
Method Article
Ein optimiertes und standardisiertes Verfahren zur Generierung von hochtiternden, qualitativ hochwertigen Adeno-assoziierten Virusvektoren unter Verwendung einer Zellfabrikplattform
In diesem Artikel
Zusammenfassung
Da sich das Gebiet der Gentherapie ständig weiterentwickelt, besteht ein wachsender Bedarf an innovativen Methoden, die diese Herausforderungen bewältigen können. Hier wird eine einzigartige Methode vorgestellt, die den Prozess der Generierung von hochreinen und hochreinen AAV-Vektoren unter Verwendung einer Zellfabrikplattform rationalisiert und die Qualitätsstandards für In-vivo-Studien erfüllt.
Zusammenfassung
Die präklinische Gentherapieforschung, insbesondere in Nagetier- und Großtiermodellen, erfordert die Herstellung von AAV-Vektoren mit hoher Ausbeute und Reinheit. Traditionelle Ansätze in Forschungslabors beinhalten oft den umfangreichen Einsatz von Zellkulturschalen zur Kultivierung HEK293T Zellen, ein Prozess, der sowohl mühsam als auch problematisch sein kann. Hier wird eine einzigartige hauseigene Methode vorgestellt, die diesen Prozess mit einer spezifischen Cell Factory (oder Cell Stacks, CF10) Plattform vereinfacht. Eine Integration von Polyethylenglykol/wässriger Zweiphasenpartitionierung mit Iodixanol-Gradienten-Ultrazentrifugation verbessert sowohl die Ausbeute als auch die Reinheit der erzeugten AAV-Vektoren. Die Reinheit der AAV-Vektoren wird durch SDS-PAGE und Silberfärbung verifiziert, während das Verhältnis von vollen zu leeren Partikeln mittels Transmissionselektronenmikroskopie (TEM) bestimmt wird. Dieser Ansatz bietet eine effiziente Zellfabrikplattform für die Herstellung von AAV-Vektoren mit hohen Ausbeuten, gepaart mit einer verbesserten Aufreinigungsmethode, um die Qualitätsanforderungen für In-vivo-Studien zu erfüllen.
Einleitung
Adeno-assoziierte Virusvektoren (AAV) sind zu einem unverzichtbaren Werkzeug in der Gentherapieforschung geworden und bieten eine einzigartige Kombination aus Wirksamkeit und Sicherheit für die Genübertragung1. Traditionelle Methoden zur Erzeugung von AAVs im Labor waren entscheidend für das Verständnis und die Anwendung der Gentherapie2. Diese Methoden sind zwar grundlegend, weisen jedoch gewisse Einschränkungen und Herausforderungen auf, insbesondere in Bezug auf die Ausbeute, die Zeiteffizienz und die Qualität der erzeugten Vektoren, insbesondere das Verhältnis von vollen zu leeren Partikeln3.
Das konventionelle Verfahren zur AAV-Herstellung beinhaltet in erster Linie die Transfektion von HEK293-Zellen4. Dieser Prozess, der typischerweise in Zellkulturschalen durchgeführt wird, erfordert, dass die Zellen mit einem Plasmid transfiziert werden, das das interessierende Gen zusammen mit dem Helferplasmid und dem AAV-Kapsidplasmidenthält 5,6. Nach der Transfektion produzieren die Zellen AAV-Partikel, die dann geerntet und gereinigt werden 5,6. Der Aufreinigungsprozess beinhaltet häufig eine Ultrazentrifugation, ein kritischer Schritt bei der Gewinnung hochreiner AAV-Vektoren7. Die Ultrazentrifugation, insbesondere unter Verwendung von Cäsiumchlorid (CsCl) oder Iodixanol-Gradienten, ist eine Standardmethode zur Trennung von AAV-Partikeln von Zelltrümmern und anderen Verunreinigungen8. Dieser Schritt ist entscheidend für das Erreichen der gewünschten Reinheit und Konzentration von AAV-Vektoren, was sich direkt auf ihre Wirksamkeit bei der Genübertragung auswirkt8. Trotz ihrer weit verbreiteten Anwendung hat die traditionelle Ultrazentrifugation ihre Nachteile. So kann beispielsweise die Ausbeute an AAV-Vektoren aus dieser Methode variabel und oft gering sein, was eine große Herausforderung darstellt, wenn große Mengen an Vektoren mit hohem Titer benötigt werden, insbesondere für In-vivo-Studien oder große Tiermodelle9.
Ein weiterer kritischer Aspekt der AAV-Vektorqualität ist das Verhältnis von vollen zu leeren Partikeln10. AAV-Präparate enthalten oft ein Gemisch dieser Partikel; Allerdings enthalten nur die vollen Partikel das therapeutische Erbgut. Das Vorhandensein eines hohen Anteils leerer Partikel kann die Effizienz der Genübertragung erheblich verringern10. Die Bewertung und Optimierung des Verhältnisses von vollen zu leeren Partikeln ist daher ein wichtiger Parameter bei der Bewertung der Wirksamkeit von AAV-Vektoren. Herkömmliche Methoden sind zwar in der Lage, AAV-Vektoren zu erzeugen, haben aber oft Schwierigkeiten, dieses Verhältnis konsistent zu kontrollieren, was zu Schwankungen in der Vektorpotenz führt10.
Hier wird eine einzigartige Methode vorgestellt, die den Prozess der Erzeugung von hochreinen und hochreinen AAV-Vektoren unter Verwendung einer Zellfabrikplattform rationalisiert, die ohne arbeitsintensive HEK293T Zellkulturen in Zellschalen auskommt und die zweiphasige Verteilung von Polyethylenglykol/wässrigem Wasser mit der Iodixanol-Gradienten-Ultrazentrifugation integriert. Die Reinheit des AAV-Vektors wird durch SDS-PAGE und Silberfärbung bestätigt, und das Verhältnis von vollen zu leeren Partikeln wird mittels Transmissionselektronenmikroskopie (TEM) bestimmt, was den Qualitätsstandards für in vivo-Studien entspricht11.
Protokoll
Die Einzelheiten zu den in der Studie verwendeten Reagenzien, Plasmiden und Geräten sind in der Materialtabelle aufgeführt. Die Zusammensetzung der verwendeten Puffer ist in der Zusatzdatei 1 enthalten.
1. Vorbereitung des Plasmids
- Transformieren Sie Plasmide (pAAV-GOI, pHelper, pAAV-Cap) in E. coli.
HINWEIS: Jeder E.coli-Stamm kann für die Plasmidamplifikation verwendet werden. Bei einigen Plasmiden können spezielle kompetente Zellen wie z.B. NEB stable die Plasmidausbeute verbessern. Die drei in diesem Protokoll verwendeten Plasmide sind pAAV-GOI: pAAV-CMV-GFP, pHelper: pAdDeltaF6 und pAAV-Cap: pAAV-RC6. - Züchten Sie Bakterien in LB-Medien, die die entsprechenden selektiven Antibiotika in optimalem Volumen bei 37 °C für 16-18 h enthalten.
HINWEIS: Bei Verwendung von stabilen NEB-Zellen 18-20 h bei 30 °C kultivieren. - Ernten Sie Plasmid-DNA, indem Sie die E. coli zentrifugieren. Nährmedium bei 4000 x g, 4 °C, für 15 min.
- Gießen Sie die überstehende Flüssigkeit aus der Zentrifugenflasche vorsichtig in einen Abfallbehälter und reinigen Sie die Plasmid-DNA aus dem Pellet mit einem groß angelegten endotoxinfreien Plasmid-Aufreinigungskit.
HINWEIS: Achten Sie darauf, es langsam zu gießen und Spritzer zu vermeiden. - Messen Sie die DNA-Konzentration und -Reinheit mit einem Spektralphotometer.
HINWEIS: Überprüfen Sie die DNA-Reinheit, indem Sie das Verhältnis OD260/OD280 überprüfen. Das Verhältnis für reine DNA liegt bei etwa 1,8. Die DNA-Konzentration sollte für den späteren Co-Transfektionsschritt über 1 μg/μl liegen. Stellen Sie bakterielle Glycerinvorräte her, indem Sie 500 μl der Übernachtkultur zu 500 μl 50 % Glycerin in einem Kryofläschchen hinzufügen und bei -80 °C lagern.
2. Vorbereitung HEK293T Zellen
- Säen Sie HEK293T Zellen mit DMEM-Medien mit hohem Glukosegehalt von 10 % FBS in eine 10-Schicht-Zellfabrik (CF10) und lassen Sie die Zellen über Nacht im Inkubator wachsen.
HINWEIS: Die Durchgänge der 293T-Zellen sollten weniger als 10 betragen, um den optimalen Zustand für eine robuste AAV-Produktion zu gewährleisten. Wenn möglich, fügen Sie den Nährmedien zu diesem Zeitpunkt keine Antibiotika hinzu. CF10 hat etwa die 42-fache Wachstumsoberfläche einer 150-mm-Zellkulturschale. Die gleiche Zelldichte in eine 150-mm-Zellkulturschale aussäen, um das Zellwachstum unter dem Mikroskop überprüfen zu können. Bereiten Sie 1075 ml Zellsuspension vor, geben Sie 25 ml Zellsuspension in eine 150-mm-Schale und geben Sie den Rest in den CF10. - Überprüfen Sie die Zellkonfluenz am nächsten Tag vor der Transfektion.
HINWEIS: Die Zellen sollten zum Zeitpunkt der Transfektion eine Konfluenz von 80 % bis 90 % erreichen, indem sie unter einem Mikroskop beobachtet werden.
3. Dreifache Transfektion von AAV-Plasmiden
- Berechnen Sie die Menge jedes Plasmids, die benötigt wird (pAAV-GOI, pHelper, pAAV-Serotyp), um ein molares Verhältnis von 1,2: 1: 1 mit 2,5-5 mg Gesamt-DNA pro CF10 zu erhalten.
HINWEIS: Das molare Verhältnis der drei Plasmide kann für die optimale Wirksamkeit der Transfektion variabel sein. Es ist besser, es in einer kleinen Umgebung zu testen, bevor Sie zu dieser CF10-Einstellung wechseln. pAAV-CMV-GFP, pAdDeltaF6 und pAAV-RC6 werden verwendet, um das AAV6-CMV-GFP-Virus als Beispiel zu verwenden. Die Formel des PEI-Rechners ist in Tabelle 1 aufgeführt. - Aliquotieren Sie 350 mL OptiMEM in eine sterile 500 mL Flasche.
- Geben Sie alle drei Plasmid-DNA in die Flasche mit dem Opti-MEM. Gut mischen.
- Fügen Sie 15 ml 1 mg/ml Polyethylenimin (PEI)-Lösung hinzu (Verhältnis 1:3 μg DNA zu μg PEI). Schütteln Sie das OptiMEM/DNA/PEI-Gemisch 30 s lang kräftig auf und ab.
HINWEIS: Es ist in Ordnung, Blasen zu machen. - Inkubieren Sie die Mischung bei Raumtemperatur für 10-15 Minuten.
HINWEIS: Eine längere Inkubation kann die Transfektionseffizienz verringern. - Geben Sie die OptiMEM/DNA/PEI-Lösung in die 1-l-Flasche mit 700 mL DMEM-Niederglukose mit 10 mM HEPES und 2 % FBS. Gut mischen.
HINWEIS: In dieser Studie zeigte die niedrige Glukose im DMEM eine bessere AAV-Vektorproduktion nach Co-Transfektion. Die Zugabe von HEPES hilft, die Zellen in einem besseren Zustand zu halten. Mischen Sie, indem Sie die Flasche langsam drehen. Vermeiden Sie es, zu viele Blasen zu erzeugen. - Nehmen Sie den CF10 aus dem Inkubator und saugen Sie das Medium in einen Abfallbehälter ab.
HINWEIS: Legen Sie den CF10 auf die Oberfläche in der Haube und heben Sie nach und nach eine Seite nach oben, saugen Sie das Medium langsam ab, ohne die Zellen zu lösen. - Geben Sie die Transfektantenmischung vorsichtig in den CF10. Stellen Sie sicher, dass alle zehn Schichten mit Medien bedeckt sind.
HINWEIS: Geben Sie 25 mL Transfektantenmischung in die 150-mm-Schale. Überwachen Sie die Zellen täglich bis zur Ernte. Die restliche Transfektantenmischung durch langsames Aufgießen in den CF10 geben. Drehen Sie den CF10 sehr vorsichtig und sorgen Sie für das gleiche Volumen für jede Schicht. - Bringen Sie den CF10 für 72-96 h in den Inkubator zurück.
HINWEIS: Überprüfen Sie die 150-mm-Monitorschüssel, um zu entscheiden, wann die AAV-Vektoren geerntet werden sollen. Wenn sich die Zellen mit einer vollen Ladung AAV sehr leicht ablösen, ist es Zeit für die Ernte.
4. Ernten von AAV-Vektoren
- Ernten Sie das Virus 3-4 Tage nach der Transfektion, indem Sie das CF10 kräftig schütteln. Gießen Sie das Medium in die konischen 250-ml-Röhrchen und schleudern Sie das Virus bei 4000 x g für 20 Minuten bei 4 °C herunter.
HINWEIS: Bei dem in diesem Protokoll verwendeten AAV6-Serotyp bleiben 80 % AAV größtenteils an zelluläres Material im Pellet gebunden. Und die 20% AAV wurden in die Medien verheimlicht. Diese Anteile können bei anderen AAV-Serotypen unterschiedlich sein. - Den geklärten Überstand mit einer 0,45 μm Filtereinheit filtrieren und für den Niederschlag speichern.
- Spülen Sie den CF10 mit 500 mL DPBS 1x Puffer und zentrifugieren Sie ihn bei 4000 x g für 20 min bei 4 °C.
- Der Überstand wird mit einem Vakuumsystem und einer Aspirierpipette entsorgt. 20 ml AAV-Lysepuffer pro CF10 bei -80 °C bis zur Reinigung zugeben.
HINWEIS: Übertragen Sie AAV-Lysat in ein konisches 50-ml-Röhrchen für die spätere AAV-Extraktion. - Der filtrierte Überstand wird herausgenommen, 40 % PEG-2,5 M NaCl-Lösung für eine Endkonzentration von 8 % PEG zugegeben und in einem Kühlraum auf einem Orbitalrotator mindestens 3 h oder über Nacht inkubiert.
HINWEIS: Für 100 mL Überstand 25 mL 40% PEG-2,5 M NaCl-Lösung hinzufügen. - Das Virus wird durch Zentrifugieren bei 4000 x g für 30 min bei 4 °C ausgefällt.
HINWEIS: Füllen Sie die Mischung zur Zentrifugation in konische 250-ml-Röhrchen. - Aspirieren, um den Überstand zu entfernen, und 5-10 ml AAV HEPES-Resuspensionspuffer hinzufügen, um die Pellets zu suspendieren. In ein konisches 50-ml-Röhrchen umfüllen, um die nachgeschaltete Reinigung fortzusetzen, oder bei -80 °C lagern.
5. AAV-Extraktion
- Tauen Sie das Viruspellet in einem 37 °C warmen Wasserbad für 10-15 min mit einem Schüttler auf.
HINWEIS: Wirbeln Sie das AAV-Lysat vor, bis es vollständig schmilzt. - In -80 °C 100%igem Ethanolbad für 20-30 min einfrieren.
HINWEIS: Alternativ kann der Gefrierzyklus mit einem kalten Becherglas aus 100 % Ethanol durchgeführt werden, das von Trockeneis umgeben ist. - 3-4 Mal einfrieren/auftauen und bei Bedarf zwischendurch vortexen.
HINWEIS: Dieser Zyklus kann beim Einfrieren pausiert werden. Lagern Sie das AAV-Lysat bei -80 °C, bis die nachgeschaltete Arbeit beginnt. - Tauen Sie AAV-Lysat (aus Schritt 5.3) und resuspendiertes AAV (aus Schritt 4.7)" in einem 37 °C warmen Wasserbad für 10-15 min mit einem Shaker auf und wirbeln Sie es vortexen.
HINWEIS: Achten Sie darauf, dass es vollständig schmilzt und mischen Sie es. - Fügen Sie Benzonase-Nuklease (250 Einheiten/μl) zu AAV-Lysat und resuspendiertem AAV für eine Endkonzentration von 50 Einheiten/ml hinzu. Vortex die Lösung.
- In einem 37 °C warmen Wasserbad unter Schütteln 30 min inkubieren.
HINWEIS: Nehmen Sie 10% Natriumdesoxycholat-Aliquote in einem Wasserbad heraus, um Zeit zum Auflösen, Aufwärmen und gründlichen Mischen zu haben. - 10 % Natriumdesoxycholat für eine Endkonzentration von 0,5 % zugeben und bei 37 °C mit eingeschaltetem Shaker 30 Minuten inkubieren.
HINWEIS: Natriumdesoxycholat wurde verwendet, um die AAV-Freisetzung aus den Zellen zu verbessern. - Die rohe AAV-Lösung in 2 mL Zentrifugenröhrchen verteilen und bei 14.000 x g für 10 min bei 4 °C zentrifugieren.
- Den Überstand in ein konisches 50-ml-Röhrchen überführen.
HINWEIS: Verwenden Sie eine 1-ml-Pipette, um den Überstand zu übertragen. Entsorgen Sie die Pellets. - Fügen Sie eine gleiche Menge Chloroform hinzu und extrahieren Sie AAV, indem Sie es 1-2 Minuten lang vortexen.
HINWEIS: Die Lösung sollte undurchsichtig, weiß und milchartig werden. - Die Milchlösung in 2 mL Zentrifugenröhrchen füllen und gleichmäßig verteilen. Zentrifugieren Sie bei 14.000 x g für 10 min bei 4 °C.
- Pipettieren Sie die oberste rosa Schicht vorsichtig heraus und übertragen Sie sie in ein neues konisches 50-ml-Röhrchen.
HINWEIS: Stören Sie nicht die Chloroform-/Proteinschichten. - Messen Sie das endgültige Volumen mit einer Pipette. Gemäß der rohen AAV-PEG-Sulfat-Volumentabelle (Tabelle 2) mischen Sie geeignete Volumina aus 50 % (NH4)2SO4 und 40 % PEG-Lösung. 2 Min. Vortexen.
- Übertragen Sie die Mischung in 2 mL Zentrifugenröhrchen, um sie gleichmäßig zu verteilen. Zentrifugieren bei 14.000 x g für 10 min bei 4 °C
- Entnehmen Sie die untere Schicht mit einer 22-G-Nadel und einer 3-ml-Spritze. Sammeln Sie das AAV in einem konischen 50-ml-Röhrchen und lagern Sie es bei 4 °C für die spätere Iodixanol-Gradienten-Ultrazentrifugation.
6. AAV-Aufreinigung durch Iodixanol-Gradienten-Ultrazentrifugation
- Bereiten Sie die Iodixanol-Gradienten frisch vor, bevor Sie das AAV entsprechend der Anzahl der verwendeten Ultrazentrifugationsröhrchen beladen (Tabelle 3).
HINWEIS: Zur Beobachtung der Schichten wurde Phenolrot verwendet. - Überlagern Sie jede Lösung langsam mit einer 10-ml-Spritze, die mit einer langen 16-G-Nadel befestigt ist, in ein Schnellverschlussröhrchen aus Polypropylen-Ultrazentrifuge mit runder Oberseite. Vermeiden Sie Blasen.
- Geben Sie vorsichtig bis zu 17 ml AAV-Lösung mit einer 22-G-Spritze auf den Gradienten. Verwenden Sie AAV-Dialysepuffer, um den Schlauch aufzufüllen.
HINWEIS: Fügen Sie die AAV-Lösung hinzu, indem Sie Tropfen gegen die Wand in das Ultrazentrifugenröhrchen laden. Stören Sie nicht die Verlaufsebenen. - Verschließen Sie die Ultrazentrifugenröhrchen mit einem elektrischen Versiegelungsgerät.
HINWEIS: Gleichen Sie die Röhrchen aus, bevor Sie sie zur Ultrazentrifuge schicken, mit einer Differenz von ±0,2 g. - Zentrifugieren Sie bei 350.000 x g für 2 h in einem Ti70-Rotor bei 4 °C.
- Nehmen Sie die Röhrchen vorsichtig vom Rotor und legen Sie sie in das Gestell der Ultrazentrifugenröhrchen.
HINWEIS: Achten Sie darauf, dass die Farbverläufe nicht gestört werden. - Sammeln Sie AAV-Fraktionen, indem Sie die folgenden Schritte ausführen:
- Stabilisieren Sie das Rohr auf einem Stativhalter.
- Punktieren Sie die Ultrazentrifugenröhrchen etwas unterhalb der 40%-60%-Grenzfläche mit einer 19-G-Nadel, die an einer 10-ml-Spritze befestigt ist.
HINWEIS: Die Öffnung der Nadel sollte nach oben zeigen und der Steigung von 40 % zugewandt sein. - Stanzen Sie mit einer 16 G Nadel ein Loch in die Oberseite der Tube.
- Sammeln Sie bis zu 5 mL pro Röhrchen. Vermeiden Sie es, die Proteinkontamination an der Grenzfläche von 25 % bis 40 % zu sammeln.
- Wiederholen Sie den Vorgang für jedes Ultrazentrifugenröhrchen.
HINWEIS: Übertragen Sie die AAV-Fraktionen in ein konisches 50-ml-Röhrchen.
7. Ultrazentrifugation der zweiten Runde der Iodixanol-Gradienten
HINWEIS: Dieser Schritt ist optional. Dieser Schritt dient dazu, das leere AAV-Verhältnis zu reduzieren, um ein vollständiges AAV-Kapsid von höherer Qualität zu erhalten.
- AAV >1:1 mit AAV-Dialysepuffer verdünnen.
HINWEIS: AAV, das aus der ersten Runde der Ultrazentrifugation entnommen wurde, lag bei 40 % Iodixanolschicht. Es sollte um mindestens 50 % verdünnt werden, um auf die 30 %ige Iodixanolschicht geladen werden zu können. - Überlagern Sie jede Lösung (Tabelle 4) mit einer 16-G-Nadel in ein Ultrazentrifugationsröhrchen und befestigen Sie langsam eine 10-ml-Spritze. Vermeiden Sie Blasen.
- Geben Sie vorsichtig 20 ml verdünnte AAV-Lösung mit einer 22-G-Spritze auf den Gradienten. Verwenden Sie AAV-Dialysepuffer, um den Schlauch aufzufüllen.
- Wiederholen Sie die Schritte 6.4 bis 6.7.
8. AAV-Dialyse und Konzentration
- Übertragen Sie das AAV-Virus mit einer neuen 18-G-Nadel und einer 10-ml-Spritze in die Dialysekassette. Injizieren Sie das Virus langsam in die Kassette und entfernen Sie die Luft.
- Geben Sie einen Rührstab in das Becherglas mit AAV-Dialysepuffer und legen Sie ihn auf eine Rührplatte.
- Setzen Sie die Dialysekassette mit einer Schwimmboje in den Dialysepuffer ein. Bei 4 °C umrühren. Wechseln Sie den AAV-Dialysepuffer 2-3 mal.
HINWEIS: Verwenden Sie genügend Puffer, damit die Kassette schwimmen kann, und führen Sie den Puffertausch durch. Decken Sie den Becher mit Alufolie ab. - Sammeln Sie das Virus mit einer 10-ml-Spritze, die mit einer 18-G-Nadel befestigt ist, aus der Kassette in eine Zentrifugalfiltereinheit.
- Zentrifugieren Sie bei 4.000 x g für 15-30 min bei 4 °C.
HINWEIS: Spülen Sie das AAV 2-3 Mal mit AAV-Dialysepuffer. Konzentrieren Sie das AAV, bis ~200 μl des verbleibenden Proteins auf dem Filter liegen. - Sammeln Sie das konzentrierte AAV aus der Filtereinheit. Spülen Sie den Filter mit weiteren 300 μl AAV-Dialysepuffer und geben Sie ihn auf den konzentrierten AAV.
- Filtrieren Sie das Virus durch einen 0,22-μm-Spritzenvorsatzfilter mit einer 1-ml-Tuberkulin-Luer-Slip-Spritze. Um den geringsten Volumenverlust zu gewährleisten, verwenden Sie die 10-ml-Spritze, um 2-3 Mal Luft durch den Filter zu drücken.
- Lagern Sie das gereinigte AAV-Virus zur Titration vorübergehend bei 4 °C.
HINWEIS: Titrieren Sie das Virus, aliquotieren Sie es und lagern Sie es innerhalb von 1 Woche bei -80 °C.
9. Titration von AAV-Viren
HINWEIS: Die quantitative Polymerase-Kettenreaktion (qPCR) von TaqMan wurde verwendet, um gereinigtes AAV zu titrieren.
- AAV-Vektoren mit DNase I behandeln und 30 min bei 37 °C inkubieren, dann 10 min bei 95 °C inkubieren.
HINWEIS: Als Beispiel für eine Reaktion von 10 μl wird eine 2 μl AAV-Virusprobe, 1 μl DNase, 1 μl DNase-Puffer und 6 μl H2O verwendet. Sie kann in einem Thermocycler mit PCR-Röhrchen durchgeführt werden. - Bereiten Sie die Primer-Sets vor, die auf den AAV-Einsatzbereich abzielen.
HINWEIS: Ziele können der Promotor, das Transgen und der Reporter im AAV-Konstrukt sein. Die Grundierungen sind in Tabelle 5 aufgeführt. - Bereiten Sie DNA-Plasmidstandards (10, 1, 0,1, 0,01, 0,001 und 0,0001 pg/μl) und eine Negativkontrolle (H2O) vor.
- Bereiten Sie den qPCR-Mastermix einschließlich der Primer gemäß den Anweisungen des Herstellers vor.
- Legen Sie die Standards und Proben in dreifacher Ausfertigung auf eine PCR-Platte. Geben Sie die qPCR-Mischung in die Standards und Proben. Verschließen Sie die Platte und führen Sie die qPCR-Reaktion gemäß den Anweisungen des Herstellers durch.
HINWEIS: Die Reaktionsbedingungen hängen von den Materialien/Reagenzien und dem Instrument ab. Schnelle und konventionelle PCR-Zyklen sind anwendbar. - Berechnen Sie den AAV-Virustiter.
HINWEIS: Die Probenkonzentration in diesem Protokoll beträgt 1/10 Verdünnung gegenüber den Originalproben. - Aliquotieren Sie das AAV-Virus in 1,5-ml-Röhrchen und lagern Sie es bis zu einem Monat lang bei 4 °C und langfristig bei -80 °C.
HINWEIS: Vermeiden Sie Gefrier-/Auftauzyklen für die Lagerung. Bei Verwendung für In-vivo-Versuche ist ein Auftauen von mehr als doppelten Aliquoten zu verwenden.
10. Qualitätskontrolle von AAV
HINWEIS: AAV-Viren wurden durch SDS-PAGE-Silberfärbung mit einem SDS-PAGE-Gel auf Reinheit charakterisiert und mit einem handelsüblichen Färbekit gefärbt.
- Denaturierung von 3 x 109 vg AAV-Virusprobe mit Laemmli-Puffer.
HINWEIS: Verwenden Sie einen AAV-Referenzvirus als Kontrolle. Es wird das AAV6-CMV-GFP-Referenz-Vollkapsid verwendet. - Laden Sie das vergällte AAV auf ein SDS-PAGE-Gel mit 4%-20% Steigung und lassen Sie es 50 Minuten lang bei 180 V laufen.
- Entfernen Sie das Gel von der Elektrophoreseplatte. Färben Sie das Gel mit einem Silberfärbeset gemäß den Anweisungen des Herstellers.
- Überprüfen Sie die AAV-Kapsidproteine VP1, VP2 und VP3.
HINWEIS: Das reine AAV-Virus enthält nur das Kapsidprotein VP1, VP2 und VP3. Wenn sie nicht rein sind, wären andere Proteinbanden auf dem Gel sichtbar.
HINWEIS: Die Transmissionselektronenmikroskopie (TEM) von negativ gefärbten rekombinanten AAV-Virionen wurde durchgeführt, um die morphologische Integrität und das Voll/Leer-Verhältnis zu beurteilen. - Nehmen Sie mit einer Pinzette ein EM-Gitter und legen Sie es mit der glänzenden Seite des Gitters nach oben auf die Bank.
- Pipettieren Sie 5 μl AAV-Probe auf das Gitter und lassen Sie sie durch Verdampfen trocknen.
HINWEIS: Verdünnen Sie das AAV bei Bedarf. 1012 vg/mL ist ein durchschnittlicher Titer. Es kann 30-60 Minuten dauern, bis das Gitter getrocknet ist. - Waschen Sie das Gitter, indem Sie es Tropfen für Tropfen mit etwa 200 μl H2O auf das Gitter pipettieren.
- Entfernen Sie das überschüssige Wasser, indem Sie langsam ein Chromatographiepapier senkrecht neben das Gitter legen.
- Pipettieren Sie 5 μl 2%ige Uranylacetatlösung auf den Rost. 5 min inkubieren und wie oben erwähnt abtropfen lassen. Lassen Sie den Rost trocknen.
- Visualisieren Sie die AAV-Partikel unter einem Transmissionselektronenmikroskop (bei 50.000-facher Vergrößerung). Virale Kapside mit einem viralen Genom erscheinen als homogene weiße Sechsecke, während leere Kapside als Sechsecke mit weißem Rand, aber dunklem Zentrum erscheinen.
- Zählen Sie nach dem Zufallsprinzip mindestens 100 Partikel, um den ungefähren Prozentsatz von vollen vs. leere AAV-Partikel.
Ergebnisse
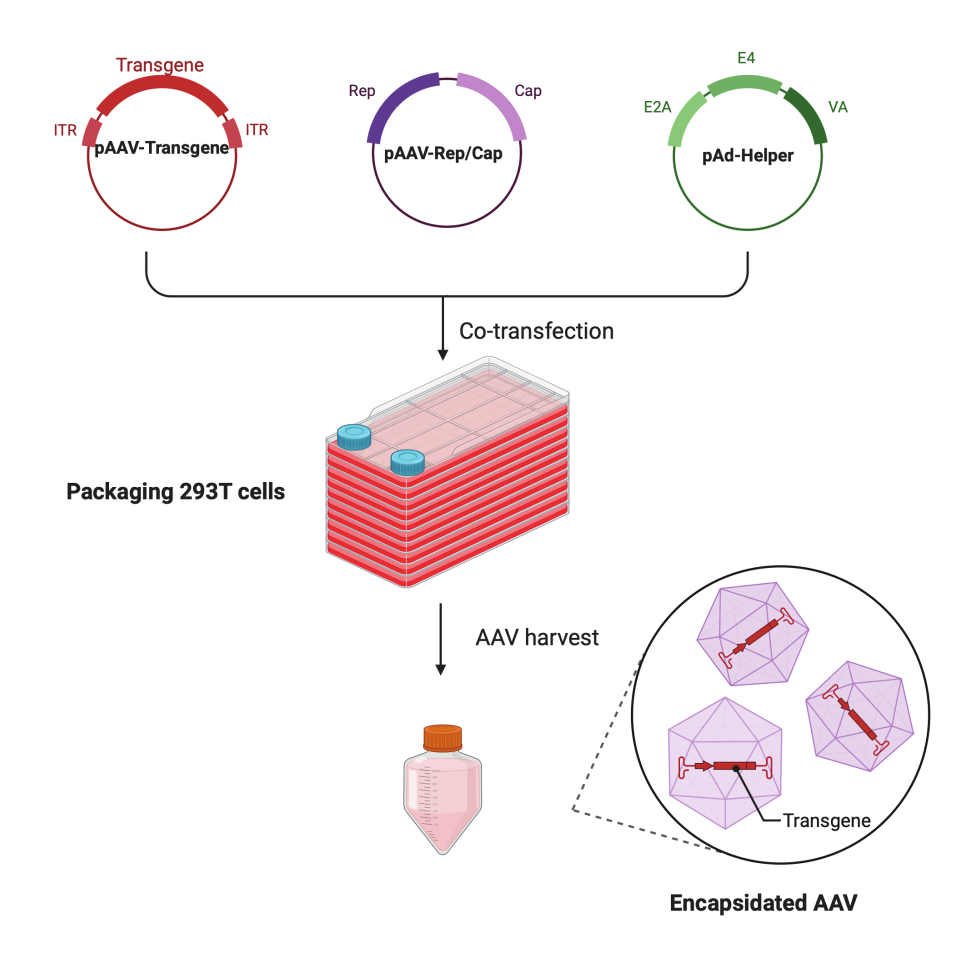
In diesem detaillierten Schritt-für-Schritt-Protokoll wird eine standardisierte Plattform demonstriert, um mit dem CF10 in einem groß angelegten Forschungslabor hochtiternde und qualitativ hochwertige AAV-Viren herzustellen. Im Vergleich zu herkömmlichen Zellkulturschalen bietet das CF10 eine bequeme Möglichkeit, große Mengen an Zellen zu kultivieren und AAV-Viren zu produzieren (Abbildung 1). Es wurden mehrere Kulturbedingungen getestet, um festzustellen, ob Zellen in einer optimalen Umgebung die Virusproduktion fördern können. Ein niedriges Glukose-DMEM, ergänzt mit 10 mM HEPES und 2% FBS, zeigte die beste AAV-Produktion.
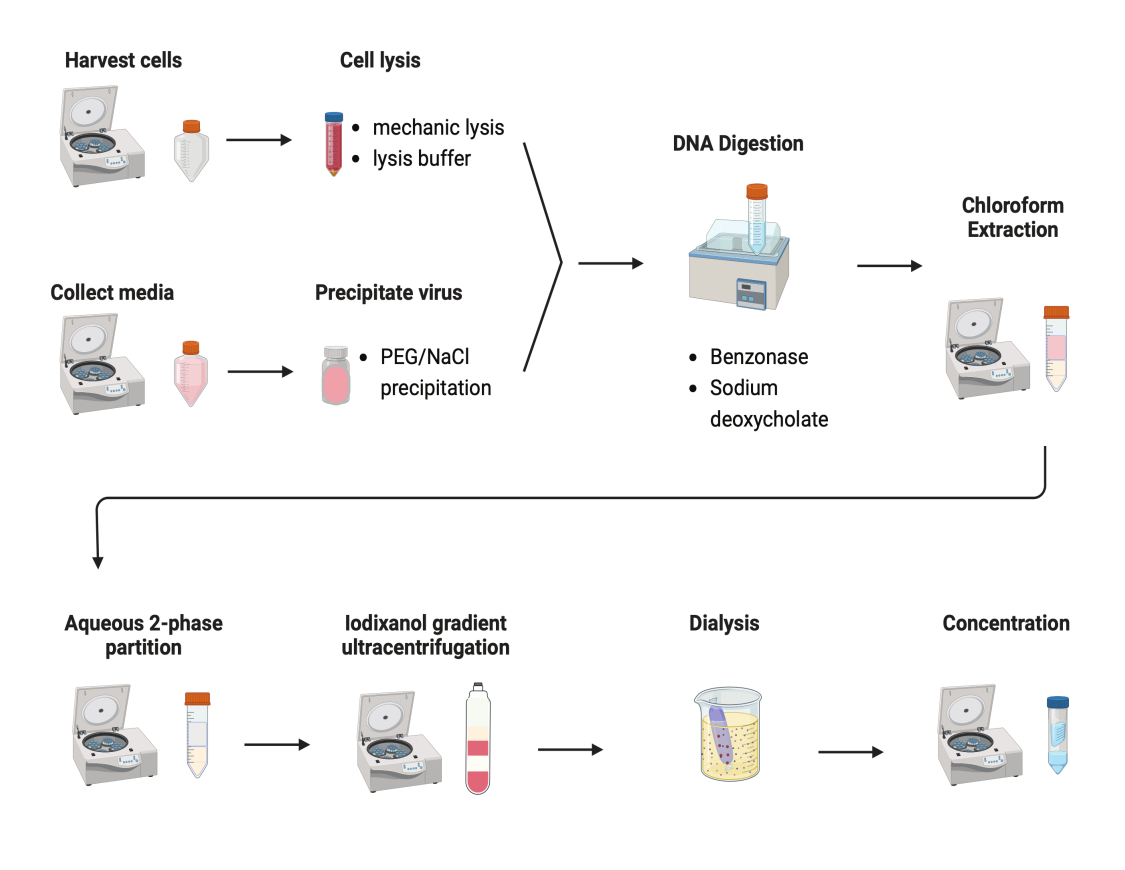
Es wurden mehrere Protokolle zur Aufreinigung von AAVs getestet. Die meisten Verfahren weisen eine geringe Virusausbeute und eine geringe Verunreinigung der AAV-Kapside auf. Hier wurde ein überarbeitetes Aufreinigungsprotokoll entwickelt, bei dem das AAV sowohl aus Zellpellets als auch aus Kulturmedien kombiniert wird (Abbildung 2). Wir haben festgestellt, dass sich 80% des AAV in den Zellen befanden und weitere 20% des AAV in den Kulturmedien, die aus den Zellen sezerniert wurden. Beide Teile des AAV wurden mit DNase behandelt, um die freie DNA zu entfernen. Natriumdesoxycholat wurde verwendet, um AAV weiter aus den Zellen freizusetzen. Die AAVs wurden dann mit Chloroform-Extraktion extrahiert, gefolgt von einer wässrigen Zweiphasen-Partitionierung. Auf diese Weise konnten die meisten Proteinverunreinigungen entfernt werden. AAV bleibt in der Ammoniumsulfat-Phase löslich.
Die verbleibenden Verunreinigungen wurden mit einer diskontinuierlichen Iodixanol-Gradienten-Ultrazentrifugation entfernt (Abbildung 3A). Der Gradient war auch hilfreich bei der Entfernung der leeren AAV-Kapside, insbesondere bei der zweiten Runde der Iodixanol-Gradienten-Ultrazentrifugation.
Die Reinheit des AAV-Virus wurde durch Silberfärbung bestimmt. Wenn drei Hauptbanden, die dem AAV-Kapsidprotein entsprechen, VP1, VP2 und VP3, mit einer Reinheit von mehr als 90 % erhalten wurden, war das AAV-Virus für die In-vivo-Anwendung geeignet (Abbildung 3B). Das Verhältnis von AAV-Vollkapsid zu leerem Kapsid wurde mittels TEM abgerufen (Abbildung 3C). Nur ein vollständiges Kapsid mit einem Transgen-Insert würde die Expression des Transgens im Zielgewebe ermöglichen. Ein hoher Anteil des leeren Kapsids könnte auch eine Immunantwort auf das AAV-Kapsid induzieren. Diese Qualitätskontrollen sind für jedes AAV-Virus erforderlich, das vor der Verwendung hergestellt und gereinigt wird.

Abbildung 1: Schematische Darstellung der AAV-Produktion durch HEK293T Zell-Dreifach-Transfektionsmethode. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Schematische Darstellung der AAV-Aufreinigung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: AAV-Aufreinigung durch Iodixanol-Gradienten-Ultrazentrifugation und Überprüfung der AAV-Virusreinheit. (A) Iodixanol-Gradientenschichten und die Position der Nadel für die Ernte. (B) Repräsentatives Gel zur Beurteilung des Kapsidgehalts und der Reinheit. M: molekularer Marker; R: Referenz AAV6 Vollkapsid; S1: hauseigenes AAVDJ-Kapsid mit einer Runde Ultrazentrifugation; S2: hauseigenes AAVDJ-Kapsid mit zwei Runden Ultrazentrifugation; S3: hausgemachtes AAV6-Kapsid. (C) Elektronenmikroskopische Aufnahme von AAV. Virus, das nach der Reinigung gesammelt wurde. Virale Kapside, die ein virales Genom enthalten, erscheinen als homogene weiße Sechsecke, während leere Kapside als Sechsecke mit weißem Rand, aber dunklem Zentrum erscheinen. Maßstab: 100 nm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Tabelle 1: PEI-Rechner für AAV-Verpackungen. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 2: Rohes AAV-PEG-Sulfat-Volumendiagramm. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 3: Herstellung des Iodixanol-Gradienten. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 4: Vorbereitung der zweiten Runde des Iodixanol-Gradienten. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 5: AAV-Titrationsprimer. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Ergänzende Datei 1: Zusammensetzung der für die Studie verwendeten Puffer. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Hier wird ein technisch fortschrittliches Protokoll für die großtechnische Produktion von hochtitern und hochwertigen AAV-Vektoren unter Verwendung einer Zellfabrikplattform (CF10) vorgestellt, was eine signifikante Verbesserung gegenüber herkömmlichen Zellkulturschalenmethoden darstellt. Der Einsatz von Zellfabriken vereinfacht den Prozess der Kultivierung großer Zellmengen und erleichtert die Produktion von AAV-Viren effizienter1. Durch die Optimierung der Kulturbedingungen, insbesondere bei niedrigem Glukose-DMEM, ergänzt mit 10 mM HEPES und 2% FBS, wurde eine signifikant erhöhte Virusproduktion bestätigt, was auf die entscheidende Rolle der zellulären Umgebung bei der Virusausbeute hinweist.
Das überarbeitete Aufreinigungsprotokoll, das AAV sowohl aus Zellpellets als auch aus Kulturmedien kombiniert, befasst sich mit dem häufigen Problem der geringen Virusausbeute und der Verunreinigung, das in vielen bestehenden Protokollen zu finden ist. Die Schritte der Chloroform-Extraktion und der wässrigen Zweiphasen-Partitionierung entfernen effektiv die meisten Proteinverunreinigungen, wobei AAV in der Ammoniumsulfatphase löslich bleibt. Die Verbesserung sowohl der Ausbeute als auch der Reinheit von AAV-Vektoren unter Verwendung der PEG/wässrigen Zweiphasen-Partitionierung in Kombination mit der Iodixanol-Gradienten-Ultrazentrifugation im Gegensatz zu herkömmlichen Gradienten-Ultrazentrifugationsmethoden kann auf eine verbesserte Anfangstrennung mit PEG/wässriger Zweiphasen-Partitionierung, eine verfeinerte Reinheit durch Iodixanol-Gradienten-Ultrazentrifugation und eine Verringerung der Kontaminanten-Co-Reinigung zurückgeführtwerden 12. Erstens verbessert die Einführung einer zweiphasigen PEG/wässrigen Partitionierung vor der Ultrazentrifugation die anfängliche Trennung von AAV-Partikeln von Zelltrümmern und anderen Verunreinigungen erheblich. PEG, ein Polymer mit hohem Molekulargewicht, bildet, wenn es mit einer wässrigen Lösung gemischt wird, zwei unterschiedliche Phasen13. AAV-Vektoren neigen dazu, bevorzugt in eine dieser Phasen (üblicherweise die PEG-reiche Phase) aufzuteilen, während viele Kontaminanten und Verunreinigungen in die anderen13 aufgeteilt werden. Durch diese selektive Aufteilung werden die AAV-Partikel effektiv konzentriert und ein erheblicher Teil der Verunreinigungen bereits vor der Ultrazentrifugation entfernt, wodurch die Ausbeute erhöht und die in die Ultrazentrifugationsstufe13 eintretende Verunreinigungsfracht reduziert wird. Zweitens verfeinert die Iodixanol-Gradienten-Ultrazentrifugation die Reinheit von AAV-Vektoren weiter. Iodixanol, ein nichtionisches, isoosmolares Gradientenmedium, ermöglicht eine schonendere und kontrolliertere Trennung im Vergleich zu herkömmlichen CsCl-Gradienten14. In diesem Gradienten wandern AAV-Partikel zu einer Position im Gradienten, die ihrer Auftriebsdichte14 entspricht. Wichtig ist, dass dieser Prozess bei der Trennung von vollständigen AAV-Kapsiden (die die genetische Nutzlast enthalten) von leeren Kapsiden (ohne genetisches Material) wirksam ist, was eine entscheidende Determinante für die Vektorqualität ist. Die iso-osmolare Natur von Iodixanol bewahrt auch die Integrität der AAV-Kapside besser als hyperosmolarer Wirkstoffe wie CsCl, was möglicherweise zu einer höheren Ausbeute an intakten, funktionellen Vektoren führt14. Schließlich können herkömmliche Ultrazentrifugationsmethoden, insbesondere solche mit CsCl-Gradienten, manchmal Kontaminanten mitreinigen, die eine ähnliche Auftriebsdichte wie AAV-Vektorenaufweisen 15. Durch die Verwendung der PEG-Partitionierung als Vorstufe wird die Belastung mit solchen Verunreinigungen vor der Ultrazentrifugation stark reduziert13. Diese Verringerung der Schadstoffbelastung bedeutet, dass der Iodixanol-Gradient effektiver und selektiver bei der Reinigung von AAV-Vektoren arbeiten kann, was zu einer höheren Reinheitführt 15.
Die Reinheit und Qualität der AAV-Vektoren wird durch Silberfärbung und TEM11 streng bewertet. Die Beobachtung von drei Hauptbanden, die den AAV-Kapsidproteinen VP1, VP2 und VP3 entsprechen, mit einer Reinheit von mehr als 90 %, deutet auf die Eignung dieser AAV-Vektoren für die In-vivo-Verwendung hin. Die TEM-Analyse zur Bestimmung des Verhältnisses von vollen zu leeren Kapsiden ist besonders wichtig, da ein hoher Anteil an leeren Kapsiden zu einer verminderten Effizienz der Genverabreichung und potenziellen Immunantworten führen kann11. Diese Qualitätsprüfung ist zwar unerlässlich, erhöht aber die verfahrenstechnische Komplexität und erfordert möglicherweise zusätzliches technisches Fachwissen.
Zusammenfassend lässt sich sagen, dass das Protokoll erhebliche technische Fortschritte bei der Herstellung von AAV-Vektoren bietet, insbesondere in Bezug auf Skalierbarkeit und Reinheit. Die mit dem Reinigungsprozess verbundene Komplexität und der Bedarf an spezialisierten Geräten und Fachkenntnissen können jedoch immer noch eine geringfügige Einschränkung für die Anwendung in bestimmten Forschungsumgebungen darstellen. Eine weitere Verfeinerung und Vereinfachung dieser Techniken könnte diesen Ansatz im Bereich der Gentherapieforschung zugänglicher und breiter anwendbar machen.
Offenlegungen
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagungen
Das TZ hat die Experimente konzipiert. TZ, VD, SB und JP führten die Experimente durch. TZ und VD generierten Daten und analysierten die Daten. TZ und YX schrieben das Manuskript. TZ und GG überarbeiteten das Manuskript. Diese Arbeit wurde vom UPMC Children's Hospital of Pittsburgh unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 293T/17 cells | ATTC | CRL-11268 | |
| 0.5 M EDTA | MilliporeSigma | 324506-100ml | |
| 1 mL Henke-Ject syringe | Fisher Scientific | 14-817-211 | |
| 10% pluronic F68 solution | Fisher Scientific | 24-040-032 | |
| 10x Tris/Glycine/SDS Buffer | Biorad | 1610732 | |
| 1M HEPES | Fisher Scientific | 15-630-080 | |
| 2% Uranyl Acetate Solution | Electron Microscopy Sciences | 22400-2 | |
| 4%–20% Precast Protein Gels | biorad | 4561094 | |
| 40% PEG solution | Sigma | P1458-50ML | |
| AAV6 reference full capsids | Charles River Laboratories | RS-AAV6-FL | |
| Accutase Cell Detachment Solution | Fisher Scientific | A6964-100ML | |
| Benzonase | Sigma | E1014-25KU | |
| BioLite Cell Culture Treated Dishes 150 mm | Fisher Scientific | 12-556-003 | |
| Centrifugal Filter Unit | MilliporeSigma | UFC905024 | |
| Corning PES Syringe Filters | Fisher Scientific | 09-754-29 | |
| Dialysis Cassettes, 10 K MWCO | Fisher Scientific | PI66810 | |
| Disposable PES Filter Units 1 L 0.2 µm | Fisher Scientific | FB12566506 | |
| Disposable PES Filter Units 1 L 0.45 µm | Fisher Scientific | FB12566507 | |
| Disposable PES Filter Units 500 mL 0.2 µm | Fisher Scientific | FB12566504 | |
| DMEM high glucose | Fisher Scientific | 10-569-044 | |
| DMEM low glucose | Fisher Scientific | 10567022 | |
| DNase | NEB | M0303S | |
| DPBS 1x | Fisher Scientific | 14-190-250 | |
| Fetal Bovin Serum (FBS) | Biowest | S1620 | |
| Formvar/Carbon 300 Mesh, Cu | Electron Microscopy Sciences | FCF300-Cu-50 | |
| glycerol | Sigma | G5516-1L | |
| KCl | Sigma | P9541-500G | |
| LB agar | Sigma | L2897-250G | |
| LB broth | Fisher Scientific | BP9732-500 | |
| MgCL2·6H2O | Sigma | M9272-100G | |
| NEB stable competent cells | NEB | C3040H | |
| Nest Biofactory 10 chamber | MidSci | 771302 | |
| NucleoBond Xtra Maxi EF | Macherey-Nagel | 740424 | |
| Opti-MEM | Fisher Scientific | 31-985-088 | |
| OptiPrep Density Gradient Medium | Millipore Sigma | D1556-250ml | |
| pAAV-CMV-GFP | Addgene | 105530 | |
| pAAV-DJ | Cell BioLab | VPK-420-DJ | |
| pAAV-RC6 | Cell BioLab | VPK-426 | |
| pAdDeltaF6 | Addgene | 112867 | |
| PEG 8000 | Promega | V3011 | |
| PEI Max | Polysciences, Inc | 49553-93-7 | |
| Pen-Strep | Fisher Scientific | 15-140-163 | |
| Phenol red | Millipore Sigma | 1.07242.0100 | |
| Pierce Silver Stain Kit | Thermo Fisher Scientific | 24612 | |
| QuickSeal tube | Fisher Scientific | NC9144589 | |
| Sodium Chloride | Sigma | 1162245000 | |
| sodium deoxycholate | Millipore Sigma | D6750-100G | |
| Taqman Fast Advanced Master Mix | Thermo Fisher Scientific | 4444557 | |
| Type 70 Ti Fixed-Angle Titanium Rotor | Beckman Coulter | 337922 | |
| Western Blotting Substrate | ThermoFisher | 32209 |
Referenzen
- Arjomandnejad, M., Dasgupta, I., Flotte, T. R., Keeler, A. M. Immunogenicity of recombinant adeno-associated virus (AAV) vectors for gene transfer. BioDrugs. 37 (3), 311-329 (2023).
- Liu, Y., Siriwon, N., A Rohrs, J., Wang, P. Generation of targeted adeno-associated virus (AAV) vectors for human gene therapy. Curr Pharm Des. 21 (22), 3248-3256 (2015).
- Bilal, A. S., et al. Optimization of large-scale Adeno-Associated Virus (AAV) production. Curr Protoc. 3 (5), e757(2023).
- Rashnonejad, A., Chermahini, G. A., Li, S., Ozkinay, F., Gao, G. Large-scale production of adeno-associated viral vector serotype-9 carrying the human survival motor neuron gene. Mol Biotechnol. 58, 30-36 (2016).
- Challis, R. C., et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat Protoc. 14 (2), 379-414 (2019).
- Challis, R. C., et al. Publisher Correction: Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat Protoc. 14 (8), 2597-2597 (2019).
- Mueller, C., Ratner, D., Zhong, L., Esteves-Sena, M., Gao, G. Production and discovery of novel recombinant adeno-associated viral vectors. Curr Protoc Microbiol. 26 (1), 14(2012).
- Guo, P., Wiersch, J., Xiao, X., Gittes, G. Simplified purification of AAV and delivery to the pancreas by intraductal administration. Methods Mol Biol. 1950, 373-387 (2019).
- Berns, K. I., Srivastava, A. Next generation of adeno-associated virus vectors for gene therapy for human liver diseases. Gastroenterol Clin North Am. 48 (2), 319-330 (2019).
- Khasa, H., Kilby, G., Chen, X., Wang, C. Analytical band centrifugation for the separation and quantification of empty and full AAV particles. Mol Ther Methods Clin Dev. 21, 585-591 (2021).
- Chen, H. Comparative observation of the recombinant adeno-associated virus 2 using transmission electron microscopy and atomic force microscopy. Microsc Microanal. 13 (5), 384-389 (2007).
- Burnham, B., et al. Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Hum Gene Ther Methods. 26 (6), 228-242 (2015).
- Kato, M., et al. In situ-formable, dynamic crosslinked poly (ethylene glycol) carrier for localized adeno-associated virus infection and reduced off-target effects. Commun Biol. 6 (1), 508(2023).
- Sena-Esteves, M., Gao, G. Enrichment of fully packaged virions in column-purified recombinant adeno-associated virus (rAAV) preparations by iodixanol gradient centrifugation followed by anion-exchange column chromatography. Cold Spring Harb Protoc. 2020 (2), 095638(2020).
- Matsumoto, M., Wangelin, J. R., Murphy, M. L. Purification of avian encephalomyelitis virus by ultracentrifugation in a nonlinear cesium chloride gradient. Avian Dis. 22 (3), 496-502 (1978).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten