
Method Article
Une procédure rationalisée et standardisée pour générer des vecteurs viraux adéno-associés de haute qualité et à forte teneur en titres à l’aide d’une plate-forme d’usine cellulaire
Dans cet article
Résumé
Alors que le domaine de la thérapie génique continue d’évoluer, il existe un besoin croissant de méthodes innovantes capables de relever ces défis. Ici, une méthode unique est présentée, qui rationalise le processus de génération de vecteurs AAV à haut rendement et de haute pureté à l’aide d’une plate-forme d’usine de cellules, répondant aux normes de qualité des études in vivo .
Résumé
La recherche préclinique en thérapie génique, en particulier sur des modèles de rongeurs et de grands animaux, nécessite la production de vecteurs AAV à haut rendement et pureté. Les approches traditionnelles dans les laboratoires de recherche impliquent souvent une utilisation intensive de boîtes de culture cellulaire pour cultiver HEK293T cellules, un processus qui peut être à la fois laborieux et problématique. Ici, une méthode interne unique est présentée, qui simplifie ce processus avec une plate-forme spécifique d’usine de cellules (ou d’empilements de cellules, CF10). L’intégration d’une séparation biphasique polyéthylèneglycol/aqueuse avec une ultracentrifugation à gradient d’iodixanol améliore à la fois le rendement et la pureté des vecteurs AAV générés. La pureté des vecteurs AAV est vérifiée par SDS-PAGE et coloration à l’argent, tandis que le rapport entre les particules pleines et vides est déterminé à l’aide de la microscopie électronique à transmission (MET). Cette approche offre une plate-forme d’usine cellulaire efficace pour la production de vecteurs AAV à haut rendement, associée à une méthode de purification améliorée pour répondre aux exigences de qualité des études in vivo .
Introduction
Les vecteurs de virus adéno-associés (AAV) sont devenus un outil indispensable dans la recherche en thérapie génique, offrant une combinaison unique d’efficacité et d’innocuité pour l’administration de gènes1. Les méthodes traditionnelles de génération d’AAV en laboratoire ont joué un rôle essentiel dans l’avancement de notre compréhension et de notre application de la thérapie génique2. Cependant, ces méthodes, bien que fondamentales, présentent certaines limites et défis, notamment en termes de rendement, d’efficacité temporelle et de qualité des vecteurs produits, notamment le rapport entre les particules pleines et les particules vides3.
Le procédé conventionnel de production d’AAV implique principalement la transfection de cellules HEK2934. Ce processus, généralement mené dans des boîtes de culture cellulaire, nécessite que les cellules soient transfectées avec un plasmide contenant le gène d’intérêt ainsi qu’un plasmide auxiliaire et un plasmide de capside AAV 5,6. Après la transfection, les cellules produisent des particules d’AAV, qui sont ensuite récoltées et purifiées 5,6. Le processus de purification implique souvent l’ultracentrifugation, une étape critique dans l’obtention de vecteurs AAV de haute pureté7. L’ultracentrifugation, en particulier à l’aide de chlorure de césium (CsCl) ou d’un gradient d’iodixanol, est une méthode standard pour séparer les particules d’AAV des débris cellulaires et autres impuretés8. Cette étape est cruciale pour atteindre la pureté et la concentration souhaitées des vecteurs AAV, ce qui a un impact direct sur leur efficacité dans l’administration des gènes8. Malgré son utilisation répandue, l’ultracentrifugation traditionnelle a ses inconvénients. Par exemple, le rendement des vecteurs AAV de cette méthode peut être variable et souvent faible, ce qui pose des défis importants lorsque de grandes quantités de vecteurs à titre élevé sont nécessaires, en particulier pour les études in vivo ou les grands modèles animaux9.
Un autre aspect critique de la qualité du vecteur AAV est le rapport entre les particules pleines et les particules videsde 10. Les préparations AAV contiennent souvent un mélange de ces particules ; Cependant, seules les particules complètes contiennent le matériel génétique thérapeutique. La présence d’une forte proportion de particules vides peut réduire considérablement l’efficacité de l’administration des gènes10. L’évaluation et l’optimisation du rapport entre les particules pleines et les particules vides est donc un paramètre clé dans l’évaluation de l’efficacité des vecteurs AAV. Les méthodes traditionnelles, bien qu’elles soient capables de produire des vecteurs AAV, ont souvent du mal à contrôler ce rapport de manière constante, ce qui entraîne des variations de la puissance du vecteur10.
Ici, une méthode unique est présentée, qui rationalise le processus de génération de vecteurs AAV à haut rendement et de haute pureté à l’aide d’une plate-forme d’usine cellulaire exempte d’utilisation de cultures cellulaires HEK293T à forte intensité de main-d’œuvre dans des boîtes cellulaires, intégrant une séparation biphasique polyéthylène glycol/aqueuse avec ultracentrifugation à gradient d’iodixanol. La pureté du vecteur AAV est confirmée par SDS-PAGE et coloration à l’argent, et le rapport entre les particules pleines et vides est déterminé à l’aide de la microscopie électronique à transmission (MET), ce qui répond aux normes de qualité des études in vivo 11.
Protocole
Les détails des réactifs, des plasmides et de l’équipement utilisés dans l’étude sont répertoriés dans la table des matériaux. La composition des tampons utilisés est fournie dans le Fichier supplémentaire 1.
1. Préparation du plasmide
- Plasmides transformants (pAAV-GOI, pHelper, pAAV-Cap) chez E. coli.
REMARQUE : N’importe quelle souche d’E. coli peut être utilisée pour l’amplification plasmidique. Pour certains plasmides, des cellules compétentes spéciales telles que NEB stable peuvent améliorer le rendement plasmidique. Les trois plasmides utilisés dans ce protocole sont pAAV-GOI : pAAV-CMV-GFP, pHelper : pAdDeltaF6, et pAAV-Cap : pAAV-RC6. - Cultivez des bactéries dans un milieu LB contenant les antibiotiques sélectifs appropriés dans un volume optimal à 37 °C pendant 16 à 18 h.
REMARQUE : En cas d’utilisation de cellules stables NEB, cultiver à 30 °C pendant 18 à 20 h. - Récoltez l’ADN plasmidique en centrifugeant E . coli. milieu de culture à 4000 x g, 4 °C, pendant 15 min.
- Versez avec précaution le liquide surnageant de la bouteille de centrifugation dans un récipient à déchets et purifiez l’ADN plasmidique de la pastille avec un kit de purification de plasmide sans endotoxines à grande échelle.
REMARQUE : Assurez-vous de le verser lentement et d’éviter les éclaboussures. - Mesurez la concentration et la pureté de l’ADN à l’aide d’un spectrophotomètre.
REMARQUE : Vérifiez la pureté de l’ADN en vérifiant le rapport OD260/OD280 . Le rapport pour l’ADN pur est d’environ 1,8. La concentration d’ADN doit être supérieure à 1 μg/μL pour l’étape de co-transfection ultérieure. Préparez des stocks de glycérol bactérien en ajoutant 500 μL de la culture de nuit à 500 μL de glycérol à 50 % dans un flacon cryogénique et conservez-les à -80 °C.
2. Préparation des cellules HEK293T
- Ensemez HEK293T cellules avec un milieu à haute teneur en glucose DMEM contenant 10 % de FBS dans une usine de cellules à 10 couches (CF10) et laissez les cellules se développer dans l’incubateur pendant la nuit.
REMARQUE : Les passages des cellules 293T doivent être inférieurs à 10 pour avoir les conditions optimales pour une production robuste d’AAV. Si possible, n’ajoutez pas d’antibiotiques dans le milieu de culture pour le moment. CF10 a environ 42 fois la surface de croissance d’une boîte de culture cellulaire de 150 mm. Ensemencez la même densité cellulaire dans une boîte de culture cellulaire de 150 mm pour permettre de vérifier la croissance cellulaire au microscope. Préparez une suspension cellulaire de 1075 ml, ajoutez une suspension cellulaire de 25 ml dans un plat de 150 mm et ajoutez le reste dans le CF10. - Vérifiez la confluence des cellules le lendemain avant la transfection.
REMARQUE : Les cellules doivent atteindre une confluence de 80 à 90 % au moment de la transfection en observant au microscope.
3. Triple transfection des plasmides AAV
- Calculez la quantité de chaque plasmide nécessaire (pAAV-GOI, pHelper, sérotype pAAV) pour avoir un rapport molaire de 1,2 : 1:1 avec 2,5-5 mg d’ADN total par CF10.
REMARQUE : Le rapport molaire des trois plasmides peut être variable pour une efficacité de transfection optimale. Il est préférable de le tester à petite échelle avant de passer à ce réglage CF10. pAAV-CMV-GFP, pAdDeltaF6 et pAAV-RC6 sont utilisés pour fabriquer le virus AAV6-CMV-GFP à titre d’exemple. La formule du calculateur de l’Île-du-Prince-Édouard est indiquée au tableau 1. - Aliquote 350 mL d’OptiMEM dans un flacon stérile de 500 mL.
- Ajoutez les trois ADN plasmidiques dans la bouteille contenant l’Opti-MEM. Bien mélanger.
- Ajouter 15 mL d’une solution de polyéthylénimine (PEI) à 1 mg/mL (rapport ADN/μg d’ADN/μg PEI). Agitez vigoureusement le mélange OptiMEM/DNA/PEI de haut en bas pendant 30 s.
REMARQUE : Il n’y a pas de mal à faire des bulles. - Incuber le mélange à température ambiante pendant 10-15 min.
REMARQUE : Une incubation plus longue peut réduire l’efficacité de la transfection. - Ajoutez la solution OptiMEM/DNA/PEI à la bouteille de 1 L contenant 700 mL de DMEM à faible teneur en glucose avec 10 mM d’HEPES et 2 % de FBS. Bien mélanger.
REMARQUE : Dans cette étude, le faible taux de glucose dans le DMEM a montré une meilleure production de vecteurs AAV après co-transfection. L’ajout de HEPES aide à maintenir les cellules dans un meilleur état. Mélangez en tournant lentement la bouteille. Évitez de créer trop de bulles. - Sortez le CF10 de l’incubateur et aspirez le média dans un conteneur à déchets.
REMARQUE : Posez le CF10 sur la surface à l’intérieur du capot et soulevez progressivement un côté, aspirez lentement le support sans détacher les cellules. - Ajoutez délicatement le mélange transfectant au CF10. Assurez-vous que les dix couches sont recouvertes de support.
REMARQUE : Ajouter 25 ml de mélange transfectant dans le plat de 150 mm. Surveillez les cellules quotidiennement jusqu’à la récolte. Ajouter le reste du mélange transfectant dans le CF10 en versant lentement. Faites pivoter le CF10 très soigneusement et assurez-vous d’un volume égal pour chaque couche. - Remettez le CF10 dans l’incubateur pendant 72 à 96 h.
REMARQUE : Vérifiez la parabole du moniteur de 150 mm pour décider quand récolter les vecteurs AAV. Lorsque les cellules se détachent très facilement avec une charge complète d’AAV, il est temps de récolter.
4. Récolte des vecteurs AAV
- Récoltez le virus 3 à 4 jours après la transfection en secouant vigoureusement le CF10. Verser le milieu dans les tubes coniques de 250 mL et faire tourner le virus à 4000 x g pendant 20 min à 4 °C.
REMARQUE : Pour le sérotype AAV6 utilisé dans ce protocole, 80 % des AAV resteront principalement liés au matériel cellulaire dans la cuffe. Et les 20 % d’AAV ont été dissimulés dans les médias. Ces proportions peuvent différer pour d’autres sérotypes AAV. - Filtrez le surnageant clarifié avec une unité de filtration de 0,45 μm et conservez-le pour les précipitations.
- Rincez le CF10 avec 500 mL de tampon DPBS 1x et centrifugez-le à 4000 x g pendant 20 min à 4 °C.
- Jeter le surnageant à l’aide d’un système de vide et d’une pipette d’aspiration. Ajouter 20 mL de tampon de lyse AAV par CF10 à -80 °C jusqu’à purification.
REMARQUE : Transférez le lysat d’AAV dans un tube conique de 50 mL pour une extraction ultérieure de l’AAV. - Retirer le surnageant filtré, ajouter une solution de NaCl 40 % de PEG-2,5 M pour une concentration finale de 8 % de PEG, et incuber dans une chambre froide sur un rotateur orbital pendant au moins 3 h ou toute la nuit.
REMARQUE : Pour un surnageant de 100 mL, ajouter 25 mL de solution de NaCl PEG-2,5 M à 40 %. - Précipiter le virus en le centrifugeant à 4000 x g pendant 30 min à 4 °C.
REMARQUE : Transférer le mélange dans des tubes coniques de 250 ml pour la centrifugation. - Aspirer pour éliminer le surnageant et ajouter 5 à 10 ml de tampon de remise en suspension AAV HEPES pour suspendre les granulés. Transvasez-le dans un tube conique de 50 mL pour poursuivre la purification en aval, ou stockez-le à -80 °C.
5. Extraction AAV
- Décongelez la pastille de virus dans un bain-marie à 37 °C pendant 10 à 15 minutes avec un agitateur.
REMARQUE : Vortex le lysat AAV pour fondre complètement. - Congeler dans un bain 100% éthanol à -80 °C pendant 20-30 min.
REMARQUE : Alternativement, le cycle de congélation peut être effectué avec un bécher froid 100% éthanol entouré de glace sèche. - Effectuez la congélation/décongélation 3 à 4 fois et faites un tourbillon entre les deux si nécessaire.
REMARQUE : Ce cycle peut être interrompu à l’étape de congélation. Stockez le lysat AAV à -80 °C jusqu’au travail en aval. - Décongelez le lysat d’AAV (à partir de l’étape 5.3) et l’AAV à nouveau en suspension (à partir de l’étape 4.7) dans un bain-marie à 37 °C pendant 10 à 15 minutes avec un agitateur, puis vortex.
REMARQUE : Assurez-vous de fondre complètement et de le mélanger. - Ajouter la nucléase de benzonase (250 unités/μL) au lysat d’AAV et à l’AAV remis en suspension pour une concentration finale de 50 unités/mL. Vortex la solution.
- Incuber au bain-marie à 37 °C en secouant pendant 30 min.
REMARQUE : Retirez les aliquotes de désoxycholate de sodium à 10% dans un bain-marie pour laisser le temps de se dissoudre, de se réchauffer et de bien mélanger. - Ajouter du désoxycholate de sodium à 10 % pour une concentration finale de 0,5 % et incuber à 37 °C avec un agitateur pendant 30 min.
REMARQUE : Le désoxycholate de sodium a été utilisé pour améliorer la libération d’AAV par les cellules. - Répartir la solution brute d’AAV dans des tubes à centrifuger de 2 mL et centrifuger à 14 000 x g pendant 10 min à 4 °C.
- Transvasez le surnageant dans un tube conique de 50 ml.
REMARQUE : À l’aide d’une pipette de 1 ml, transférez le surnageant. Jetez les granulés. - Ajouter une quantité égale de chloroforme et extraire l’AAV en tourbillonnant pendant 1 à 2 min.
REMARQUE : La solution doit devenir opaque, blanche, comme du lait. - Transférez la solution lactée dans des tubes à centrifuger de 2 ml et répartissez-la uniformément. Centrifugeuse à 14 000 x g pendant 10 min à 4 °C.
- Pipetez soigneusement la couche rose supérieure et transférez-la dans un nouveau tube conique de 50 ml.
REMARQUE : Ne dérangez pas les couches de chloroforme/protéines. - Mesurez le volume final à l’aide d’une pipette. Selon le tableau des volumes bruts AAV-PEG-Sulfate (tableau 2), mélanger les volumes appropriés de 50 % (NH4)2SO4 et de 40 % de solution PEG. Vortex pendant 2 min.
- Transférer le mélange dans des tubes à centrifuger de 2 mL pour répartir uniformément. Centrifugeuse à 14 000 x g pendant 10 min à 4 °C
- Prélever la couche inférieure à l’aide d’une aiguille de 22 g et d’une seringue de 3 ml. Prélever l’AAV dans un tube conique de 50 mL et le stocker à 4 °C pour une ultracentrifugation ultérieure des gradients d’iodixanol.
6. Purification AAV par ultracentrifugation par gradient d’iodixanol
- Préparez les gradients d’iodixanol fraîchement avant de charger l’AAV en fonction du nombre de tubes d’ultracentrifugation utilisés (tableau 3).
REMARQUE : Du rouge de phénol a été utilisé pour observer les couches. - Recouvrez lentement chaque solution dans un tube à fermeture rapide en polypropylène à bouchon rond à l’aide d’une seringue de 10 ml fixée à l’aide d’une aiguille longue de 16 g. Évitez les bulles.
- Ajouter délicatement jusqu’à 17 ml de solution d’AAV sur le dessus du gradient avec une seringue de 22 G. Utilisez un tampon de dialyse AAV pour remplir le tube.
REMARQUE : Ajoutez la solution AAV en chargeant les gouttes contre la paroi dans le tube de l’ultracentrifugeuse. Ne dérangez pas les couches de dégradé. - Scellez les tubes d’ultracentrifugation avec une soudeuse électrique.
REMARQUE : Équilibrez les tubes avant de les envoyer à l’ultracentrifugeuse avec une différence de ±0,2 g. - Centrifugeuse à 350 000 x g pendant 2 h dans un rotor Ti70 à 4 °C.
- Retirez délicatement les tubes du rotor et placez-les dans le rack des tubes d’ultracentrifugation.
REMARQUE : Assurez-vous que les pentes ne sont pas perturbées. - Collectez les fractions AAV en suivant les étapes ci-dessous :
- Stabilisez le tube sur un support de support.
- Percez les tubes de l’ultracentrifugeuse légèrement en dessous de l’interface 40 %-60 % avec une aiguille de 19 G attachée à une seringue de 10 ml.
REMARQUE : L’ouverture de l’aiguille doit être vers le haut, face à la pente de 40%. - Percez un trou dans le haut du tube avec une aiguille de 16 G.
- Recueillir jusqu’à 5 ml par tube. Évitez de collecter la contamination protéique à l’interface 25 %-40 %.
- Répétez l’opération pour chaque tube d’ultracentrifugation.
REMARQUE : Transférez les fractions AAV dans un tube conique de 50 ml.
7. Ultracentrifugation des gradients d’iodixanol du deuxième tour
REMARQUE : Cette étape est facultative. Cette étape consiste à réduire le rapport AAV vide pour une capside AAV complète de meilleure qualité.
- Diluer l’AAV >1:1 avec un tampon de dialyse AAV.
REMARQUE : L’AAV recueilli lors de la première série d’ultracentrifugation était à 40 % de couche d’iodixanol. Il doit être dilué à au moins 50 % pour pouvoir être chargé sur la couche d’iodixanol à 30 %. - Superposer chaque solution (tableau 4) dans un tube d’ultracentrifugation à l’aide d’une aiguille de 16 G et fixer lentement une seringue de 10 mL. Évitez les bulles.
- Ajouter délicatement 20 ml de solution d’AAV diluée sur le gradient à l’aide d’une seringue de 22 G. Utilisez un tampon de dialyse AAV pour remplir le tube.
- Répétez les étapes 6.4 à 6.7.
8. Dialyse AAV et concentration
- Transférez le virus AAV dans la cassette de dialyse à l’aide d’une aiguille neuve de 18 G et d’une seringue de 10 ml. Injectez lentement le virus dans la cassette et retirez-en l’air.
- Ajoutez une barre d’agitation dans le bécher contenant un tampon de dialyse AAV et placez-la sur une plaque d’agitation.
- Insérez la cassette de dialyse dans le tampon de dialyse à l’aide d’une bouée à flotteur. Remuez à 4 °C. Changez 2 à 3 fois le tampon de dialyse AAV.
REMARQUE : Utilisez suffisamment de tampon pour permettre à la cassette de flotter et effectuez le remplacement de tampon. Couvrez le bécher de papier d’aluminium. - À l’aide d’une seringue de 10 mL munie d’une aiguille de 18 G, recueillir le virus de la cassette dans une unité de filtration centrifuge.
- Centrifugeuse à 4 000 x g pendant 15 à 30 min à 4 °C.
REMARQUE : Rincez l’AAV avec le tampon de dialyse AAV 2-3 fois. Concentrez l’AAV jusqu’à ce que ~200 μL de liquide restant se trouvent sur le dessus du filtre. - Récupérez l’AAV concentré de l’unité de filtration. Rincez le filtre avec 300 μL supplémentaires de tampon de dialyse AAV et transférez-le dans l’AAV concentré.
- Filtrez le virus à travers un filtre de seringue de 0,22 μm à l’aide d’une seringue à lamelle Luer Slip de tuberculine de 1 mL. Pour assurer le moins de perte de volume possible, utilisez la seringue de 10 ml pour pousser l’air à travers le filtre 2 à 3 fois.
- Conservez le virus AAV purifié à 4 °C temporellement pour le titrage.
REMARQUE : Titrez le virus, aliquote et conservez-le à -80 °C dans un délai de 1 semaine.
9. Titrage du virus AAV
REMARQUE : La réaction en chaîne quantitative par polymérase (qPCR) de TaqMan a été utilisée pour titrer l’AAV purifié.
- Traiter les vecteurs AAV avec de la DNase I et incuber à 37 °C pendant 30 min, puis incuber à 95 °C pendant 10 min.
REMARQUE : À titre d’exemple de réaction de 10 μL, 2 μL d’échantillon de virus AAV, 1 μL de DNase, 1 μL de tampon DNase et 6 μLde H 2O sont utilisés. Il peut être réalisé dans un thermocycleur avec des tubes PCR. - Préparez les jeux d’amorces ciblant la zone d’insertion AAV.
REMARQUE : Les cibles pourraient être le promoteur, le transgène et le rapporteur dans la construction AAV. Les amorces sont énumérées dans le tableau 5. - Préparez des étalons de plasmide d’ADN (10, 1, 0,1, 0,01, 0,001 et 0,0001 pg/μL) et un témoin négatif (H2O).
- Préparez le master mix qPCR, y compris les amorces, conformément aux instructions du fabricant.
- Placez les étalons et les échantillons en trois exemplaires sur une plaque PCR. Ajoutez le mélange qPCR dans les étalons et les échantillons. Fermez la plaque et effectuez la réaction qPCR conformément aux instructions du fabricant.
REMARQUE : Les conditions de réaction dépendent des matériaux/réactifs et de l’instrument. Des cycles PCR rapides et conventionnels sont applicables. - Calculer le titre du virus AAV.
REMARQUE : La concentration de l’échantillon dans ce protocole est de 1/10 de dilution par rapport aux échantillons d’origine. - Aliquote le virus AAV dans des tubes de 1,5 mL et conserver à 4 °C pendant un mois au maximum et à -80 °C à long terme.
REMARQUE : Évitez les cycles de gel/dégel pour le stockage. Lorsqu’il est utilisé pour des expériences in vivo , ne pas utiliser de décongélation sur deux aliquotes.
10. Contrôle de la qualité de l’AAV
REMARQUE : La pureté des virus AAV a été caractérisée par coloration à l’argent SDS-PAGE à l’aide d’un gel SDS-PAGE et colorée à l’aide d’un kit de coloration disponible dans le commerce.
- Dénaturer 3 x 109 vg d’échantillon de virus AAV avec tampon Laemmli.
REMARQUE : Utilisez un virus AAV de référence comme contrôle. La capside complète de référence AAV6-CMV-GFP est utilisée. - Chargez l’AAV dénaturé sur un gel SDS-PAGE à gradient de 4 % à 20 % et faites-le fonctionner à 180 V pendant 50 min.
- Retirez le gel de la plaque d’électrophorèse. Teindre le gel avec un kit de coloration à l’argent selon les instructions du fabricant.
- Vérifiez les protéines de capside AAV VP1, VP2 et VP3.
REMARQUE : Le virus AAV pur ne contient que les protéines de capside VP1, VP2 et VP3. S’ils ne sont pas purs, d’autres bandes de protéines seraient visibles sur le gel.
REMARQUE : La microscopie électronique à transmission (MET) de virions AAV recombinants colorés négativement a été réalisée pour évaluer l’intégrité morphologique et le rapport plein/vide. - Prenez une grille EM à l’aide d’une pince à épiler et posez-la sur le banc avec le côté brillant de la grille vers le haut.
- Placez 5 μL d’échantillon AAV sur la grille et laissez-le sécher par évaporation.
REMARQUE : Diluez l’AAV si nécessaire. 1012 vg/mL est un titre moyen. Cela peut prendre 30 à 60 minutes pour sécher la grille. - Lavez la grille par pipetage, goutte à goutte, avec environ 200 μL de H2O sur la grille.
- Retirez l’excès d’eau en plaçant lentement un papier de chromatographie verticalement à côté de la grille.
- Pipeter 5 μL de solution d’acétate d’uranyle à 2 % sur la grille. Incuber pendant 5 min et retirer la mèche comme mentionné ci-dessus. Laissez sécher la grille.
- Visualisez les particules AAV au microscope électronique à transmission (à un grossissement de 50 000 fois). Les capsides virales avec un génome viral apparaîtront comme des hexagones blancs homogènes, tandis que les capsides vides apparaîtront comme des hexagones avec un bord blanc mais un centre sombre.
- Comptez au hasard au moins 100 particules pour déterminer le pourcentage approximatif de pleins vs. particules AAV vides.
Résultats
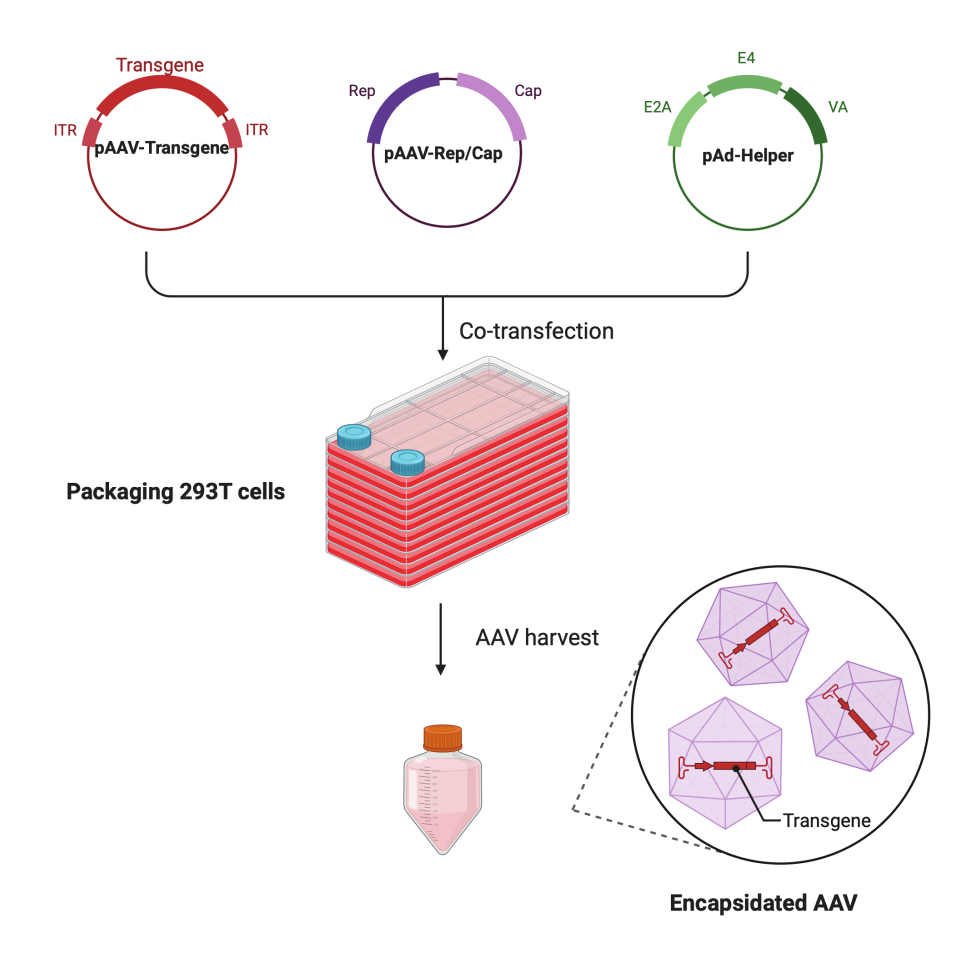
Dans ce protocole détaillé étape par étape, il est démontré qu’une plateforme standardisée permet de fabriquer un virus AAV à titre élevé et de haute qualité avec le CF10 dans un laboratoire de recherche à grande échelle. Par rapport aux boîtes de culture cellulaire conventionnelles, le CF10 offre un moyen pratique de cultiver de grandes quantités de cellules et de produire le virus AAV (Figure 1). Plusieurs conditions de culture ont été testées pour déterminer si les cellules dans un environnement optimal peuvent favoriser la production virale. Un DMEM à faible teneur en glucose complété par 10 mM d’HEPES et 2% de FBS a montré la meilleure production d’AAV.
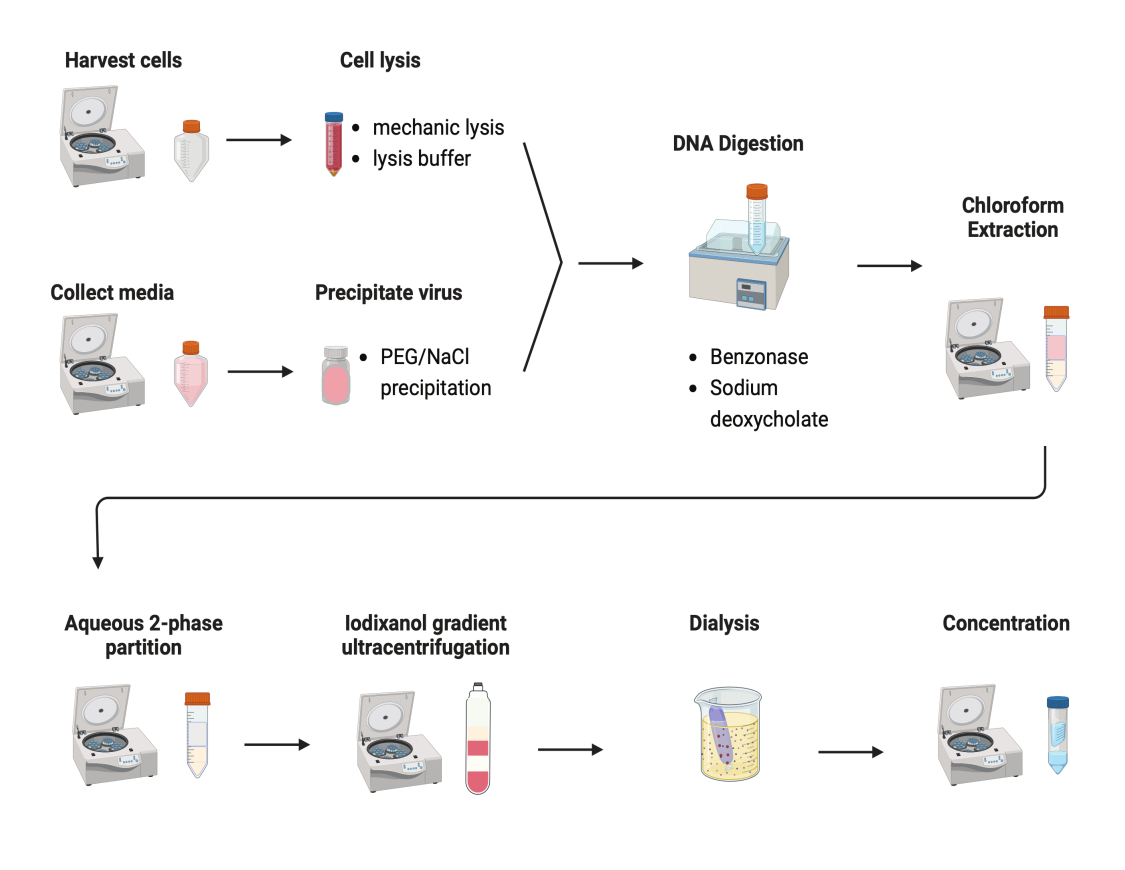
Plusieurs protocoles ont été testés pour purifier les AAV. La plupart des procédures ont de faibles rendements en virus et une impureté des capsides AAV. Ici, un protocole de purification révisé a été développé, combinant l’AAV à partir de pastilles cellulaires et de milieux de culture (Figure 2). Nous avons constaté que 80 % des AAV se trouvaient dans les cellules, et 20 % des AAV dans les milieux de culture, qui étaient sécrétés par les cellules. Les deux parties de l’AAV ont été traitées avec de la DNase pour éliminer l’ADN libre. Le désoxycholate de sodium a été utilisé pour libérer davantage d’AAV des cellules. Les AAV ont ensuite été extraits par une extraction au chloroforme suivie d’une répartition aqueuse en deux phases. Ces étapes ont permis d’éliminer la plupart des contaminants protéiques. L’AAV reste soluble dans la phase sulfate d’ammonium.
Les contaminants restants ont été éliminés par ultracentrifugation discontinue à gradient d’iodixanol (figure 3A). Le gradient a également été utile pour éliminer les capsides AAV vides, en particulier lors de la deuxième série d’ultracentrifugation du gradient d’iodixanol.
La pureté du virus AAV a été déterminée par coloration à l’argent. Lorsque trois bandes principales correspondant à la protéine de capside de l’AAV, VP1, VP2 et VP3, avec une pureté supérieure à 90 %, ont été obtenues, le virus AAV était adapté à une utilisation in vivo (figure 3B). Le rapport de la capside pleine à la capside vide de l’AAV a été consulté par TEM (figure 3C). Seule une capside complète avec un insert de transgène permettrait l’expression du transgène dans le tissu ciblé. Une partie élevée de la capside vide pourrait également induire une réponse immunitaire à la capside AAV. Ces contrôles de qualité sont nécessaires pour chaque virus AAV produit et purifié avant utilisation.

Figure 1 : Illustration schématique de la production d’AAV par la méthode de triple transfection à HEK293T cellules. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Illustration schématique de la purification AAV. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Purification de l’AAV par ultracentrifugation du gradient d’iodixanol et vérification de la pureté du virus AAV. (A) Couches du gradient d’iodixanol et position de l’aiguille pour la récolte. (B) Gel représentatif évaluant la teneur et la pureté de la capside. M : marqueur moléculaire ; R : référence AAV6 pleine capside ; S1 : capside AAVDJ fabriquée en interne avec un cycle d’ultracentrifugation ; S2 : capside AAVDJ fabriquée en interne avec deux cycles d’ultracentrifugation ; S3 : capside AAV6 fabriqué en interne. (C) Image au microscope électronique d’AAV. Virus collecté après purification. Les capsides virales contenant un génome viral apparaissent sous la forme d’hexagones blancs homogènes, tandis que les capsides vides apparaissent sous la forme d’hexagones avec un bord blanc mais un centre sombre. Barre d’échelle : 100 nm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Tableau 1 : Calculateur de l’IPE pour les emballages AAV. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 2 : Tableau des volumes bruts d’AAV-PEG-Sulfate. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 3 : Préparation du gradient d’iodixanol. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 4 : Préparation du deuxième gradient d’iodixanol rond. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 5 : Amorces de titrage AAV. Veuillez cliquer ici pour télécharger ce tableau.
Dossier supplémentaire 1 : Compositions des tampons utilisés pour l’étude. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Ici, un protocole techniquement avancé pour la production à grande échelle de vecteurs AAV à titre élevé et de haute qualité à l’aide d’une plate-forme d’usine cellulaire (CF10) est introduit, représentant une amélioration significative par rapport aux méthodes conventionnelles de culture cellulaire. L’utilisation d’usines cellulaires simplifie le processus de culture de grands volumes de cellules, facilitant ainsi la production plus efficace de virus AAV1. De plus, en optimisant les conditions de culture, en particulier avec du DMEM à faible teneur en glucose complété par 10 mM d’HEPES et 2% de FBS, une production virale significativement accrue a été confirmée, indiquant le rôle crucial de l’environnement cellulaire dans le rendement du virus.
Le protocole de purification révisé, qui combine l’AAV des granules de cellules et des milieux de culture, résout le problème commun des faibles rendements de virus et des impuretés observés dans de nombreux protocoles existants. Les étapes de l’extraction au chloroforme et de la répartition aqueuse en deux phases éliminent efficacement la plupart des contaminants protéiques, l’AAV restant soluble dans la phase sulfate d’ammonium. L’amélioration du rendement et de la pureté des vecteurs AAV à l’aide de la séparation diphasique PEG/aqueuse combinée à l’ultracentrifugation à gradient d’iodixanol, par opposition aux méthodes traditionnelles d’ultracentrifugation à gradient d’iodixanol, peut être attribuée à une séparation initiale améliorée avec une séparation biphasique PEG/aqueuse, à une pureté raffinée avec l’ultracentrifugation à gradient d’iodixanol et à une réduction de la co-purification des contaminants12. Tout d’abord, l’introduction d’une séparation biphasique PEG/aqueuse avant l’ultracentrifugation améliore considérablement la séparation initiale des particules d’AAV des débris cellulaires et autres contaminants. Le PEG, un polymère de haut poids moléculaire, lorsqu’il est mélangé à une solution aqueuse, crée deux phases distinctes13. Les vecteurs AAV ont tendance à se répartir préférentiellement dans l’une de ces phases (généralement la phase riche en PEG), tandis que de nombreux contaminants et impuretés sont répartis dans les13 autres. Cette répartition sélective concentre efficacement les particules d’AAV et élimine une partie substantielle des impuretés avant même l’ultracentrifugation, augmentant ainsi le rendement et réduisant la charge de contaminants entrant dans l’étape d’ultracentrifugation13. Deuxièmement, l’ultracentrifugation par gradient d’iodixanol affine davantage la pureté des vecteurs AAV. L’iodixanol, un milieu à gradient iso-osmolaire non ionique, permet une séparation plus douce et contrôlée par rapport aux gradients CsCl traditionnels14. Dans ce gradient, les particules AAV migrent vers une position dans le gradient qui correspond à leur densité flottante14. Il est important de noter que ce processus est efficace pour séparer les capsides AAV complètes (contenant la charge utile génétique) des capsides vides (manquant de matériel génétique), ce qui est un déterminant crucial de la qualité du vecteur. La nature iso-osmolaire de l’iodixanol préserve également l’intégrité des capsides AAV mieux que les agents hyperosmolaires comme le CsCl, ce qui pourrait conduire à des rendements plus élevés de vecteurs intacts et fonctionnels14. Enfin, les méthodes traditionnelles d’ultracentrifugation, en particulier celles utilisant des gradients de CsCl, peuvent parfois co-épurer des contaminants qui ont des densités de flottabilité similaires à celles des vecteurs AAV15. En utilisant le cloisonnement PEG comme étape préliminaire, la charge de ces contaminants est considérablement réduite avant l’ultracentrifugation13. Cette réduction de la charge de contaminants signifie que le gradient d’iodixanol peut fonctionner plus efficacement et de manière plus sélective dans la purification des vecteurs AAV, ce qui conduit à une pureté plus élevée15.
La pureté et la qualité des vecteurs AAV sont rigoureusement évaluées par coloration à l’argent et TEM11. L’observation de trois bandes principales correspondant aux protéines de capside AAV VP1, VP2 et VP3, avec une pureté supérieure à 90%, indique l’adéquation de ces vecteurs AAV pour une utilisation in vivo . L’analyse TEM pour déterminer le rapport capside plein/vide est particulièrement cruciale, car une proportion élevée de capsides vides peut entraîner une réduction de l’efficacité de l’administration de gènes et des réponses immunitaires potentielles11. Ce contrôle de qualité, bien qu’essentiel, ajoute à la complexité de la procédure et peut nécessiter une expertise technique supplémentaire.
En conclusion, le protocole offre des avancées techniques significatives dans la production de vecteurs AAV, notamment en termes d’évolutivité et de pureté. Cependant, les complexités associées au processus de purification et la nécessité d’un équipement et d’une expertise spécialisés peuvent encore constituer une limitation mineure de son application dans certains contextes de recherche. Le raffinement et la simplification de ces techniques pourraient rendre cette approche plus accessible et largement applicable dans le domaine de la recherche en thérapie génique.
Déclarations de divulgation
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière qui pourrait être interprétée comme un conflit d’intérêts potentiel.
Remerciements
TZ a conçu les expériences. TZ, VD, SB et JP ont réalisé les expériences. TZ et VD ont généré des données et les ont analysées. TZ et YX ont écrit le manuscrit. TZ et GG ont révisé le manuscrit. Ce travail a été soutenu par l’hôpital pour enfants UPMC de Pittsburgh.
matériels
| Name | Company | Catalog Number | Comments |
| 293T/17 cells | ATTC | CRL-11268 | |
| 0.5 M EDTA | MilliporeSigma | 324506-100ml | |
| 1 mL Henke-Ject syringe | Fisher Scientific | 14-817-211 | |
| 10% pluronic F68 solution | Fisher Scientific | 24-040-032 | |
| 10x Tris/Glycine/SDS Buffer | Biorad | 1610732 | |
| 1M HEPES | Fisher Scientific | 15-630-080 | |
| 2% Uranyl Acetate Solution | Electron Microscopy Sciences | 22400-2 | |
| 4%–20% Precast Protein Gels | biorad | 4561094 | |
| 40% PEG solution | Sigma | P1458-50ML | |
| AAV6 reference full capsids | Charles River Laboratories | RS-AAV6-FL | |
| Accutase Cell Detachment Solution | Fisher Scientific | A6964-100ML | |
| Benzonase | Sigma | E1014-25KU | |
| BioLite Cell Culture Treated Dishes 150 mm | Fisher Scientific | 12-556-003 | |
| Centrifugal Filter Unit | MilliporeSigma | UFC905024 | |
| Corning PES Syringe Filters | Fisher Scientific | 09-754-29 | |
| Dialysis Cassettes, 10 K MWCO | Fisher Scientific | PI66810 | |
| Disposable PES Filter Units 1 L 0.2 µm | Fisher Scientific | FB12566506 | |
| Disposable PES Filter Units 1 L 0.45 µm | Fisher Scientific | FB12566507 | |
| Disposable PES Filter Units 500 mL 0.2 µm | Fisher Scientific | FB12566504 | |
| DMEM high glucose | Fisher Scientific | 10-569-044 | |
| DMEM low glucose | Fisher Scientific | 10567022 | |
| DNase | NEB | M0303S | |
| DPBS 1x | Fisher Scientific | 14-190-250 | |
| Fetal Bovin Serum (FBS) | Biowest | S1620 | |
| Formvar/Carbon 300 Mesh, Cu | Electron Microscopy Sciences | FCF300-Cu-50 | |
| glycerol | Sigma | G5516-1L | |
| KCl | Sigma | P9541-500G | |
| LB agar | Sigma | L2897-250G | |
| LB broth | Fisher Scientific | BP9732-500 | |
| MgCL2·6H2O | Sigma | M9272-100G | |
| NEB stable competent cells | NEB | C3040H | |
| Nest Biofactory 10 chamber | MidSci | 771302 | |
| NucleoBond Xtra Maxi EF | Macherey-Nagel | 740424 | |
| Opti-MEM | Fisher Scientific | 31-985-088 | |
| OptiPrep Density Gradient Medium | Millipore Sigma | D1556-250ml | |
| pAAV-CMV-GFP | Addgene | 105530 | |
| pAAV-DJ | Cell BioLab | VPK-420-DJ | |
| pAAV-RC6 | Cell BioLab | VPK-426 | |
| pAdDeltaF6 | Addgene | 112867 | |
| PEG 8000 | Promega | V3011 | |
| PEI Max | Polysciences, Inc | 49553-93-7 | |
| Pen-Strep | Fisher Scientific | 15-140-163 | |
| Phenol red | Millipore Sigma | 1.07242.0100 | |
| Pierce Silver Stain Kit | Thermo Fisher Scientific | 24612 | |
| QuickSeal tube | Fisher Scientific | NC9144589 | |
| Sodium Chloride | Sigma | 1162245000 | |
| sodium deoxycholate | Millipore Sigma | D6750-100G | |
| Taqman Fast Advanced Master Mix | Thermo Fisher Scientific | 4444557 | |
| Type 70 Ti Fixed-Angle Titanium Rotor | Beckman Coulter | 337922 | |
| Western Blotting Substrate | ThermoFisher | 32209 |
Références
- Arjomandnejad, M., Dasgupta, I., Flotte, T. R., Keeler, A. M. Immunogenicity of recombinant adeno-associated virus (AAV) vectors for gene transfer. BioDrugs. 37 (3), 311-329 (2023).
- Liu, Y., Siriwon, N., A Rohrs, J., Wang, P. Generation of targeted adeno-associated virus (AAV) vectors for human gene therapy. Curr Pharm Des. 21 (22), 3248-3256 (2015).
- Bilal, A. S., et al. Optimization of large-scale Adeno-Associated Virus (AAV) production. Curr Protoc. 3 (5), e757(2023).
- Rashnonejad, A., Chermahini, G. A., Li, S., Ozkinay, F., Gao, G. Large-scale production of adeno-associated viral vector serotype-9 carrying the human survival motor neuron gene. Mol Biotechnol. 58, 30-36 (2016).
- Challis, R. C., et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat Protoc. 14 (2), 379-414 (2019).
- Challis, R. C., et al. Publisher Correction: Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat Protoc. 14 (8), 2597-2597 (2019).
- Mueller, C., Ratner, D., Zhong, L., Esteves-Sena, M., Gao, G. Production and discovery of novel recombinant adeno-associated viral vectors. Curr Protoc Microbiol. 26 (1), 14(2012).
- Guo, P., Wiersch, J., Xiao, X., Gittes, G. Simplified purification of AAV and delivery to the pancreas by intraductal administration. Methods Mol Biol. 1950, 373-387 (2019).
- Berns, K. I., Srivastava, A. Next generation of adeno-associated virus vectors for gene therapy for human liver diseases. Gastroenterol Clin North Am. 48 (2), 319-330 (2019).
- Khasa, H., Kilby, G., Chen, X., Wang, C. Analytical band centrifugation for the separation and quantification of empty and full AAV particles. Mol Ther Methods Clin Dev. 21, 585-591 (2021).
- Chen, H. Comparative observation of the recombinant adeno-associated virus 2 using transmission electron microscopy and atomic force microscopy. Microsc Microanal. 13 (5), 384-389 (2007).
- Burnham, B., et al. Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Hum Gene Ther Methods. 26 (6), 228-242 (2015).
- Kato, M., et al. In situ-formable, dynamic crosslinked poly (ethylene glycol) carrier for localized adeno-associated virus infection and reduced off-target effects. Commun Biol. 6 (1), 508(2023).
- Sena-Esteves, M., Gao, G. Enrichment of fully packaged virions in column-purified recombinant adeno-associated virus (rAAV) preparations by iodixanol gradient centrifugation followed by anion-exchange column chromatography. Cold Spring Harb Protoc. 2020 (2), 095638(2020).
- Matsumoto, M., Wangelin, J. R., Murphy, M. L. Purification of avian encephalomyelitis virus by ultracentrifugation in a nonlinear cesium chloride gradient. Avian Dis. 22 (3), 496-502 (1978).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.