiliser les protéines des corps d’inclusion. 2.1. La protéine Hisx6-pro-MMP-3cd dénaturée a été purifiée par purification par chromatographie d’affinité. 3. L’Hisx6-pro-MMP-3cd élué a été lentement replié pendant la dialyse par élimination progressive de l’urée du tampon. 4. Enfin, la protéine MMP-3cd repliée a été activée à l’aide de l’APMA en supprimant le domaine pro-peptidique N-terminal. L’APMA est ensuite retiré de la solution par dessalement. Les numéros correspondent aux sections du protocole décrivant ces étapes. Abréviations : MMP-3cd = Matrice métalloprotéinase-3 domaine catalytique ; APMA = acétate de 4-aminophénylmercurique. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocole
1. Expression MMP
- Clonage et transformation de pET-3a-Hisx6-pro-MMP-3cd en cellules R2DP
- Digérer le plasmide pET-3a (voir le tableau des matériaux) avec les enzymes de restriction NdeI et BamHI dans digest buffer (voir le tableau des matériaux). Dans un volume de réaction total de 40 μL, ajouter 4 μL de tampon de digestion, 33 μL de 100 ng/μL de plasmide et 1,5 μL de chaque enzyme de restriction et laisser la réaction se poursuivre pendant environ 2 h jusqu’à son achèvement à 37 °C.
- Effectuez une réaction PCR sur la séquence MMP-3cd pour insérer une étiquette His-terminalE N. Utilisez 25 μL de mélange PCR (voir le tableau des matériaux), 2,5 μL d’amorces de 10 μM (figure supplémentaire S1) et 1,25 μL de la séquence d’insertion de 100 ng/μL. Ajouter de l’eau stérile à un volume de réaction final de 50 μL.
- Exécutez le produit PCR et le vecteur digéré sur un gel d’agarose à 1%. Purifiez les bandes de gel à l’aide d’un kit de récupération de gel (voir le tableau des matériaux) selon le protocole du fabricant.
- Clonez le produit de PCR amplifié dans le vecteur digéré entre les sites de restriction NdeI et BamHI à l’aide du mélange d’assemblage d’ADN (voir la table des matériaux). Utilisez des outils en ligne pour déterminer le volume requis de l’insert et du vecteur de coupe pour un volume de réaction total de 15 μL.
- Décongeler une aliquote de 50 μL de cellules à haute efficacité de transformation (voir le tableau des matériaux) sur de la glace jusqu’à ce qu’elles soient décongelées. Milieu de croissance SOC préwarme (voir le tableau des matériaux) jusqu’à 37 °C et plaques de LB-ampicilline (LB Amp) (voir le tableau des matériaux).
- Ajouter 1 à 2 μL de la réaction d’assemblage pET-3a-Hisx6-pro-MMP-3cd à l’aliquote de 50 μL. Incuber sur de la glace pendant 30 min.
- Choquez thermiquement les cellules en incubant à 42 °C pendant 30 s. Incuber sur de la glace pendant 2 min.
- Ajouter 950 μL de milieu de croissance SOC à chaque mélange transformant. Agiter pendant 1 h à 250 tr/min et 37 °C.
- Plaque 100 μL des transformants sur des plaques LB Amp et incuber pendant la nuit à 37 °C.
- Inoculer chaque colonie isolée dans 10 mL de milieu LB Amp. Agiter toute la nuit à 250 tr/min et à 37 °C.
- Extraire l’ADN plasmidique selon le protocole du fabricant pour le kit miniprep (voir la table des matériaux). Confirmez la séquence de la construction à l’aide d’amorces avant et arrière T7 (figure supplémentaire S1).
REMARQUE: L’ADN de construction pET-3a-Hisx6-pro-MMP-3cd peut être stocké à -20 ° C. Lorsque vous êtes prêt, procédez à la transformation en cellules R2DP. - Décongeler une aliquote de 20 μL de cellules R2DP (voir la table des matériaux) sur de la glace pendant 2 à 5 minutes. Préchaudissement SOC croissance milieu à température ambiante et LB Amp CamR plaques à 37 °C (voir le tableau des matériaux).
- Ajouter 1 μL de pET-3a-Hisx6-pro-MMP-3cd confirmé par séquence de 100 ng/μL à l’aliquote de 50 μL. Remuer doucement pour mélanger et remettre le tube sur la glace.
- Incuber le tube sur de la glace pendant 5 min.
- Choquez thermiquement les cellules en incubant à 42 °C pendant exactement 30 s. Ne secouez pas.
- Placez les cellules sur de la glace pendant 2 min.
- Ajouter 80 μL de milieu SOC à température ambiante au mélange transformant. Agiter pendant 1 h à 250 tr/min et 37 °C.
- Plaquer les transformants sur des plaques LB Amp CamR et incuber pendant la nuit à 37 °C.
- Croissance et induction
- Inoculer une seule colonie isolée de R2DP pET-3a-Hisx6-pro-MMP-3cd transformant à partir d’une plaque LB Amp CamR dans 10 mL de support LB Amp CamR à 37 °C. Agiter à 250 tr/min pendant la nuit (~16 h). Économisez les aliquotes de chaque culture et préparez des stocks de glycérol à 40% (v/ v) (voir le tableau des matériaux) si vous le souhaitez.
- Par culture de nuit, inoculer une fiole de 1 L contenant 500 mL de milieu LB Amp CamR à une densité optique à 600 nm (OD600) de 0,05-0,1.
REMARQUE: Cela devrait ramener les cellules à la croissance logarithmique. - Mesurez l’OD600 à plusieurs moments, généralement pendant 3-4 h, jusqu’à ce qu’il tombe entre 0,4 et 0,6.
- Avant l’induction, aliquoter une fraction de culture dans un tube de microfuge de 1,5 mL (voir le tableau des matériaux) et l’étiqueter Fraction non induite. Conservez-le à -80 °C pour l’analyse du gel. Si vous n’exécutez pas de gel SDS-PAGE, ignorez cette étape et passez à l’étape 1.2.5.
- Induire les cultures à une concentration finale de 1 mM en utilisant 1 M d’isopropyl-ß-D-thiogalactopyranoside (IPTG) (voir le Tableau des matériaux). Continuer à incuber dans le shaker à 37 °C pendant 3-4 h supplémentaires.
REMARQUE: Pendant l’expression, le lecteur doit déterminer l’OD600 optimal au moment de l’induction et la concentration IPTG. Si le rendement diminue considérablement après la purification, la concentration d’imidazole dans les tampons de purification peut nécessiter un ajustement, ou la pastille de cellule peut avoir besoin d’être soniquer davantage. - Avant de centrifuger les cultures, aliquotez une fraction de culture dans un deuxième tube de microfuge de 1,5 mL et étiquetez-le Fraction induite. Conservez-le à -80 °C pour l’analyse du gel. Si vous n’exécutez pas de gel SDS-PAGE, ignorez cette étape et passez à l’étape 1.2.7.
- Centrifuger la culture cellulaire dans des bouteilles coniques de 250 mL (voir le tableau des matériaux) à vitesse maximale et à 4 °C pendant 10 min.
- Répétez l’étape 1.2.7 jusqu’à ce que les cultures soient complètement granulées.
REMARQUE: PAUSE: Les granulés de cellules peuvent être congelés à -80 ° C et décongelés plus tard pour un traitement ultérieur. Sinon, ignorez cette étape et passez à l’étape 1.3.1.
- Extraction et solubilisation du corps d’inclusion
REMARQUE: Préparer de l’urée fraîche de 10 M, de préférence au plus tôt un jour à l’avance, en remuant soigneusement jusqu’à dissolution complète. Ne pas chauffer ou autoclaver l’urée; conservez-le à température ambiante.- Remettre en suspension la pastille (à partir de l’étape 1.2.8) dans le tampon de lyse (voir le tableau des matériaux). Par gramme de granulé, ajouter 3 mL de tampon de lyse et remettre en suspension par vortex ou pipetage. Agiter toute la nuit à 4 °C.
- Ajouter 1,25 mL de désoxycholate de sodium à 10 % (p/v) (voir le tableau des matériaux) par 1 L de culture. Agiter à température ambiante pendant 30 min à 150 tr/min.
- Ajouter 10 μL de DNase I (voir le tableau des matériaux) pour 1 L de culture. Agiter à température ambiante pendant 30 min à 150 tr/min.
- Centrifuger pendant 10 min à 13 000 × g et 4 °C.
- Réservez une fraction de MMP Lysed pour l’analyse du gel. Conservez-le à -80 °C. Si vous n’effectuez pas d’analyse de gel, passez à l’étape 1.3.6.
REMARQUE: Après centrifugation, la pastille peut être filandreuse et non compacte, ce qui rend risqué de jeter le surnageant. Si tel est le cas, ignorez l’étape 1.3.6 et passez à l’étape 1.3.7. - Jeter le surnageant des échantillons centrifugés.
REMARQUE: Le protocole peut être mis en pause à ce stade et les granulés de cellule congelés à -80 ° C et décongelés plus tard. Sinon, ignorez cette étape et passez à l’étape 1.3.7. - Remettre en suspension la pastille dans une culture de 100 mL/L de tampon pour corps d’inclusion (voir le tableau des matériaux) en pipetant de haut en bas.
- Pendant la sonication, gardez les échantillons sur la glace pour éviter la surchauffe. Soniquez chaque échantillon pendant 6 cycles de 15 s, émettez 5 et 50 % d’impulsion. Prévoyez des périodes de repos de 15 s pour le refroidissement entre les cycles.
REMARQUE: Si nécessaire, transférer les échantillons dans des tubes coniques de 50 mL pour une centrifugation plus poussée (voir le tableau des matériaux). Centrifuger pendant 10 min à 13 000 × g et 4 °C. - Réservez une fraction de MMP sonicated pour l’analyse du gel. Conservez-le à -80 °C. Si vous n’effectuez pas d’analyse de gel, passez à l’étape 1.3.11.
- Vérifiez la pastille. Si elle est filandreuse, répétez les étapes 1.3.8 à 1.3.10. Si la pastille est compacte, jetez le surnageant et passez à l’étape 1.3.12.
REMARQUE: La sonication dans le tampon corporel d’inclusion peut être répétée pour récupérer plus de protéines des débris cellulaires lysés. Cependant, trop de sonication peut provoquer un cisaillement, ce qui nuit au rendement MMP. Le protocole peut être mis en pause à ce stade, et les granulés cellulaires peuvent être congelés à -80 ° C et décongelés plus tard. - Remettre en suspension chaque pastille d’une culture de 1 L dans 5 mL de tampon de solubilisation (voir le tableau des matériaux) par pipetage. Incuber pendant au moins 30 min sur de la glace pour permettre aux protéines de se solubiliser.
- Mettez de côté une fraction de MMP solubilisée pour l’analyse du gel. Conservez-le à -80 °C. Si vous n’effectuez pas d’analyse de gel, passez à l’étape 1.3.14.
- Centrifuger les cellules pendant 10 min à 13 000 × g et 4 °C. NE PAS JETER LE SURNAGEANT.
- Si une pastille se forme/reste après centrifugation, versez le surnageant dans un tube conique séparé de 50 mL. Remettre en suspension la pastille dans un autre tampon de solubilisation de 5 mL (pour 1 L de culture) en pipetant de haut en bas.
- Centrifuger pendant 10 min à 13 000 × g et 4 °C. NE PAS JETER LE SURNAGEANT.
- Répétez les étapes 1.3.13 et 1.3.14 jusqu’à ce qu’il ne se forme qu’un peu ou pas de granulés après centrifugation ou qu’il ne reste que du précipité gris. Mettez en commun les surnageants. Jeter ou stocker la pastille à -80 °C pour une sonication supplémentaire.
2. Purification et repliement MMP
- Purification d’affinité His-tag (HT)
- Selon le protocole du fabricant, remplissez une colonne d’écoulement gravitaire (voir le tableau des matériaux) avec de la résine Ni-NTA bien mélangée (voir le tableau des matériaux). Laissez la résine se déposer et se séparer du tampon de stockage de sorte qu’une ligne distincte se forme entre les deux couches.
REMARQUE: Ne laissez jamais la résine sécher, car l’air pénétrera dans la résine et nuira au rendement en protéines. Entre les utilisations, effectuer la procédure de régénération de la résine décrite à la section 2.2. - Laissez le tampon de stockage s’écouler. Remplissez la colonne avec deux volumes de lit de résine de tampon d’équilibrage HT.
- Videz le tampon d’équilibrage HT et jetez-le. Pendant que la colonne s’écoule, centrifugez l’extrait de protéine à 13 000 × g pendant 1 min et filtrez-stérilisez à l’aide d’un filtre de 0,22 μm (voir le tableau des matériaux).
- Remplacez le conteneur à déchets par un tube conique de 50 mL étiqueté HT Flowthrough. Ajouter l’extrait de protéine préparé à la colonne.
- Réappliquez le flowthrough pour maximiser la liaison.
- Réservez une fraction de Flowthrough Fraction pour l’analyse du gel. Conservez-le à -80 °C. Si vous n’effectuez pas d’analyse de gel, passez à l’étape 2.1.7.
- Lavez immédiatement la résine avec 15 mL de tampon de lavage HT (voir le tableau des matériaux). Collectez le débit dans des tubes coniques de 15 mL (voir la Table des matériaux) étiquetés HT Wash.
NOTE: Les valeurs d’absorbance à 280 nm (A280) ont été obtenues par spectrophotométrie et utilisées avec le poids moléculaire et le coefficient d’extinction, ε, pour estimer les concentrations de protéines. Pour Hisx6-pro-MMP-3cd dénaturé, le poids moléculaire est de 29,86 kDa et ε est de 34,38 M-1 cm-1. - Occultation contre LE tampon de lavage HT, mesurez et enregistrez l’A280. Répétez les étapes 2.1.7 et 2.1.8 avec des fractions de lavage supplémentaires. Une fois que l’A280 s’approche de la ligne de base et que les impuretés ont été réduites au minimum, passez à l’étape 2.1.9.
- Réservez une fraction de Wash Fraction pour l’analyse du gel. Répétez l’opération pour plusieurs fractions de lavage. Conserver les fractions à -80 °C. Si vous n’effectuez pas d’analyse de gel, passez à l’étape 2.1.10.
- Éluez immédiatement les protéines marquées His en ajoutant 5 mL de tampon d’élution HT (voir le tableau des matériaux). Collectez le débit sous forme de fractions de 0,5 à 1 mL dans des tubes de microfuge étiquetés HT Elution.
- Réservez une fraction d’Elution Fraction pour l’analyse du gel. Répétez l’opération pour plusieurs fractions d’élution. Conserver les fractions à -80 °C. Si vous n’effectuez pas d’analyse de gel, passez à l’étape 2.1.12.
- Si l’A280 est >0,3 mg/mL, diluer la fraction avec un tampon d’équilibrage HT (voir le tableau des matériaux).
REMARQUE: La fraction éluée doit être diluée dans un A280 de 0,3 mg / mL ou moins pour éviter les précipitations pendant la dialyse. Le protocole peut être mis en pause ici et les fractions regroupées congelées à -80 °C et décongelées plus tard. Sinon, ignorez cette étape et passez à l’étape 2.2.1.
- Selon le protocole du fabricant, remplissez une colonne d’écoulement gravitaire (voir le tableau des matériaux) avec de la résine Ni-NTA bien mélangée (voir le tableau des matériaux). Laissez la résine se déposer et se séparer du tampon de stockage de sorte qu’une ligne distincte se forme entre les deux couches.
- Régénération de la résine
- Lavez la résine avec dix volumes de lit de résine de tampon de régénération HT (voir le tableau des matériaux) et dix volumes de lit de résine d’eau stérile.
- Conservez la résine sous forme de boue à 50 % dans de l’éthanol à 20 % (v/v) dans de l’eau.
3. Repliement des protéines
REMARQUE: Pour les petits volumes, les cassettes de dialyse peuvent être utilisées avec un risque plus faible de perte d’échantillon. Des tubes de dialyse sont nécessaires si de plus grands volumes sont utilisés (voir le tableau des matériaux).
- Dialyse
REMARQUE: La concentration optimale en protéines pour la dialyse est d’environ 0,3 mg / mL. Si des précipitations importantes se produisent pendant la dialyse, réduire le gradient de concentration d’urée entre chaque dialyse en utilisant une méthode de dialyse par étapes et ajouter plus d’étapes intermédiaires (par exemple, de 6 M à 5 M, puis de 5 M à 4 M, plutôt que de sauter l’étape de 5 M). Comme les cycles de gel-dégel endommagent la structure cellulaire et protéique, il est essentiel de minimiser les pauses dans le protocole.- Selon le protocole du fabricant, utilisez la quantité appropriée de tubes de dialyse en fonction du volume d’échantillons de fraction d’élution .
- Immerger les fractions MMP éluées dans un tube de dialyse dans 1 L de tampon de dialyse 1 (voir la table des matériaux). Remuer le tube et son contenu sur un agitateur magnétique pendant au moins 8 h à 4 °C.
- Transférer à 1 L du tampon de dialyse 2 (voir le tableau des matériaux). Remuer le tube et son contenu sur un agitateur magnétique pendant au moins 8 h à 4 °C.
- Transfert à 1 L du tampon de dialyse 3 (voir le tableau des matériaux). Remuer le tube et son contenu sur un agitateur magnétique pendant au moins 8 h à 4 °C.
- Transférer l’échantillon dans de nouveaux tubes coniques de 50 mL et les étiqueter comme MMP dialyzés.
- Examinez le tube à la recherche de tout précipité. Si un précipité s’est formé, centrifuger l’échantillon pendant 1 min à 13 000 × g et 4 °C.
- Transférer le surnageant dans de nouveaux tubes coniques de 15 mL et les étiqueter comme MMP repliés.
- Réservez une fraction pour l’analyse du gel et étiquetez-la comme MMP repliée. Conservez-le à -80 °C. Si vous n’effectuez pas d’analyse de gel, passez à l’étape 3.1.9.
REMARQUE: Si les rendements sont faibles, le précipité peut être dissous dans le tampon d’équilibrage HT et les étapes de la section 3.1 répétées avec des tubes de dialyse. Si l’analyse du gel ne doit pas être effectuée ou si le protocole doit être interrompu ici, congeler les échantillons à -80 °C et les décongeler plus tard. Si les rendements se situent dans la fourchette souhaitée, passez à l’étape 3.2.1.
- Reconcentration
REMARQUE: Les coefficients d’extinction pour Hisx6-pro-MMP-3cd replié et dénaturé devraient être les mêmes; par conséquent, les calculs A280 ne sont pas affectés.- Reconcentrer l’échantillon jusqu’à 0,5 mg/mL. Utilisez une cellule agitée de 400 mL (voir le tableau des matériaux) pour concentrer l’échantillon à 15 mL. Pour éviter la formation de mousse, utilisez un tube de reconcentration de 50 mL pour vous concentrer davantage si nécessaire.
REMARQUE: Si un précipité se forme, il peut être granulé et dissous dans le tampon d’équilibrage HT. Ensuite, répétez les sections 3.1 et 3.2.1. Sinon, passez à l’étape 3.2.2. - Réservez une fraction pour l’analyse du gel et étiquetez-la comme MMP concentrée.
REMARQUE: Le protocole peut être mis en pause ici et les échantillons congelés à -80 ° C et décongelés plus tard.
- Reconcentrer l’échantillon jusqu’à 0,5 mg/mL. Utilisez une cellule agitée de 400 mL (voir le tableau des matériaux) pour concentrer l’échantillon à 15 mL. Pour éviter la formation de mousse, utilisez un tube de reconcentration de 50 mL pour vous concentrer davantage si nécessaire.
4. Activation
- Activation de l’acétate de 4-aminophénylmercurique (APMA)
REMARQUE: L’APMA est très toxique. Préparez une solution mère fraîche de 20 mM d’APMA avant l’activation et travaillez toujours sous une hotte lorsque vous utilisez l’APMA. Jetez les déchets de l’APMA dans son conteneur.- Par aliquote de 1 mL de MMP (1 mg/mL), ajouter 50 μL d’APMA de 20 mM (voir le Tableau des matériaux) pour atteindre une concentration finale d’APMA de 1 mM. Incuber toute la nuit à 37 °C.
- Si un précipité se forme, centrifugez-le à la vitesse maximale pendant 10 min à 4 °C. Conservez le surnageant dans un tube de microfuge de 1,5 mL portant la mention MMP activée. Jeter le précipité dans un récipient marqué pour les déchets APMA.
- Réservez une fraction pour l’analyse du gel et étiquetez-la MMP activée.
REMARQUE: Le protocole peut être mis en pause ici et les échantillons congelés à -80 ° C et décongelés plus tard. Si vous n’effectuez pas d’analyse de gel, passez à l’étape 4.2.1. Après activation, le poids moléculaire et le coefficient d’extinction de MMP-3cd sont respectivement de 19,40 kDa et 28,42 M-1 cm-1.
- Dessalage
- Retirez l’APMA de l’échantillon MMP-3cd activé à l’aide d’une colonne de dessalement de 2 mL (voir la table des matériaux), conformément au protocole du fabricant.
- Réservez une fraction pour l’analyse du gel et étiquetez-la MMP dessalée. Conservez-le à -80 °C. Si vous n’effectuez pas d’analyse de gel, passez à la rubrique 4.3 avec les échantillons restants.
REMARQUE: Le protocole peut être mis en pause ici et les échantillons congelés à -80 ° C et décongelés plus tard.
- Exécution des gels SDS-PAGE
- Exécutez toutes les fractions de protéines sur les gels SDS-PAGE : Fraction non induite, Fraction induite, MMP lysée, MMP sonique, MMP solubilisée, Fraction fluide, Fraction de lavage, Fraction d’élution, MMP repliée, MMP concentrée, MMP activée et MMP dessalée.
- Stockage à long terme du MMP-3
- Ajouter un tensioactif non ionique à 0,05 % (v/v) (voir le tableau des matériaux) aux échantillons MMP-3cd dessalés et les conserver à -80 °C.
Access restricted. Please log in or start a trial to view this content.
Résultats
Lors de l’exécution d’échantillons sur SDS-PAGE, parce que la protéine est exprimée sous la forme de corps d’inclusion insolubles, les fractions lysées et soniquées doivent contenir peu ou pas d’extrait Hisx6-pro-MMP-3cd, car la protéine n’a pas encore été résolubilisée dans l’urée. La figure 3 compare les fractions d’élution de purification His-tag de Hisx6-pro-MMP-3cd à partir de cellules BL21(DE3) et de cellules R2DP. Les fractions d’élution ont été regrou...
Access restricted. Please log in or start a trial to view this content.
Discussion
La production à grande échelle de MMP solubles, humains et recombinants reste une tâche difficile. Les cellules de mammifères peuvent exprimer des MMP fonctionnels à des coûts élevés et de longs temps d’attente, tandis que E. coli produit rapidement de grandes quantités de corps d’inclusion MMP qui doivent être purifiés et repliés11,16. Les cellules R2DP augmentent considérablement le rendement des corps d’inclusion MMP, permettant un ...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
Les auteurs déclarent qu’ils n’ont pas d’intérêts financiers concurrents.
Remerciements
Les auteurs aimeraient remercier le Dr Evette Radisky et Alexandra Hockla de la Mayo Clinic de Jacksonville, en Floride, pour avoir fourni le plasmide pET-3a-pro-MMP-3cd comme modèle de clonage du gène Hisx6pro-MMP-3cd, ainsi que le Dr Paul Hartley du Nevada Genomics Center de l’Université du Nevada, Reno, pour le séquençage de l’ADN. Les auteurs aimeraient également remercier Cassandra Hergenrader pour avoir aidé à une partie de l’expression des protéines. M.R.-S. tient à remercier la subvention NIH-P20 GM103650-COBRE Integrative Neuroscience et le prix UNR R&D mICRO SEED Grant Award.
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| 0.22 µm sterile filter | Sigma Aldrich | SLGP033RS | Used to remove some contaminants from the protein extract before purification, and prevent the Ni-NTA column from clogging |
| 1 L Erlenmeyer flasks | Thermo Fisher Scientific | S76106F | n/a |
| 1 L glass bottles | Thermo Fisher Scientific | 06-414-1D | n/a |
| 1.5 mL microfuge tubes | Thermo Fisher Scientific | 02-682-002 | n/a |
| 15 mL conical tubes | Thermo Fisher Scientific | 339650 | n/a |
| 18 G, 1-in. beveled needle | Amazon | B07S7VBHM2 | Used in combination with the dialysis casette |
| 2 mL desalting column | Thermo Fisher Scientific | 89890 | Removes APMA following activation |
| 2-(N-Morpholino)ethanesulfonic acid (MES) | Thermo Fisher Scientific | AAA1610422 | n/a |
| 250 mL conical bottle cushions | Thermo Fisher Scientific | 05-538-53A | Stabilize conical bottles during large-volume centrifugation |
| 250 mL conical bottles | Thermo Fisher Scientific | 05-538-53 | n/a |
| 400 mL stirred cell | Sigma Aldrich | UFSC40001 | Re-concentrates a much larger volume than the centrifugal filter unit. Rosetta2(DE3)pLysS cells produce high volumes of protein that may exceed the 15 mL limit of the centrifugal filter unit |
| 4-aminophenylmercuric acetate (APMA) | Sigma Aldrich | A9563-5G | Activates MMP-3 by cleaving the propeptide |
| 5 mL syringe | Thermo Fisher Scientific | NC0829167 | Used in combination with the dialysis casette |
| 50 mL conical tubes | Thermo Fisher Scientific | 339650 | Used for storage in many purification steps |
| 50 mL re-concentration tube | Sigma Aldrich | UFC901024D | Used for re-concentrating protein samples after dialysis or removing contaminants |
| Agar | Thermo Fisher Scientific | BP1423-500 | Buffer ingredient that solidifies autoclaved LB media upon cooling |
| Ampicillin | Thermo Fisher Scientific | BP1760-25 | Antibiotic used with pET3a vector; used at 100 µg/mL in LB media |
| BamHI | NEB | R3136S | Restriction enzyme to be used with the pET3a vector |
| Calcium chloride (CaCl2) | Thermo Fisher Scientific | 600-30-23 | The calcium ion stabilizes MMP structure |
| Cell spreaders | Thermo Fisher Scientific | 50-189-7544 | Can be used to spread cells across a petri dish after transformation |
| Chloramphenicol | Thermo Fisher Scientific | 22-055-125GM | Antibiotic used with pET3a vector; used at 34 µg/mL in LB media |
| Dialysis Buffer 1 | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM CaCl2, 1 µM ZnCl2, 4 M Urea. |
| Dialysis Buffer 2 | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM CaCl2, 1 µM ZnCl2, 2 M Urea. |
| Dialysis Buffer 3 | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM CaCl2 , 1 µM ZnCl2. |
| Dialysis clips | Thermo Fisher Scientific | 68011 | Used in combination with snakeskin dialysis tubing |
| Dialysis tubing | Thermo Fisher Scientific | 88243 | Alternative dialysis method that holds much larger sample volumes, but with higher risk of sample loss |
| Digest buffer | NEB | B7204S | Buffer used in digesting the pET3a vector |
| Disposable cuvettes | Thermo Fisher Scientific | 21-200-257 | Used to measure the bacterial culture OD during growth and expression |
| Dithiothreitol (DTT) | Thermo Fisher Scientific | D107125G | Assists with protein denaturation by reducing any disulfide bonds |
| DNA assembly mix | NEB | E2621S | Used to ligate the Hisx6-pro-MMP-3cd PCR product and digested pET3a vector |
| DNase I | NEB | M0303S | Endonuclease for degrading unfavorable DNA contaminants that could later affect protein purification |
| Ethanol | Thermo Fisher Scientific | A995-4 | n/a |
| Ethylenediaminetetraacetic acid (EDTA) | Thermo Fisher Scientific | J15694-AE | Used in denaturation. Prevents oxidation and subsequent formation of disulfide bonds |
| Gel recovery kit | Promega | A9281 | Isolates and purifies DNA from agarose gels |
| Glycerol | Thermo Fisher Scientific | G33-500 | Used for making glycerol stocks, which are frozen at -80 °C |
| Gravity flow column | BioRad | 7321010 | Used for Ni-NTA purification of recombinantly His-tagged proteins |
| High-transformation efficiency cells | NEB | C2987 | High-transformation efficiency cells with greater chance of success for cloning the N-terminal His-tag into the pET3a-pro-MMP-3cd construct |
| HT Elution Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 6 M urea, 250 mM imidazole. Adjust pH to 7.4 |
| HT Equilibration Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 6 M urea. Adjust pH to 7.4 |
| HT Regeneration Buffer | n/a | n/a | 20 mM MES, 0.1 M NaCl. Adjust pH to 5.0 |
| HT Wash Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 6 M urea, 25 mM imidazole. Adjust pH to 7.4 |
| Hydrochloric acid (HCl) | Thermo Fisher Scientific | A144C-212 | Used to pH buffers |
| Imidazole | Thermo Fisher Scientific | AAA1022122 | Mimics the histidine side group. Used to separate non-specifically binding proteins from the his-tagged target protein |
| Inclusion Body Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 1 mM EDTA, 100 mM NaCl, 5 mM DTT, 2% v/v Triton X 100, 0.5 M Urea. Adjust pH to 8.0 |
| Isopropyl-ß-D-thiogalactopyranoside (IPTG) | Thermo Fisher Scientific | FERR0392 | A reagent that induces target gene expression in pET3a. Make 0.5 mL 1 M aliquots, filter sterilize and store in -20 °C |
| LB Amp CamR media | n/a | n/a | To be poured into a sterible 1 L bottle or 1 L flask. For 1 L, add 25 g LB Broth. Sterilize by autoclaving. Once cooled to below 50 °C, add ampicillin to 100 µg/mL and chloramphenicol to 34 µg/mL |
| LB Amp CamR plates | n/a | n/a | To be poured into sterile petri dishes. Pour until the petri dish lid is completely covered. 1 L of media yields 40-60 plates. For 1 L: 25 g LB Broth, 16 g Agar. Sterilize by autoclaving. Once cooled to below 50 °C, add ampicillin to 100 µg/mL and chloramphenicol to 34 µg/mL |
| LB Broth | Thermo Fisher Scientific | BP1426-2 | Pre-mixed with tryptone, yeast extract, and sodium chloride |
| Lysis Buffer | n/a | n/a | 50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 100 mM NaCl, 0.133 g/mL lysozyme, 0.49% v/v Triton X-100. Adjust pH to 8.0 |
| Lysozyme | MP Biomedicals | 195303 | Used in protein extraction. Enzyme that lyses bacterial cell walls |
| Miniprep kit | Promega | A1330 | If successful, extracts the pET3a-pro-MMP-3cd construct from transformants |
| NdeI | NEB | R0111S | Restriction enzyme to be used with the pET3a vector |
| Ni-NTA resin | Thermo Fisher Scientific | PI88221 | Used to bind recombinant his-tagged proteins. This strong interaction can be displaced with higher concentrations of imidazole |
| Nonionic surfactant | Thermo Fisher Scientific | PI28316 | Storage detergent for preventing MMP aggregation. Minimizes interactions between hydrophobic residues on the MMP surface and water molecules, without disrupting catalytic activity. |
| PCR mix | NEB | M0492S | A PCR reagent for inserting an N-terminal his-tag into the pET3a-pro-MMP-3cd vector |
| pET plasmid | Addgene | n/a | The pET3a vector offers ampicillin resistance, inducible expression of a target gene, and sequencing with T7 primers |
| Petri dishes | VWR | 25384-342 | Used for plating transformants on LB agar media |
| R2DP cells | Novagen | 714033 | BL21 derivatives with enhanced expression of eukaryotic proteins. Contain tRNAs of codons found to be rare in e. coli |
| SOC growth media | NEB | B9020S | Non-selective growth media for rapid growth during transformation |
| Sodium chloride (NaCl) | Thermo Fisher Scientific | BP358-1 | Used in buffers and helps with protein stability |
| Sodium deoxycholate | Thermo Fisher Scientific | PI89905 | Detergent used in protein extraction. Lyses cell walls |
| Solubilization Buffer | n/a | n/a | 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 10 mM DTT, 6 M Urea. Adjust pH to 8.0 |
| Tris base | Thermo Fisher Scientific | BP152-1 | Common buffer used in the physiological pH range. Temperature-sensitive |
| Triton X-100 | Thermo Fisher Scientific | M1122980101 | Detergent used for cell lysis |
| Urea | Thermo Fisher Scientific | AAJ75826A7 | First chaotropic agent for disrupting protein secondary structure |
| Zinc chloride (ZnCl2) | Thermo Fisher Scientific | AAA162810E | Stabilizes MMP structure. The zinc ion is found in the catalytic site of MMP-3 |
Références
- Portolano, N., et al. Recombinant protein expression for structural biology in HEK 293F suspension cells: A novel and accessible approach. Journal of Visualized Experiments: JoVE. (92), e51897(2014).
- Subedi, G. P., Johnson, R. W., Moniz, H. A., Moremen, K. W., Barb, A. High yield expression of recombinant human proteins with the transient transfection of HEK293 cells in suspension. Journal of Visualized Experiments: JoVE. (106), e53568(2015).
- Nilvebrant, J., Alm, T., Hober, S. Orthogonal protein purification facilitated by a small bispecific affinity tag. Journal of Visualized Experiments: JoVE. (59), e3370(2012).
- Yang, Z., et al. Highly efficient production of soluble proteins from insoluble inclusion bodies by a two-step-denaturing and refolding method. PLoS One. 6 (7), 22981(2011).
- Hu, X., Beeton, C. Detection of functional matrix metalloproteinases by zymography. Journal of Visualized Experiments: JoVE. (45), e2445(2010).
- Radisky, E. S., Raeeszadeh-Sarmazdeh, M., Radisky, D. C. Therapeutic potential of matrix metalloproteinase inhibition in breast cancer. Journal of Cellular Biochemistry. 118 (11), 3531-3548 (2017).
- Raeeszadeh-Sarmazdeh, M., Do, L. D., Hritz, B. G. Metalloproteinases and their inhibitors: Potential for the development of new therapeutics. Cells. 9 (5), 1313(2020).
- Nagase, H., Visse, R., Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovascular Research. 69 (3), 562-573 (2006).
- Raeeszadeh-Sarmazdeh, M., et al. Directed evolution of the metalloproteinase inhibitor TIMP-1 reveals that its N- and C-terminal domains cooperate in matrix metalloproteinase recognition. Journal of Biological Chemistry. 294 (24), 9476-9488 (2019).
- Batra, J., et al. Matrix metalloproteinase-10 (MMP-10) interaction with tissue inhibitors of metalloproteinases TIMP-1 and TIMP-2. Journal of Biological Chemistry. 287 (19), 15935-15946 (2012).
- Singh, K. K., Jain, R., Ramanan, H., Saini, D. K. Expression and purification of matrix metalloproteinases in Escherichia coli. Matrix Metalloproteases. Galea, C. A. , Humana Press. New York, NY. 3-16 (2017).
- Manka, S. W., et al. Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proceedings of the National Academy of Sciences of the United States of America. 109 (31), 12461-12466 (2012).
- Gomis-Ruth, F. X., et al. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature. 389 (6646), 77-81 (1997).
- Shirian, J., et al. Converting a broad matrix metalloproteinase family inhibitor into a specific inhibitor of MMP-9 and MMP-14. FEBS Letters. 592 (7), 1122-1134 (2018).
- Li, C., et al. Purification of recombinant histidine-tagged catalytic domain of MMP-13 in one step using affinity column and renaturation of it with histidine tag. Journal of Liquid Chromatography & Related Technologies. 37 (15), 2118-2130 (2014).
- Aydin, H., Azimi, F. C., Cook, J. D., Lee, J. E. A convenient and general expression platform for the production of secreted proteins from human cells. Journal of Visualized Experiments: JoVE. (65), e4041(2012).
- McNiff, M. L., Haynes, E. P., Dixit, N., Gao, F. P., Laurence, J. S. Thioredoxin fusion construct enables high-yield production of soluble, active matrix metalloproteinase-8 (MMP-8) in Escherichia coli. Protein Expression and Purification. 122, 64-71 (2016).
- Maity, R., et al. GST-His purification: A two-step affinity purification protocol yielding full-length purified proteins. Journal of Visualized Experiments: JoVE. (80), e50320(2013).
- Stefan, A., Ceccarelli, A., Conte, E., Montón Silva, A., Hochkoeppler, A. The multifaceted benefits of protein co-expression in Escherichia coli. Journal of Visualized Experiments: JoVE. (96), e52431(2015).
- Yadavalli, R., Sam-Yellowe, T. HeLa based cell free expression systems for expression of Plasmodium rhoptry proteins. Journal of Visualized Experiments: JoVE. (100), e52772(2015).
- Zeytuni, N., Zarivach, R. Purification of the M. magneticum strain AMB-1 magnetosome associated protein MamAΔ41. Journal of Visualized Experiments: JoVE. (37), e1844(2010).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.