
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Utilizzo dell'elettroporazione postnatale in vivo per studiare la morfologia dei granuli cerebellari e lo sviluppo delle sinapsi
In questo articolo
Erratum Notice
Riepilogo
Qui descriviamo un metodo per visualizzare la sinaptogenesi dei neuroni granuli nel cervelletto del topo nel corso dello sviluppo postnatale del cervello quando queste cellule perfezionano le loro strutture sinaptiche e formano sinapsi per integrarsi nel circuito cerebrale generale.
Abstract
I neuroni subiscono cambiamenti dinamici nella loro struttura e funzione durante lo sviluppo del cervello per formare connessioni appropriate con altre cellule. Il cervelletto dei roditori è un sistema ideale per tracciare lo sviluppo e la morfogenesi di un singolo tipo di cellula, il neurone granulo cerebellare (CGN), nel tempo. Qui, l'elettroporazione in vivo dei progenitori dei neuroni granuli nel cervelletto di topo in via di sviluppo è stata impiegata per marcare scarsamente le cellule per le successive analisi morfologiche. L'efficacia di questa tecnica è dimostrata nella sua capacità di mostrare le fasi chiave dello sviluppo della maturazione del CGN, con un focus specifico sulla formazione di artigli dendritici, che sono strutture specializzate in cui queste cellule ricevono la maggior parte dei loro input sinaptici. Oltre a fornire istantanee delle strutture sinaptiche CGN durante lo sviluppo cerebellare, questa tecnica può essere adattata per manipolare geneticamente i neuroni granuli in modo autonomo dalla cellula per studiare il ruolo di qualsiasi gene di interesse e il suo effetto sulla morfologia CGN, sullo sviluppo degli artigli e sulla sinaptogenesi.
Introduzione
Lo sviluppo del cervello è un processo prolungato che si estende dall'embriogenesi alla vita postnatale. Durante questo periodo, il cervello integra una combinazione di stimoli intrinseci ed estrinseci che scolpiscono il cablaggio delle sinapsi tra dendriti e assoni per guidare in definitiva il comportamento. Il cervelletto dei roditori è un sistema modello ideale per studiare come si sviluppano le sinapsi perché lo sviluppo di un singolo tipo di neurone, il neurone granulo cerebellare (CGN), può essere monitorato mentre passa da una cellula progenitrice a un neurone maturo. Ciò è dovuto, in parte, al fatto che la maggior parte della corteccia cerebellare si sviluppa postnatale, il che consente una facile manipolazione genetica e l'etichettatura cellulare dopo la nascita1.
Nei mammiferi, la differenziazione CGN inizia alla fine dello sviluppo embrionale quando un sottogruppo di cellule proliferative nel cervello posteriore migra sul labbro rombico per formare una zona germinale secondaria sulla superficie del cervelletto 2,3,4. Sebbene siano pienamente impegnate in un'identità progenitrice del neurone granulo (GNP), queste cellule continuano a proliferare all'interno della porzione esterna dello strato granulo esterno (EGL) fino al giorno 14 postnatale (P14). La proliferazione di questo strato provoca una massiccia espansione del cervelletto poiché queste cellule danno origine esclusivamente a CGN5. Una volta che i CGN appena nati escono dal ciclo cellulare nell'EGL, migrano verso l'interno verso lo strato di granuli interni (IGL), lasciandosi dietro un assone che si biforcherà e viaggerà nello strato molecolare del cervelletto, formando fibre parallele che sinapsi sulle cellule di Purkinje6. La posizione di queste fibre all'interno dello strato molecolare dipende dai tempi di uscita del ciclo cellulare.
I CGN che si differenziano per primi lasciano le loro fibre parallele verso il fondo dello strato molecolare, mentre gli assoni dei CGN che si differenziano in seguito sono raggruppati nella parte superiore 7,8. Una volta che i corpi delle cellule CGN raggiungono l'IGL, iniziano a elaborare dendriti e formano sinapsi con i vicini neuroni inibitori ed eccitatori. L'albero dendritico maturo di un CGN mostra un'architettura stereotipata con quattro processi principali. Nel corso della maturazione di CGN, le strutture alla fine di questi dendriti formano un artiglio che si arricchisce di proteine postsinaptiche 9,10. Queste strutture specializzate, chiamate artigli dendritici, contengono la maggior parte delle sinapsi sui neuroni granuli e sono importanti per ricevere sia input eccitatori da innervazioni di fibre muschiose provenienti dal ponte, sia input inibitori dalle cellule locali di Golgi. Una volta completamente configurate, le connessioni sinaptiche delle CGN consentono a queste cellule di trasmettere input dai nuclei pre-cerebellari alle cellule di Purkinje, che si proiettano dalla corteccia cerebellare ai nuclei cerebellari profondi.
L'elettroporazione postnatale in vivo dei PNL è vantaggiosa rispetto ad altri metodi basati sulla marcatura, come l'infezione virale e la generazione di linee di topo transgeniche, perché l'espressione dei costrutti desiderati può essere raggiunta su una linea temporale rapida e il metodo si rivolge a una piccola popolazione di cellule, utile nello studio degli effetti autonomi delle cellule. Questo metodo è stato utilizzato in studi precedenti per studiare lo sviluppo morfologico delle CGN; Tuttavia, questi studi si sono concentrati su un singolo punto temporale o su una breve finestra temporale 9,10,11,12,13. Questo metodo di etichettatura è stato abbinato all'analisi delle immagini per documentare i cambiamenti nella morfologia CGN che si verificano durante l'intero corso della differenziazione CGN nelle prime tre settimane di vita postnatale. Questi dati rivelano le dinamiche dello sviluppo di dendriti CGN che sono alla base della costruzione dei circuiti cerebellari.
Protocollo
NOTA: Tutte le procedure sono state eseguite secondo protocolli approvati dal Comitato istituzionale per la cura e l'uso degli animali della Duke University (IACUC).
1. Preparazione del DNA per elettroporazione in vivo o IVE (1 giorno prima dell'intervento)
- Raccogliere i seguenti materiali: DNA purificato (0,5-25 μg per animale), acetato di sodio 3 M, etanolo, colorante Fast Green, acqua distillata ultrapura, soluzione tampone fosfato (PBS) (vedere la tabella dei materiali).
NOTA: Per il DNA, un costrutto che esprime la proteina fluorescente verde (GFP) sotto un promotore umano dell'ubiquitina è stato ottenuto da Addgene (FUGW, https://www.addgene.org/14883/). Qualsiasi costrutto che esprima GFP o un'altra proteina fluorescente sotto il controllo di un promotore onnipresente dovrebbe funzionare. L'etichettatura specifica CGN con questa tecnica non dipende dal costrutto, ma piuttosto dall'elettroporazione. - Preparare il DNA per l'elettroporazione mescolando la quantità desiderata di DNA, il 10% in volume di acetato di sodio 3 M e il 250% in volume di etanolo ghiacciato al 100%. Si noti che il DNA precipiterà immediatamente fuori dalla soluzione.
- Continuare a precipitare la miscela di DNA durante la notte a -20 °C o per un'ora a -80 °C.
- Il pellet ha precipitato il DNA in una centrifuga da tavolo a > 16.000 × g e lavato due volte con etanolo al 70%.
- Lasciare asciugare completamente il pellet di DNA e ricostituirlo in una soluzione 1x PBS + 0,02% Fast Green.

Figura 1: Limitazione della profondità di iniezione a 1,5 mm mediante distanziatore. (A) Un segmento di 11,2 mm viene tagliato da una pipetta di carico utilizzando una lama di rasoio. (B) Il distanziatore è montato sulla punta della siringa di Hamilton (la lunghezza totale è di 1,27 cm o 0,5 pollici) e fissato con adesivo o parafilm. La punta esposta deve essere lunga 1,5 mm. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
2. Elettroporazione in vivo di progenitori di neuroni granuli in topi postnatali di sette giorni
NOTA: Tutti gli interventi chirurgici di elettroporazione sono stati eseguiti in una suite chirurgica sterile e altamente ventilata e tutto il personale indossava dispositivi di protezione individuale completi tra cui guanti, maschera facciale, cuffia per capelli, camice e copriscarpe. In alternativa, gli interventi chirurgici possono essere eseguiti in una cappa ventilata e sterile.
- Raccogliere i seguenti materiali: DNA per elettroporazione, piccole forbici chirurgiche, piccole pinzette chirurgiche, siringa Hamilton personalizzata, applicatore con punta di cotone, piastra riscaldante, betadine, etanolo al 70%, 1x PBS, parafilm, adesivo tissutale (n-butilestere cianoacrilato), isoflurano, elettroporatore ed elettrodi tipo pinzetta (vedere la tabella dei materiali).
- Tagliare un distanziatore da una punta di carico sterilizzata per adattarlo alla siringa di Hamilton per limitare la profondità di iniezione a 1,5 mm (Figura 1A,B). Fissare il distanziatore con adesivo o parafilm.
- Anestetizzare il cucciolo P7 in una camera di isoflurano ad una velocità di erogazione di 0,8 L/min. Confermare l'anestesia completa monitorando l'animale per la diminuzione della respirazione e la mancanza di una risposta al pizzico della punta o della coda (Figura 2A).
- Una volta che l'animale è completamente anestetizzato, posizionare i cuccioli su un piedistallo dotato di un cono nasale, erogando isoflurano costante al 4% ad una velocità di erogazione di 0,8 L / min. Pulire la parte superiore della testa del cucciolo 3 volte con un tampone sterile di betadine poi etanolo al 70%, alternando tra i due, per preparare il sito. Lasciare asciugare la soluzione prima di procedere.
- Usando un paio di forbici sterilizzate, fai una piccola incisione con un taglio che si estende dalla parte superiore alla base delle orecchie per rivelare il cervello posteriore (Figura 2B).
- Individuare il cervelletto (Figura 2C), inserire la punta esposta della siringa di Hamilton attraverso il cranio, perpendicolare al cervello, e iniettare 1,5 μL di miscela di DNA nel parenchima cerebellare spingendo lentamente lo stantuffo posteriore della siringa. Dopo la somministrazione della miscela di DNA, tirare lentamente indietro l'ago per evitare fuoriuscite di schiena e lasciare che la soluzione di DNA si diffonda per 30 secondi.
- Spegnere l'isoflurano e posizionare il cucciolo su un termoforo a 37 °C. Preparare l'elettrodo a pinzetta per l'elettroporazione immergendo entrambe le estremità in 1x PBS sterile.
NOTA: Bagnare l'elettrodo a pinzetta eviterà ustioni da contatto sulla pelle del cucciolo durante la somministrazione degli impulsi elettrici. - Orientare l'elettrodo a pinzetta sopra il sito di iniezione con l'estremità più rivolta verso il basso e l'estremità negativa sopra la testa dell'animale (Figura 2D). Somministrare cinque impulsi elettrici dall'elettroporatore con le seguenti impostazioni: intervallo di interimpulso di 50 ms, 130 V e 950 ms.
NOTA: Se necessario, eseguire un'iniezione di prova per assicurarsi che il sito di iniezione si trovi sul verme cerebellare (Figura 2E). - Pizzicare l'incisione chiusa e sigillare la ferita con un adesivo tissutale n-butil-estere cianoacrilato non tossico. Pulire la ferita con etanolo al 70% poiché qualsiasi traccia di sangue aumenta la probabilità di infanticidio e cannibalismo dei genitori.
- Lasciare che l'animale si riprenda su una piastra elettrica a 37 °C prima di riportare il cucciolo alla madre. Monitorare i cuccioli ogni 30 minuti per almeno 2 ore dopo l'intervento chirurgico per garantire il pieno recupero.
NOTA: L'infanticidio da parte di entrambi i genitori è abbastanza comune. Per prevenire il cannibalismo, alloggia il sire in una gabbia diversa prima di iniziare l'elettroporazione, e riporta sempre i cuccioli puliti e recuperati (cioè senza macchie di sangue, completamente mobili) nella gabbia originale sul letto originale. I cuccioli possono anche essere puliti con escrementi dalla gabbia originale per ridurre al minimo l'odore di sangue. L'uso di una diga surrogata può essere necessario se la diga originale continua a cannibalizzare i suoi cuccioli.

Figura 2: Elettroporazione cerebellare in vivo di progenitori di neuroni granuli in cuccioli di topo wildtype P7. (A) I cuccioli vengono anestetizzati con isoflurano al 4% erogato ad una velocità di 0,8 L/min per garantire l'anestesia durante l'iniezione della soluzione di DNA. L'isoflurano viene erogato ad una velocità di 0,8 L/min. (B) Dopo aver sterilizzato il topo 3 volte con betadine e etanolo al 70%, viene praticata un'incisione che copre la distanza delle orecchie, rivelando il cervello posteriore. (C) Un'immagine ingrandita di una demarcazione bianca sul cranio, un punto di riferimento per il sito di iniezione. Il costrutto di DNA deve essere iniettato entro 1 mm sopra il segno; Le linee tratteggiate delineano la demarcazione e la freccia nera indica il sito di iniezione. Le creste del verme cerebellare possono essere visibili e possono essere utili per trovare il sito di iniezione. (D) Orientamento dell'elettrodo a pinzetta per un'elettroporazione efficiente. L'estremità più (+) deve essere orientata verso il basso per attirare il DNA caricato negativamente nel parenchima cerebellare prima della somministrazione di impulsi elettrici. (E) L'iniezione di prova di 1 μL di un colorante Fast Green allo 0,02% mostra che l'iniezione è localizzata al centro del verme cerebellare tra i lobuli 5-7. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
3. Immunoistochimica delle CGN elettroporate
- Raccogliere i seguenti materiali: isoflurano, 1x PBS, 4% paraformaldeide (PFA), 30% saccarosio, siero di capra normale, detergente non ionico, vetrini, vetrini, smalti per unghie, supporti di montaggio, colorante nucleare Hoechst e anticorpi primari e secondari appropriati (vedere la tabella dei materiali).
- Anestetizzare l'animale sperimentale con isoflurano e confermare l'anestesia completa con un pizzico di punta e coda.
- Eseguire una perfusione transcardica iniettando lentamente 1x PBS e 4% PFA nel ventricolo sinistro del cuore dell'animale. Lasciare che il sangue defluisca dall'animale tagliando la vena cava.
- Fissare il cervello durante la notte immergendolo in PFA al 4% a 4 ° C. Il giorno seguente, sciacquare rapidamente il cervello con 1x PBS e trasferire il cervello in saccarosio al 30% in 1x PBS per la crioprotezione per almeno 24 ore.
- Se necessario, tagliare il cervello a metà lungo l'asse rostrale-caudale e confermare l'espressione del costrutto reporter trasfettato usando un microscopio da dissezione fluorescente verticale.
NOTA: Tenere il cervello immerso in 1x PBS in un piccolo piatto per evitare che si secchi. - Montare il cervello su un microtomo congelante, tagliare sezioni sagittali da 25 μm e consentire alle sezioni di svolgersi in una miscela 1: 1 di 1x PBS e glicerolo.
NOTA: Le sezioni possono essere conservate in questa soluzione crioprotettiva a -20 °C per la conservazione a lungo termine. - Lavare le sezioni tre volte in 1x PBS per 10 minuti ciascuna per rimuovere il crioprotettore e bloccare il tessuto in 1x PBS + 10% siero di capra normale + 0,2% detergente non ionico su uno shaker orbitale a temperatura ambiente per 1 ora.
- Preparare la soluzione anticorpale primaria: 1x PBS, siero di capra normale al 10%, detergente non ionico allo 0,2% e anticorpo anti-GFP e centrifugare la soluzione per 5 minuti a >16.000 × g. Incubare sezioni nella soluzione anticorpale a 4 °C su uno shaker orbitale per 48 ore.
- Lavare via la soluzione anticorpale primaria per 15 minuti cinque volte con 1x PBS + 0,2% detergente non ionico.
- Preparare una soluzione anticorpale secondaria: 1x PBS, siero di capra normale al 10%, detergente non ionico allo 0,2% e un anticorpo secondario appropriato per rilevare la GFP; Centrifugare la soluzione a >16.000 × g. Incubare sezioni nella soluzione anticorpale su uno shaker orbitale a temperatura ambiente per 2-3 ore. Proteggere le sezioni dall'esposizione alla luce per evitare lo sbiancamento.
- Lavare via la soluzione anticorpale secondaria tre volte con 1x PBS + 0,2% detergente non ionico per 15 minuti ogni volta. Incubare le sezioni in 1x PBS + Hoechst per 5 minuti per colorare i nuclei.
- Lavare la soluzione Hoechst con 1x PBS + 0,2% detergente non ionico e montare su vetrini. Coprire le sezioni con supporti di montaggio, coprire le guide e sigillare la slitta con smalto per unghie per evitare l'evaporazione.
4. Analisi morfologiche di CGN - ricostruzione tridimensionale (3D) e superficie e volume cellulare
- Immagini di CGN elettroporate singole su un microscopio confocale con obiettivo 63x con uno zoom 2x, scattando immagini z-stack a 0,5 μm per stack. Immagine di una cella per finestra dell'immagine per consentire una facile analisi e ricostruzione dell'immagine.
- Installare il plug-in Simple Neurite Tracer per FIJI utilizzando il seguente link (https://imagej.net/Simple_Neurite_Tracer:_Basic_Instructions) per tracciare in modo semplice ed efficiente la struttura delle CGN elettroporate nello spazio tridimensionale (3D).
NOTA: è disponibile una versione aggiornata del plug-in (https://imagej.net/SNT). - Analizza la lunghezza dei neuriti e la formazione di artigli dendritici in cieco usando Simple Neurite Tracer. Carica immagini z-stack a canale singolo di CGN elettroporate su FIJI e fai clic su Plugin | Segmentazione | Tracciante di neurite semplice (Figura 3D).
- Accedere al menu a discesa e selezionare Crea nuovo visualizzatore 3D (Figura 3D).
- Scorrere fino alla base di un dendrite, dove si collega al soma cellulare e iniziare un percorso facendo clic sulla giunzione. Traccia manualmente il percorso facendo clic sulle sezioni in cui il segnale di riempimento della cella è più luminoso, premendo [y] per mantenere la traccia. Traccia fino alla fine del dendrite se non contiene un artiglio o fino alla base dell'artiglio e conferma il percorso premendo [f] (Figura 4D).
- Quindi, traccia l'artiglio iniziando un percorso alla base della struttura e tracciando fino alla fine del neurite più lungo. Traccia i rami secondari e terziari tenendo premuto [ctrl] su Windows o [alt] su Mac OS e facendo clic sul percorso. Confermare il percorso premendo [f].
- Osservare che le misurazioni per le tracce sono visibili su una finestra separata; Sommare tutte le misure dei rami dell'artiglio (primario, secondario, terziario) per ottenere la lunghezza totale per ciascun artiglio.
- Per analizzare l'area superficiale e il volume cellulare delle CGN elettroporate, scaricare il software di analisi cellulare Imaris (https://imaris.oxinst.com/).
NOTA: FIJI può anche essere utilizzato per ricostruire celle in 3D da immagini z-stack utilizzando plug-in prontamente disponibili e gratuiti. Inoltre, c'è una funzione di rendering volumetrico in Simple Neurite Tracer, ma Imaris è stato utilizzato per i motivi descritti di seguito. - Carica l'immagine z-stack di un CGN elettropolato su Imaris. Accedi al toolkit di ricostruzione 3D premendo Oltre.
- Per ricostruire il CGN, premere Superfici e selezionare un'area di interesse che comprenda l'intera cella all'interno della finestra dell'immagine. Una volta terminato, premi la freccia blu in avanti nell'angolo in basso a destra sotto Crea.
- Se l'immagine contiene più canali per segnali diversi, selezionare il canale contenente il CGN elettropolato e premere la freccia blu in avanti.
- Utilizzando la barra di scorrimento, impostare una soglia desiderata che si adatta più accuratamente al segnale della cella elettroporata. Ingrandire la superficie della cella per determinare con precisione la soglia. Una volta terminato, premere la doppia freccia verde per ricostruire la cella e ottenere la superficie e la dimensione del volume dai metadati.

Figura 3: Analisi immunoistochimica e ricostruzione tridimensionale di neuroni granuli elettroporati. I topi P7 CD-1 sono stati elettroporati con un costrutto che esprime GFP. I cervelli sono stati raccolti e sottoposti a immunoistochimica, microscopia confocale e ricostruzione 3D per l'analisi morfologica. (A) Timeline dall'elettroporazione all'elaborazione delle immagini di un mouse da 10 DPI. (B) Immagine di proiezione massima di una sezione trasversale sagittale del cervelletto elettropolato 10-DPI; Le linee bianche delimitano gli strati cerebellari e la barra della scala è di 25 μm. (C) Immagine di proiezione massima di un singolo neurone granulo elettropolato 10-DPI e la corrispondente traccia 3D, la barra della scala è 10 μm. (D) Le ricostruzioni 3D sono state generate utilizzando il plugin FIJI Simple Neurite Tracer. Tutte le misurazioni sono state tracciate attraverso lo z-stack, seguendo il segnale di riempimento delle celle. Le misurazioni dell'albero e degli artigli sono state tracciate separatamente per ogni dendrite; La linea tratteggiata indica una porzione di dendrite all'interno del piano corrente. Abbreviazioni: 3D = tridimensionale; GFP = proteina fluorescente verde; DPI = giorni dopo l'iniezione; PSD-95 = proteina di densità postsinaptica 95; PNL = progenitori dei neuroni granuli; PFA = paraformaldeide. Fare clic qui per visualizzare una versione ingrandita di questa figura.
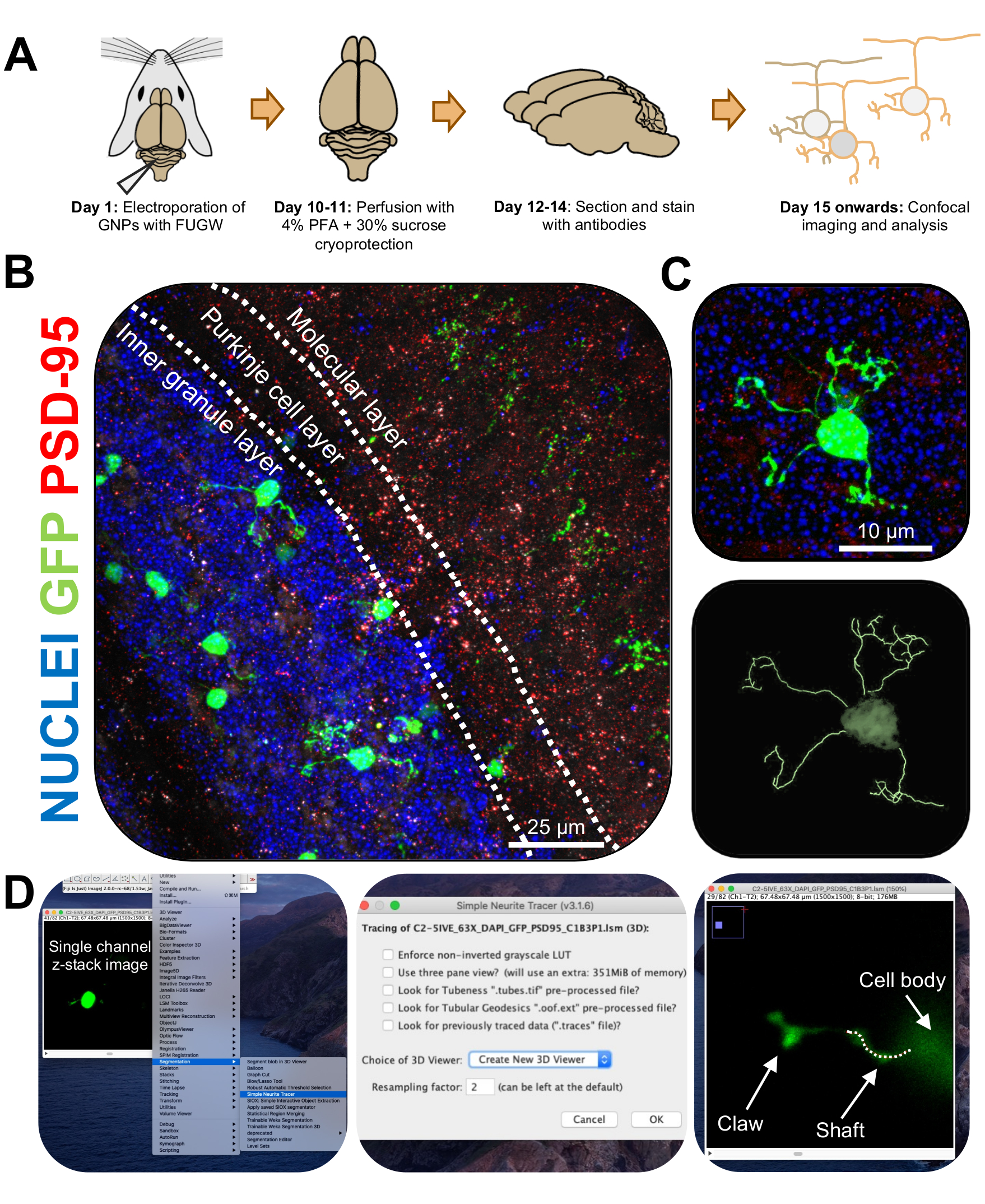{kind=link}
Risultati

Figura 4: Analisi della morfologia dei granuli neuronali durante lo sviluppo cerebellare. (A) Immagini di proiezione massima di CGN elettroporate da 3-DPI a 14-DPI (età postnatale da P10 a P21), nuclei (blu) e GFP (verde); Le punte di freccia indicano il singolo dendrite e la barra della scala è di 10 μm. (B) Numero medio di dendriti. (C...
Discussione
I neuroni dei granuli cerebellari sono i neuroni più abbondanti nel cervello dei mammiferi, costituendo quasi il 60-70% della popolazione totale di neuroni nel cervello dei roditori 1,14. Il cervelletto è stato ampiamente utilizzato per chiarire i meccanismi di proliferazione cellulare, migrazione, formazione di dendriti e sviluppo delle sinapsi. 6,9,10,11,15,16,17,18,19,20 ...
Divulgazioni
Gli autori non dichiarano conflitti di interesse.
Riconoscimenti
Il lavoro è stato supportato dalle sovvenzioni NIH R01NS098804 (A.E.W.), F31NS113394 (U.C.) e dal Summer Neuroscience Program (D.G.) della Duke University.
Materiali
| Name | Company | Catalog Number | Comments |
| Betadine | Purdue Production | 67618-150-17 | |
| Cemented 10 µL needle | Hamilton | 1701SN (80008) | 33 gauge, 1.27 cm (0.5 in), 4 point style |
| Chicken anti-GFP | Millipore Sigma | AB16901 | Our lab uses this antibody at a 1:1000 concentration |
| Cotton-tip applicator | |||
| Donkey anti-chicken Cy2 | Jackson ImmunoResearch | 703-225-155 | Our lab uses this antibody at a 1:500 concentration |
| Ethanol (200 proof) | Koptec | V1016 | |
| Electroporator ECM 830 | BTX Harvard Apparatus | 45-0052 | |
| Fast Green FCF | Sigma | F7252-5G | |
| FUGW plasmid | Addgene | 14883 | |
| Glass slides | VWR | 48311-703 | Superfrost plus |
| Glycerol | Sigma-Aldrich | G5516 | |
| Heating pad | Softheat | ||
| Hoescht 33342 fluorescent dye | Invitrogen | 62249 | |
| Imaris | Bitplane | ||
| Isoflurane | Patterson Veterinary | 07-893-1389 | |
| Micro cover glass | VWR | 48382-138 | |
| Nail polish | Sally Hansen | Color 109 | |
| Normal goat serum | Gibco | 16210064 | |
| O.C.T. embedding compound | Tissue-Tek | 4583 | |
| Olympus MVX10 Dissecting Scope | Olympus | MVX10 | |
| P200 pipette reach tip | Fisherbrand | 02-707-138 | Used for needle spacer |
| Parafilm | Bemis | PM-996 | |
| PBS pH 7.4 (10x) | Gibco | 70011-044 | |
| Simple Neurite Tracer | FIJI | https://imagej.net/Simple_Neurite_Tracer:_Basic_ Instructions | |
| Sucrose | Sigma | S0389 | |
| Surgical tools | RWD Life Science | Small scissors and tweezers | |
| Triton X-100 | Roche | 11332481001 | non-ionic detergent |
| Tweezertrodes | BTX Harvard Apparatus | 45-0489 | 5 mm, platinum plated tweezer-type electrodes |
| Ultrapure distilled water | Invitrogen | 10977-015 | |
| Vectashield mounting media | Vectashield | H1000 | |
| Vetbond tissue adhesive | 3M | 1469SB | |
| Zeiss 780 Upright Confocal | Zeiss | 780 |
Riferimenti
- Altman, J., Bayer, S. A. . Development of the cerebellar system : in relation to its evolution, structure, and functions. , (1997).
- Rahimi-Balaei, M., Bergen, H., Kong, J., Marzban, H. Neuronal migration during development of the cerebellum. Frontiers in Cellular Neuroscience. 12, 484 (2018).
- Alder, J., Cho, N. K., Hatten, M. E. Embryonic precursor cells from the rhombic lip are specified to a cerebellar granule neuron identity. Neuron. 17 (3), 389-399 (1996).
- Hatten, M. E., Heintz, N. Mechanisms of neural patterning and specification in the developing cerebellum. Annual Review of Neuroscience. 18, 385-408 (1995).
- Ben-Arie, N., et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 390 (6656), 169-172 (1997).
- Borghesani, P. R., et al. BDNF stimulates migration of cerebellar granule cells. Development. 129 (6), 1435-1442 (2002).
- Espinosa, J. S., Luo, L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. Journal of Neuroscience. 28 (10), 2301-2312 (2008).
- Markwalter, K. H., Yang, Y., Holy, T. E., Bonni, A. Sensorimotor coding of vermal granule neurons in the developing mammalian cerebellum. Journal of Neuroscience. 39 (34), 6626-6643 (2019).
- Shalizi, A., et al. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. Journal of Neuroscience. 27 (37), 10037-10046 (2007).
- Shalizi, A., et al. A Calcium-regulated MEF2 sumoylation switch controls poststynaptic differentiation. Science. 311 (5763), 1012-1017 (2006).
- Konishi, Y., Stegmuller, J., Matsuda, T., Bonni, S., Bonni, A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 303 (5660), 1026-1030 (2004).
- Holubowska, A., Mukherjee, C., Vadhvani, M., Stegmuller, J. Genetic manipulation of cerebellar granule neurons in vitro and in vivo to study neuronal morphology and migration. Journal of Visualized Experiments: JoVE. (85), e51070 (2014).
- Yang, Y., et al. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. 353 (6296), 300-305 (2016).
- Herculano-Houzel, S. Coordinated scaling of cortical and cerebellar numbers of neurons. Frontiers in Neuroanatomy. 4, 12 (2010).
- Wilson, P. M., Fryer, R. H., Fang, Y., Hatten, M. E. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. Journal of Neuroscience. 30 (25), 8529-8540 (2010).
- Kokubo, M., et al. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. Journal of Neuroscience. 29 (28), 8901-8913 (2009).
- Schwartz, P. M., Borghesani, P. R., Levy, R. L., Pomeroy, S. L., Segal, R. A. Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron. 19 (2), 269-281 (1997).
- Segal, R. A., Pomeroy, S. L., Stiles, C. D. Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. Journal of Neuroscience. 15 (7), 4970-4981 (1995).
- Zhou, P., et al. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 55 (1), 53-68 (2007).
- Dhar, M., Hantman, A. W., Nishiyama, H. Developmental pattern and structural factors of dendritic survival in cerebellar granule cells in vivo. Scientific Reports. 8 (1), 17561 (2018).
- Ito, M. Synaptic plasticity in the cerebellar cortex and its role in motor learning. Canadian Journal of Neurological Sciences. 20, 70-74 (1993).
- Jorntell, H., Hansel, C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron. 52 (2), 227-238 (2006).
- Nakanishi, S. Genetic manipulation study of information processing in the cerebellum. Neuroscience. 162 (3), 723-731 (2009).
- Chang, C. H., et al. Atoh1 controls primary cilia formation to allow for SHH-triggered granule neuron progenitor proliferation. Developmental Cell. 48 (2), 184-199 (2019).
Erratum
Formal Correction: Erratum: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development
Posted by JoVE Editors on 4/06/2023. Citeable Link.
An erratum was issued for: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development. A figure was updated.
Figure 2 was updated from:
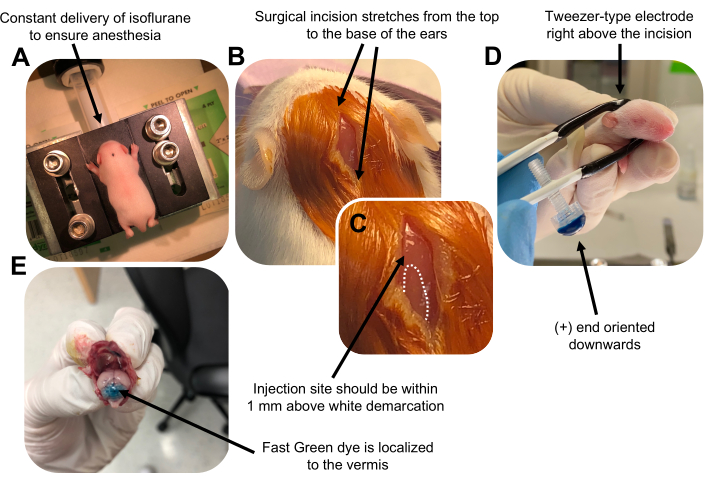
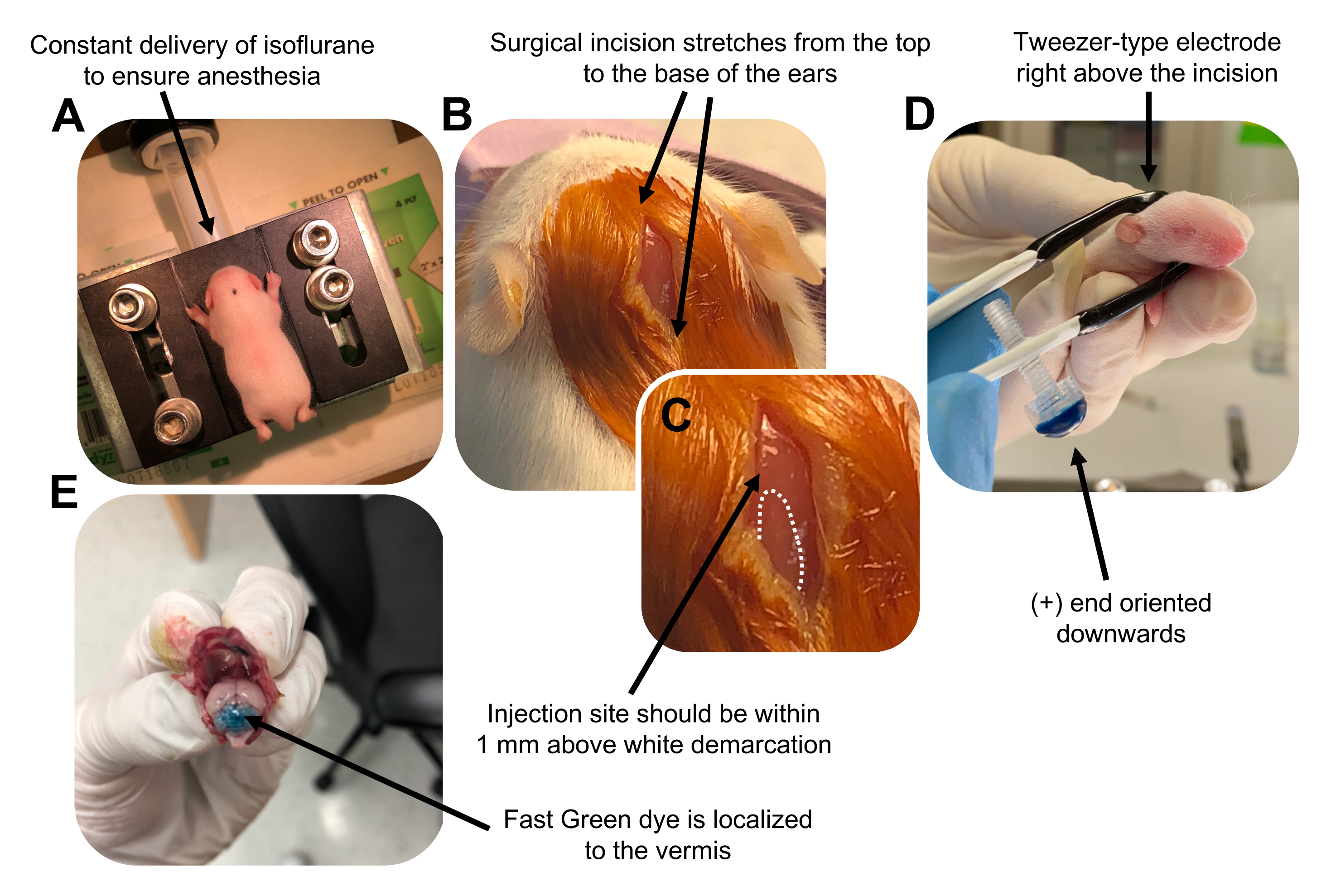
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
{kind=link}
to:
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati