
Method Article
Microdissezione laser industrializzata guidata dall'intelligenza artificiale per l'analisi proteomica microscalata del microambiente tumorale
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Questo protocollo descrive un flusso di lavoro ad alto rendimento per la segmentazione guidata dall'intelligenza artificiale delle regioni di interesse confermate dalla patologia da immagini di sezioni di tessuto sottile macchiate per l'arricchimento di popolazioni cellulari risolte in istologia utilizzando la microdissezione laser. Questa strategia include un nuovo algoritmo che consente il trasferimento di demarcazioni che denotano popolazioni cellulari di interesse direttamente ai microscopi laser.
Abstract
Il microambiente tumorale (TME) rappresenta un ecosistema complesso composto da dozzine di tipi di cellule distinte, tra cui tumori, stroma e popolazioni di cellule immunitarie. Per caratterizzare la variazione del livello del proteoma e l'eterogeneità del tumore su larga scala, sono necessari metodi ad alto rendimento per isolare selettivamente popolazioni cellulari discrete in tumori maligni solidi. Questo protocollo descrive un flusso di lavoro ad alto rendimento, abilitato dall'intelligenza artificiale (AI), che segmenta le immagini di sezioni di tessuto sottile colorate con ematossilina ed eosina (H & E) in regioni di interesse confermate dalla patologia per la raccolta selettiva di popolazioni cellulari risolte in istologia utilizzando la microdissezione laser (LMD). Questa strategia include un nuovo algoritmo che consente il trasferimento di regioni che denotano popolazioni cellulari di interesse, annotate utilizzando software di immagine digitale, direttamente ai microscopi laser, consentendo così raccolte più facili. È stata eseguita con successo l'implementazione di questo flusso di lavoro, dimostrando l'utilità di questo metodo armonizzato per raccogliere selettivamente le popolazioni di cellule tumorali dalla TME per l'analisi proteomica quantitativa e multiplexata mediante spettrometria di massa ad alta risoluzione. Questa strategia si integra pienamente con la revisione istopatologica di routine, sfruttando l'analisi delle immagini digitali per supportare l'arricchimento delle popolazioni cellulari di interesse ed è completamente generalizzabile, consentendo raccolte armonizzate di popolazioni cellulari dalla TME per analisi multiomiche.
Introduzione
La TME rappresenta un ecosistema complesso popolato da una gamma altamente diversificata di tipi di cellule, come cellule tumorali, cellule stromali, cellule immunitarie, cellule endoteliali, altri tipi di cellule mesenchimali e adipociti, insieme a una complessa matrice extracellulare1. Questo ecosistema cellulare varia all'interno e tra i diversi siti degli organi della malattia, con conseguente eterogeneità tumorale complessa 2,3. Studi recenti hanno dimostrato che tumori eterogenei e tumori con bassa cellularità tumorale (bassa purezza) spesso sono correlati con una prognosi sfavorevoledella malattia 2,3.
Per comprendere l'interazione molecolare tra popolazioni di cellule tumorali e non tumorali all'interno della TME su larga scala, sono necessarie strategie standardizzate e ad alto rendimento per raccogliere selettivamente distinte popolazioni cellulari di interesse per l'analisi multiomica a valle. La proteomica quantitativa rappresenta una tecnica in rapida evoluzione e sempre più importante per approfondire la comprensione della biologia del cancro. Ad oggi, la preponderanza di studi che impiegano la proteomica lo ha fatto con proteine estratte da interi preparati di tessuto tumorale (ad esempio, criopolverizzato), portando a una scarsità nella comprensione dell'eterogeneità a livello di proteoma nella TME 4,5,6.
Lo sviluppo di strategie di raccolta dei campioni che si integrano perfettamente e sfruttano le informazioni provenienti dai flussi di lavoro della patologia clinica consentiranno una nuova generazione di proteomica risolta in istologia che sono altamente complementari ai flussi di lavoro di patologia diagnostica gold standard. LMD consente la raccolta diretta e selettiva di sottopopolazioni cellulari o regioni di interesse (ROI) attraverso l'ispezione microscopica di sezioni sottili di tessuto istologicamente colorato7. I recenti importanti progressi nella patologia digitale e nell'analisi abilitata all'intelligenza artificiale hanno dimostrato la capacità di identificare caratteristiche compositive e ROI unici all'interno della TME in modo automatizzato, molti dei quali correlati con alterazioni molecolari e caratteristiche cliniche della malattia, come la resistenza alla terapia e la prognosi della malattia8.
Il flusso di lavoro descritto nel protocollo qui presentato sfrutta le soluzioni software commerciali per annotare selettivamente i ROI tumorali all'interno di immagini istopatologiche digitali e utilizza strumenti software sviluppati internamente per trasferire questi ROI tumorali ai microscopi laser per la raccolta automatizzata di popolazioni cellulari discrete di interesse che si integra perfettamente con i flussi di lavoro di analisi multiomica a valle. Questa strategia integrata riduce significativamente il tempo dell'operatore LMD e riduce al minimo la durata per la quale i tessuti devono essere a temperatura ambiente. L'integrazione della selezione automatizzata delle caratteristiche e della raccolta di LMD con la proteomica quantitativa ad alto rendimento è dimostrata attraverso un'analisi differenziale della TME da due sottotipi istologici rappresentativi di carcinoma ovarico epiteliale, carcinoma ovarico sieroso di alto grado (HGSOC) e carcinoma ovarico a cellule chiare (OCCC).
Protocollo
Tutti i protocolli di studio sono stati approvati per l'uso nell'ambito di un protocollo approvato dall'IRB occidentale "An Integrated Molecular Analysis of Endometrial and Ovarian Cancer to Identify and Validate Clinically Informative Biomarkers" ritenuto esente ai sensi del regolamento federale statunitense 45 CFR 46.102 (f). Tutti i protocolli sperimentali che coinvolgono dati umani in questo studio erano conformi alla Dichiarazione di Helsinki. Il consenso informato è stato ottenuto da tutti i soggetti coinvolti nello studio.
ATTENZIONE: I seguenti reagenti utilizzati in tutto il protocollo sono noti o sospetti cancerogeni e/o contengono materiali pericolosi: etanolo, acqua DEPC, soluzione di ematossilina di Mayer, soluzione di Eosina Y, metanolo, acetonitrile e acido formico. La corretta manipolazione, come descritto nelle rispettive schede di dati di sicurezza (SDS), e l'uso di adeguati dispositivi di protezione individuale (DPI) sono obbligatori.
1. Generazione del file di dati dell'elenco di forme predefinito (.sld) contenente i fiduciali del calibratore
NOTA: i passaggi del protocollo descritti in questa sezione sono specifici da utilizzare con un microscopio laser invertito e il software associato (vedere la Tabella dei materiali). La creazione di un file .sld predefinito è necessaria solo una volta per microscopio laser. Il file risultante può essere utilizzato per tagliare i fiduciali in tutte le diapositive PEN utilizzate successivamente. Tempo approssimativo: 5 minuti (una sola volta).
- Aprire il software LMD e caricare la diapositiva della membrana in polietilene naftalato (PEN) sullo stadio LMD a faccia in giù, con l'etichetta più vicina all'utente. Deselezionate la casella Chiudi riga (s) sul lato destro della finestra del programma.
- Utilizzare la funzione PtoP (point to point) ad alto ingrandimento (63x) per disegnare tre frecce "V" che fungono da fiduciali di calibrazione. Partendo da un punto esterno sulla V, disegna una linea fino al punto medio della V e fai un solo clic. Quindi, disegnate una seconda linea dal punto centrale della V alla fine del secondo punto V esterno e fate doppio clic per creare una singola forma a V non chiusa dalle due linee.
NOTA: questi fiduciali di calibrazione devono essere posizionati in tre angoli della slitta: anteriore destro, posteriore destro, posteriore sinistro. - Selezionate l'opzione AF (messa a fuoco automatica) prima del taglio . Tagliare la diapositiva in posizione 1, spostarla in ciascuna delle restanti posizioni della diapositiva e tracciare con precisione i tagli di calibrazione.
- Salvare il file .sld e selezionare l'opzione Salva senza calibrazione dalla finestra di dialogo a comparsa per evitare di tagliare i fiduciali di calibrazione nella membrana.
NOTA: un file .sld rappresentativo contenente fiduciali di calibratori standard per quattro posizioni di scorrimento è fornito nel file supplementare 1.
2. Preparazione di diapositive LMD
NOTA: i passaggi del protocollo descritti in questa sezione sono specifici da utilizzare con un microscopio laser invertito e il software associato (vedere la Tabella dei materiali). Tempo approssimativo: 5 min.
- Assicurarsi che la slitta sia completamente asciutta prima di tagliare i fiduciali di calibrazione di riferimento. Apri il software LMD e apri il file .sld di calibrazione predefinito sotto l'opzione Importa forme .
- Selezionate l'opzione AF (messa a fuoco automatica) prima del taglio . Caricare le diapositive con il tessuto rivolto verso il basso e la slitta dell'etichetta più vicina all'operatore nel supporto del vetrino sullo stadio LMD.
- Utilizzando il microscopio laser e il file .sld di calibrazione predefinito, tagliare i fiduciali di calibrazione nella membrana PEN.
- OPZIONALE: Tagliare i fiduciali di calibrazione nella membrana PEN prima o dopo che le sezioni di tessuto sono state posizionate sul vetrino. Se i fiduciali di taratura vengono tagliati prima del posizionamento del tessuto, assicurarsi che il tessuto e/o il fissativo non si sovrappongano ai calibratori quando il tessuto viene posizionato sul vetrino nel passaggio 2.5. Se i fiduciali di taratura vengono tagliati dopo il posizionamento dei tessuti, interrompere dopo il completamento del passaggio 2.4 e procedere al paragrafo 3.
- Rivedere tutti i calibratori singolarmente per assicurarsi che ogni taglio sia completo e visibile.
NOTA: utilizzare la funzione Sposta e taglia per dirigere manualmente il laser su tutti i fiduciali di calibrazione che non hanno tagliato completamente la membrana PEN. - Posizionare la sezione di tessuto congelata o fissata in formalina, incorporata in paraffina (FFPE) sul vetrino contenente i fiduciali di calibrazione.
3. Colorazione dei tessuti
NOTA: Tempo approssimativo: 30 min.
- Fissare i vetrini di tessuto LMD congelati in etanolo al 70% (EtOH) contenenti reagenti cocktail inibitori della fosfatasi per 5 minuti.
- Lavare i vetrini in acqua dietil pirocarbomato (DEPC) contenente reagenti cocktail inibitori della fosfatasi per 1 min.
- Lavare i vetrini in acqua DEPC per 1 min.
- Incubare i vetrini nella soluzione di ematossilina di Mayer per 3 minuti.
- Risciacquare i vetrini in acqua DEPC per 3 min.
- Risciacquare i vetrini in un nuovo scambio di acqua DEPC per 1 minuto.
- Incubare i vetrini in soluzione acquosa di Eosin Y per 1 s.
- Risciacquare le diapositive 2 x 5 s in 95% EtOH.
- Risciacquare i vetrini 3 x 10 s in 100% EtOH.
- Pulire l'EtOH in eccesso dal retro delle diapositive e lasciare asciugare le diapositive all'aria.
- Conservare i vetrini a -80 °C se LMD non deve essere eseguito immediatamente.
4. Imaging delle diapositive
NOTA: i passaggi del protocollo descritti in questa sezione sono specifici per le diapositive analizzate (vedere la Tabella dei materiali) e le immagini risultanti salvate come file con estensione svs. Utilizzare qualsiasi scanner e il software associato che genera file di immagine in un formato che il software di analisi delle immagini (vedere la Tabella dei materiali) può aprire. I tipi di file che utilizzano tiff piramidali supportati includono JPG, TIF, MRXS, QPTIFF, componente TIFF, SVS, AFI, SCN, LIF, DCM, OME. TIFF, ND2, VSI, NDPI, NDPIS, CZI, BIF, KFB e ISYNTAX. Tempo approssimativo: 5 min.
- Accendere lo scanner e aprire il software dello scanner di diapositive. Caricare il vetrino con il tessuto rivolto verso l'alto sul singolo stadio di scorrimento nello scanner. Assicurarsi che la diapositiva sia completamente asciutta e posizionare delicatamente una copertura sul tessuto. Non usare etanolo o olio ad immersione sotto il coperchio.
- Acquisire l'immagine micrografica utilizzando le impostazioni calibrate per regolare la membrana PEN anziché lo sfondo di vetro e ignorare la colorazione della membrana di sfondo, secondo le istruzioni del produttore.
- Regolare l'area di imaging trascinando e ridimensionando il perimetro verde interno per acquisire l'intera area della membrana PEN, in base alle esigenze. Aggiungere quattro punti di messa a fuoco sul tessuto facendo doppio clic sull'immagine panoramica dell'istantanea e tre punti di messa a fuoco sulla membrana vicino ai fiduciali di calibrazione (un punto di messa a fuoco per ciascuno dei tre fiduciali di calibrazione).
NOTA: i quattro punti di messa a fuoco possono essere posizionati quasi ovunque sulla sezione del tessuto, anche se il posizionamento su tessuto troppo scuro e che appare nero può causare il fallimento della scansione. - Nel menu Visualizza , selezionare Monitor video. Regolare manualmente la messa a fuoco utilizzando il cursore di messa a fuoco fine e/o macro, se necessario, per ogni punto intorno al tessuto LMD. Cattura la scansione dell'immagine con ingrandimento elevato (20x). Verificare che tutti i fiduciali di calibrazione siano visibili e chiari nell'immagine salvata.
5. Selezione automatica delle funzionalità tramite software di analisi delle immagini
- Per intere raccolte tumorali (tempo approssimativo: 5 min; caso-dipendente):
- Aprire il software di analisi delle immagini (vedere la Tabella dei materiali). Selezionare Apri immagini e dalla finestra pop-up selezionare il file di immagine .svs generato dalla scansione della diapositiva sullo scanner AT2.
NOTA: un file di immagine .svs rappresentativo viene fornito nel file supplementare 2. - Passare alla scheda Annotazioni . Selezionate lo strumento penna sulla barra degli strumenti Annotazione e disegnate una forma attorno al tessuto.
- Seleziona la forma e fai clic con il pulsante destro del mouse sull'immagine. Dal menu a discesa Avanzate, selezionare Partizionamento (affiancato). Impostare dimensioni e spazio tra riquadri rispettivamente su 500 e 40 e selezionare OK per generare i riquadri. Selezionare ed eliminare la forma perimetrale utilizzata per generare i riquadri nel passaggio 5.1.2.
- Selezionare il menu a discesa Azioni livello | Esportare per salvare le annotazioni affiancate come file con estensione annotation.
NOTA: un file di annotazione rappresentativo per un'intera raccolta di tessuto tumorale è fornito nel file supplementare 3. - Creare una cartella per la sessione o il progetto e salvare il file con estensione annotation all'interno di una sottocartella contrassegnata con l'identificatore univoco della diapositiva.
- Passare alla scheda Annotazioni . Selezionare il menu a discesa Azioni livello | Elimina tutti i livelli per rimuovere tutte le annotazioni dall'immagine. Selezionate lo strumento penna e disegnate una breve linea dalla punta interna della punta della freccia per ogni fiduciale di calibrazione. Disegna le linee dai segni nel seguente ordine: in alto a sinistra, in alto a destra, in basso a destra.
- Selezionare il menu a discesa Azioni livello | Esportare per salvare le annotazioni di riga come file con estensione annotation. Aggiungere _calib al nome del file e posizionare il file nella sottocartella contenente le coordinate per le forme affiancate.
NOTA: un file _calib.annotation rappresentativo è fornito nel file supplementare 4. - Copiare l'indirizzo della cartella principale del progetto o della sessione. Aprire lo script di generazione dell'importazione XML, "Malleator" (disponibile tramite https://github.com/GYNCOE/Mitchell.et.al.2022), utilizzando l'ambiente di sviluppo integrato IDLE e incollare l'indirizzo della cartella del progetto tra le virgolette nella parte inferiore dello script.
- Seleziona il menu a discesa Esegui | Eseguire Module per eseguire lo script.
NOTA: il file di importazione LMD .xml verrà generato all'interno della sottocartella creata per l'immagine/diapositiva. Un file di .xml rappresentativo è fornito nel file supplementare 5.
- Aprire il software di analisi delle immagini (vedere la Tabella dei materiali). Selezionare Apri immagini e dalla finestra pop-up selezionare il file di immagine .svs generato dalla scansione della diapositiva sullo scanner AT2.
- Solo per le collezioni arricchite con LMD (tempo approssimativo: 15 min; case-dependent):
- Aprire il software di analisi delle immagini (vedere la Tabella dei materiali). Selezionare Apri immagini e dalla finestra pop-up selezionare il file di immagine .svs generato dalla scansione della diapositiva.
- Passare alla scheda Annotazioni . Selezionate e utilizzate lo strumento di annotazione Rettangolo per disegnare una casella attorno al tessuto.
- Selezionare l'annotazione della casella e fare clic con il pulsante destro del mouse sull'immagine. Seleziona il menu a discesa Avanzate | Opzione di partizionamento (affiancato). Impostare dimensioni e spazio tra riquadri rispettivamente su 500 e 40 e selezionare OK per generare i riquadri. Selezionare ed eliminare l'annotazione della casella perimetrale utilizzata per generare i riquadri nel passaggio 5.2.2.
- Selezionare il menu a discesa Azioni livello | Esportare per salvare le annotazioni affiancate come file con estensione annotation.
- Inserisci una copia salvata dell'algoritmo Python "Dapọ" ( disponibile tramite https://github.com/GYNCOE/Mitchell.et.al.2022), sviluppato per unire i livelli di annotazione classificati AI, nella stessa cartella del file di annotazioni affiancate.
- Copiare il nome del file di annotazione affiancato. Aprire il programma Python utilizzando l'ambiente di sviluppo integrato IDLE e incollare il nome del file di annotazione affiancato tra le virgolette nella parte inferiore del programma.
- Seleziona il menu a discesa Esegui | Esegui modulo. Attendi che venga generato un nuovo file con tutte le annotazioni affiancate unite sotto un singolo livello.
- Aprire il software di analisi delle immagini e passare alla scheda Annotazioni . Selezionare il menu a discesa Azioni livello | Elimina tutti i livelli per rimuovere tutte le annotazioni dall'immagine.
- Selezionare il menu a discesa Azioni livello | Importare il file di annotazione locale. Nella finestra popup, selezionare il file di annotazione unito generato dallo script. Assicuratevi che tutti i riquadri importati si trovino sotto lo stesso livello di annotazione.
- Passare alla scheda Classificatore e seguire le istruzioni del produttore per generare forme per i ROI. Prima di eseguire il classificatore, selezionare i livelli di annotazione desiderati (ad esempio, il livello tumorale o stroma) selezionando la casella o le caselle ROI nella scheda Annotazioni , sotto Opzioni classificatore avanzato. Utilizzate l'opzione Livello annotazione dal menu Azioni classificatore per eseguire il classificatore.
- Una volta completata l'analisi del classificatore, passare alla scheda Annotazioni e selezionare il livello di annotazione generato dall'analisi. Selezionare il menu a discesa Azioni livello | Elimina tutti i livelli ma correnti per rimuovere tutti gli altri livelli di annotazione dall'immagine.
- Selezionare il menu a discesa Azioni livello | Esporta per salvare le annotazioni come file con estensione annotation. Creare una cartella per la sessione o il progetto e salvare il file con estensione annotation all'interno di una sottocartella contrassegnata con l'identificatore univoco della diapositiva.
NOTA: un file di annotazione rappresentativo per una raccolta di tessuti arricchiti con LMD classificati è fornito nel file supplementare 6. - Passare alla scheda Annotazioni , selezionare l'elenco a discesa Azioni livello | Elimina tutti i livelli per rimuovere tutte le annotazioni dall'immagine. Selezionate lo strumento penna e tracciate una breve linea da ogni fiduciale di calibrazione. Disegna linee dai segni nel seguente ordine: in alto a sinistra, in alto a destra, in basso a destra.
- Selezionare il menu a discesa Azioni livello | Esportare per salvare le annotazioni di riga come file con estensione annotation. Aggiungere _calib al nome del file e posizionare il file nella sottocartella contenente le coordinate per le forme affiancate.
- Copiare l'indirizzo della cartella principale del progetto o della sessione. Aprire lo script di generazione dell'importazione XML, "Malleator" ( disponibile tramite https://github.com/GYNCOE/Mitchell.et.al.2022), utilizzando l'ambiente di sviluppo integrato IDLE, quindi incollare l'indirizzo della cartella del progetto tra le virgolette nella parte inferiore dello script.
- Seleziona il menu a discesa Esegui | Eseguire Module per eseguire lo script.
NOTA: il file di importazione LMD .xml verrà generato all'interno della sottocartella creata per l'immagine/diapositiva.
6. Microdissezione laser
NOTA: i passaggi del protocollo descritti in questa sezione sono specifici da utilizzare con un microscopio laser invertito e il software associato (vedere la Tabella dei materiali). Tempo approssimativo: 2 h; caso dipendente.
- Caricare il vetrino a membrana contrassegnato (contenente fiduciali di calibrazione) con il tessuto rivolto verso il basso e il lato dell'etichetta più vicino all'operatore nel supporto del vetrino sullo stadio del microscopio laser.
- Selezionare Importa forme dal menu a discesa File . Selezionate il file di importazione LMD .xml generato per la diapositiva. Selezionare No nella finestra pop-up per evitare di caricare punti di riferimento dal file e No nella seconda finestra popup per evitare di utilizzare punti di riferimento memorizzati in precedenza per la calibrazione.
- Seguire le istruzioni dell'applicazione LMD e allineare la croce di calibrazione a ciascuno dei tre fiduciali di calibrazione sulla diapositiva. Cerca i fiduciali di calibrazione che appaiono in alto a sinistra, in alto a destra e in basso a destra dell'immagine della diapositiva nel software di analisi delle immagini che corrisponderanno ai punti di riferimento negli angoli anteriore destro, posteriore destro e posteriore sinistro, rispettivamente, del vetrino LMD invertito sullo stadio del microscopio. Passare dall'utilizzo dell'obiettivo 5x per localizzare all'obiettivo 63x per allineare ogni fiduciale di calibrazione. Selezionare No nella finestra popup per evitare di salvare i punti di riferimento nel file e OK nella seconda finestra popup per confermare che la diapositiva sia stata inserita.
- Spostare l'obiettivo 5x in posizione e selezionare Sì nella finestra pop-up per utilizzare l'ingrandimento effettivo. Una volta visualizzate le forme importate, focalizzare la fotocamera sul tessuto.
- Evidenziate e selezionate tutte le forme nella finestra Elenco forme , trascinatele in posizione utilizzando una o due annotazioni all'interno del campo visivo come riferimenti e allineate l'asse Z verticale per il taglio con il laser.
- Esaminare le forme importate e assegnarle alla posizione del tubo appropriata per la raccolta. Premere Start Cut per avviare il laser.
NOTA: le forme importate nel file .xml verranno automaticamente assegnate alla posizione "no cap" nella finestra Elenco forme . Per raccogliere il tessuto, le forme importate devono essere riassegnate a una posizione contenente un tubo caricato.
7. Digestione delle proteine mediante tecnologia a ciclo di pressione (PCT)
NOTA: Tempo approssimativo: 4 h (3 h senza tempo di asciugatura della centrifuga sottovuoto).
- Posizionare tubi da 0,5 mL contenenti i MicroTubi PCT chiusi contenenti tessuto raccolto LMD in 20 μL di 100 mM TEAB/acetonitrile al 10% in un termociclatore e riscaldare a 99 °C per 30 minuti, quindi raffreddare a 50 °C per 10 minuti.
- Ruotare i tubi per 30 s a 4.000 × g e quindi rimuovere i MicroTube dai tubi da 0,5 ml. Utilizzando lo strumento MicroCap, rimuovere ed eliminare i MicroCap dai MicroTube PCT. Aggiungere tripsina (vedere la tabella dei materiali) con un rapporto di 1 μg per tessuto da 30 mm2 e inserire una micropestle nel microtubo utilizzando lo strumento MicroCap.
- Trasferire i MicroTube in una cartuccia barocycler e assemblare la cartuccia completa. Posizionare la cartuccia nella camera di pressione del barociclo e fissare il coperchio. Barociclo a 45.000 psi per 50 s e pressione atmosferica per 10 s a 50 °C per 60 cicli.
- Una volta completato il barocycling, trasferire i microtubi in un tubo microcentrifuga da 0,5 mL e centrifuga per 2 minuti a 4.000 × g.
- Rimuovere il MicroTube dal tubo microcentrifuga da 0,5 mL utilizzando lo strumento tappo. Rimuovere con cura il pestello utilizzando lo strumento cappuccio e risciacquare la metà inferiore del pestello con 20 μL di acqua liquida di cromatografia-spettrometria di massa (LC-MS) e raccogliere il lavaggio in un tubo microcentrifuga pulito da 0,5 mL.
- Toccare delicatamente il MicroTube sul piano di lavoro per spostare il liquido verso il basso e trasferire tutta la soluzione dal MicroTube nel tubo microcentrifuga da 0,5 mL.
- Aggiungere 20 μL di acqua LC-MS al MicroTube e picchiettarlo delicatamente sul piano di lavoro. Trasferire la soluzione di lavaggio nel tubo da 0,5 ml e ripetere nuovamente questa fase di lavaggio.
- Centrifuga sottovuoto per asciugare i campioni a ~2 μL e aggiungere 100 μL di 100 mM TEAB, pH 8,0.
- Determinare la concentrazione peptidica utilizzando un test colorimetrico (saggio dell'acido bicinchoninico (BCA); vedere la Tabella dei materiali) secondo il protocollo del produttore.
8. Etichettatura TMT (Tandem-mass tag) e pulizia EasyPep
NOTA: Tempo approssimativo: 7 h 20 min (2 h 20 min senza tempo di asciugatura della centrifuga sottovuoto).
- Portare i reagenti isobarici di etichettatura TMT a temperatura ambiente prima dell'apertura. Aggiungere 500 μL di acetonitrile al 100% a ciascun flaconcino di TMT (5 mg). Incubare per 10 minuti con vortici occasionali.
- Sciogliere 5 μg del campione peptidico in 100 μL di 100 mM TEAB, pH 8,0, e aggiungere 10 μL di un dato reagente TMT. Costruisci e includi pool di riferimento che rappresentano ogni singolo campione nell'esperimento in ogni set di campioni multiplex TMT per facilitare la quantificazione dei campioni su più multiplex TMT9. Reazioni di incubazione per 1 ora a temperatura ambiente con occasionali agitazioni/picchiettamenti.
- Spegnere la reazione di marcatura TMT aggiungendo 10 μL di idrossilammina al 5% e incubare per 30 minuti a temperatura ambiente con occasionali picchiettamenti. Dopo la tempra, combinare i campioni etichettati TMT in un unico tubo e asciugare a circa 200 μL.
- Aggiungere 1.800 μL di acido formico allo 0,1%. Controllare il pH con carta pH: se pH ~ 3, aggiungere 1 mL di acido formico allo 0,1%; se pH >3, aggiungere 10-20 μL di acido formico al 5% fino a pH ~3. Aggiungere lo 0,1% di acido formico per portare ad un volume finale di 3 ml.
- Rimuovere la linguetta nella parte inferiore della colonna di pulizia del peptide, rimuovere il cappuccio e posizionarlo in un tubo conico da 15 ml. Trasferire il campione con etichetta TMT nella colonna e procedere con la pulizia secondo il protocollo del produttore.
- Centrifuga sottovuoto per asciugare i peptidi eluiti a ~ 20 μL, trasferire in un flaconcino LC usando bicarbonato di ammonio da 25 mM per un volume finale di 80 μL e procedere al frazionamento offline.
9. Frazionamento e pooling del campione multiplex TMT
NOTA: Tempo approssimativo: 3 h 30 min (1 h 30 min senza tempo di asciugatura della centrifuga sottovuoto).
- Frazionare i multiplex peptidici marcati TMT mediante cromatografia di base a fase inversa in 96 frazioni sviluppando un gradiente lineare crescente (0,69% min-1) di fase mobile B (acetonitrile) in fase mobile A (10 mM NH4HCO3, pH 8,0).
- Generare 36 frazioni concatenate raggruppando pozzi campione. Centrifugare sottovuoto per essiccare frazioni a ~2 μL e risospese in bicarbonato di ammonio da 25 mM (concentrazione finale 1,5 μg/10 μL), centrifugare a 15.000 × g per 10-15 min e trasferire a flaconcini LC per l'analisi MS.
10. Cromatografia liquida spettrometria di massa tandem (LC-MS/MS)
NOTA: Tempo approssimativo: metodo dello strumento e definizione sperimentale dipendenti.
- Calibrare lo spettrometro di massa secondo le istruzioni/protocolli del produttore.
- Preparare nuove fasi mobili e standard ed eseguire preparazioni LC pre-run appropriate (inclusi, a titolo esemplificativo ma non esaustivo, solventi di spurgo, aria di lavaggio e script di prova di tenuta per lo strumento di riferimento [vedere la tabella dei materiali]). Equilibrare le colonne pre- e analitiche e il ciclo di campionamento prima di iniziare le analisi.
- Prima e tra le analisi multiplex TMT seriali, convalidare che il sistema LC-MS soddisfi le metriche delle prestazioni precedentemente confrontate utilizzando digest peptidici marcati TMT per garanzia di qualità / controllo qualità (QA / QC) e (ad esempio, MSPE (vedere la tabella dei materiali), HeLa).
- Caricare i flaconcini dell'autocampionatore nelle posizioni appropriate nell'autocampionatore LC. Analizzare le singole frazioni con un metodo gradiente/MS appropriato. Intervallare una corsa di "lavaggio" con lo standard peptidico (ad esempio, calibrazione del tempo di ritenzione peptidica [PRTC]) circa una volta al giorno per valutare le prestazioni cromatografiche e spettrali di massa. Dopo l'analisi di ciascuna serie di frazioni campione multiplex TMT, eseguire gli standard di benchmark QA/QC TMT per valutare le prestazioni del sistema.
- Eseguire routine di valutazione dello spettrometro di massa secondo gli standard di benchmark QA/QC TMT per valutare le prestazioni post-campione e quindi calibrare il sistema come nel passaggio 10.1 per il set di campioni successivo.
11. Analisi dei dati bioinformatici
NOTA: Tempo approssimativo: dipendente dal progetto sperimentale.
- Trasferire tutti i dati di esempio (ad esempio, file .raw) su un'unità di archiviazione di rete/computer appropriata.
- Cerca tutte le frazioni insieme utilizzando l'applicazione di analisi dei dati desiderata (ad esempio, Proteome Discover, Mascot) utilizzando i parametri appropriati9 rispetto a un database di riferimento proteico specifico per specie per generare corrispondenze spettrali peptidiche (PSM) ed estrarre le intensità del segnale ionico reporter TMT. Filtra i PSM in base a metriche di controllo di qualità appropriate e aggrega le abbondanze del rapporto ionico reporter TMT normalizzato e mediano mediano trasformato in abbondanze di livello proteico globale, come descritto in precedenza 3,9.
- Confronta le alterazioni proteiche nelle condizioni di interesse utilizzando il software di analisi differenziale desiderato.
Risultati
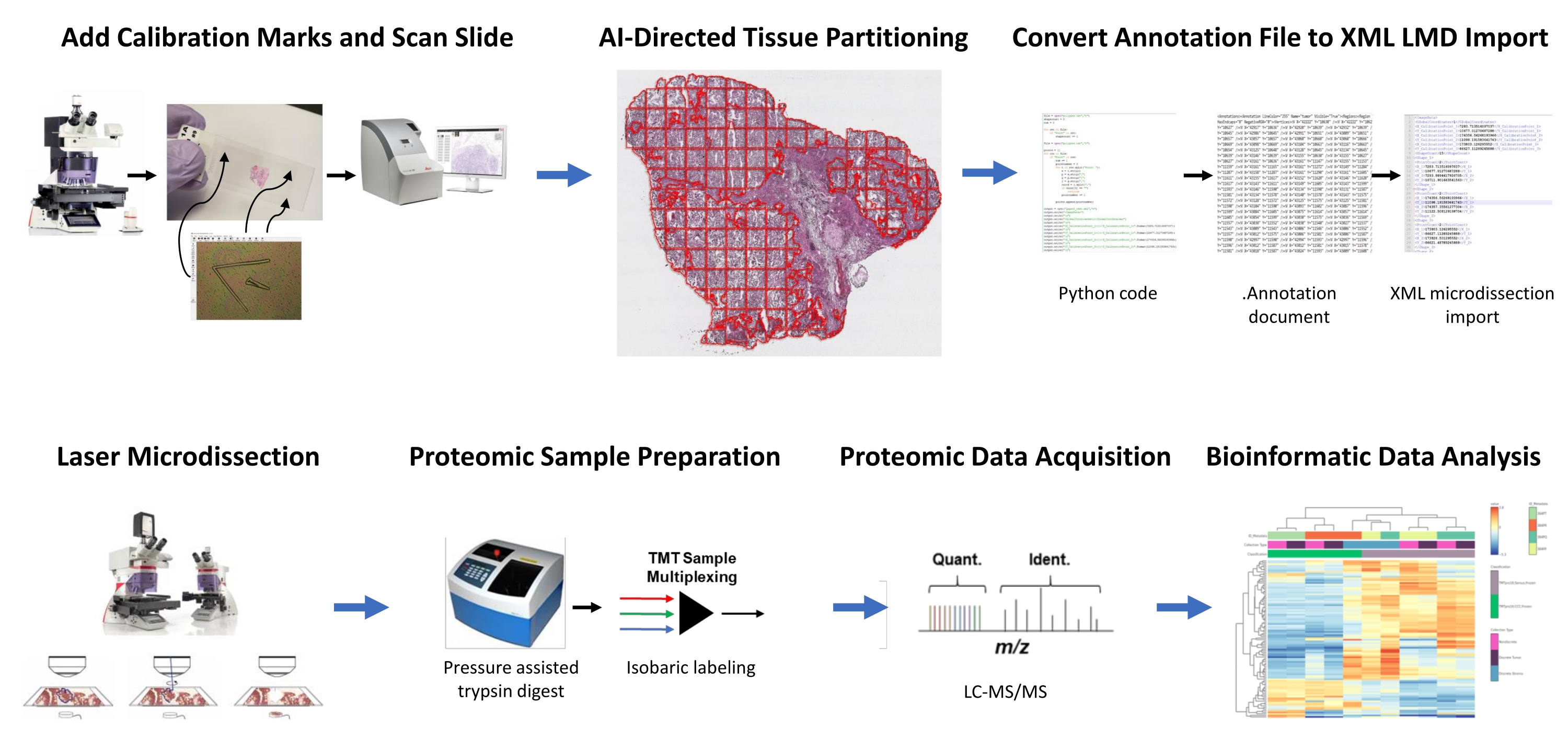
Le sezioni sottili di tessuto appena congelato di due pazienti HGSOC e due OCCC sono state analizzate utilizzando questo flusso di lavoro integrato di identificazione, segmentazione, LMD e analisi proteomica quantitativa dei tessuti basata sull'intelligenza artificiale (Figura 1). Le sezioni di tessuto rappresentative colorate di H & E per ciascun tumore sono state esaminate da un patologo certificato; la cellularità tumorale variava dal 70% al 99%. I tessuti sono stati a sezione sottile su vetrini a membrana PEN (File supplementare 2) e pretagliati con fiduciali calibratori (File supplementare 1), consentendo l'integrazione dei dati di orientamento posizionale dalle annotazioni generate nel software di analisi delle immagini (vedere la Tabella dei materiali) con l'orientamento delle coordinate cartesiane nel software LMD. Dopo la colorazione H&E, sono state catturate immagini ad alta risoluzione (20x) dei vetrini PEN contenenti il tessuto più calibratori.
Le popolazioni tumorali e di cellule stromali nelle micrografie sono state segmentate utilizzando un software di analisi delle immagini (vedere la Tabella dei materiali) per la raccolta selettiva da parte di LMD, insieme a raccolti che rappresentano l'intera sezione sottile del tessuto (ad esempio, tessuto tumorale intero) (Figura 1). Le annotazioni non discriminatorie per intere collezioni di tessuto tumorale sono state generate dividendo l'intera sezione tissutale con piastrelle di 500 μm2, lasciando uno spazio di 40 μm tra le piastrelle per mantenere l'integrità della membrana PEN e impedire alla membrana di arricciarsi durante LMD. Sulle diapositive per l'arricchimento LMD risolto in istologia, il classificatore AI nel software di analisi delle immagini (vedi la Tabella dei materiali) è stato addestrato a discriminare tra cellule tumorali e stromali, insieme allo sfondo del vetrino bianco. Le regioni rappresentative di tumore, stroma e vetro bianco sono state evidenziate manualmente e lo strumento classificatore è stato utilizzato per segmentare questi ROI in tutta la sezione tissutale. Gli strati segmentati che rappresentano l'intero tessuto, l'epitelio tumorale e lo stroma sono stati salvati separatamente come singoli file .annotation (File supplementare 3 e File supplementare 6). In una copia separata del file immagine (senza le annotazioni ROI partizionate), una breve riga dalla punta più centrale di ciascuno dei tre calibratori fiduciali è stata annotata e salvata come file .annotation utilizzando lo stesso nome di file di ciascuno dei file del livello di annotazione LMD ma aggiunta con il suffisso "_calib" (File supplementare 4). Queste linee sono state utilizzate per co-registrare la posizione dei calibratori a membrana PEN con i dati dell'elenco delle forme di annotazione disegnati nel software di analisi delle immagini.
Il presente studio fornisce due algoritmi, "Malleator" e "Dapọ" in Python per supportare questo flusso di lavoro LMD basato sull'intelligenza artificiale, che sono disponibili presso https://github.com/GYNCOE/Mitchell.et.al.2022. L'algoritmo Malleator estrae le coordinate cartesiane specifiche per tutte le singole annotazioni (ROI dei tessuti e calibratori) dai file .annotation accoppiati e li unisce in un unico file di importazione XML (Extensible Markup Language) (File supplementare 5). In particolare, l'algoritmo Malleator utilizza il nome della directory da una cartella padre come input per cercare tutte le cartelle della sottodirectory e genera file .xml per tutte le sottocartelle che non dispongono già di un file .xml unito. L'algoritmo Malleator unisce tutti i livelli di annotazione nel software di analisi delle immagini (vedere la Tabella dei materiali) in un unico livello e converte i dati dell'elenco di forme generati dall'IA, che vengono salvati come tipo di file .annotation proprietario, in .xml formato compatibile con il software LMD. Dopo aver unito i file di annotazione e calibratore, il file .xml generato dall'algoritmo viene salvato e importato nel software LMD. Sono necessarie lievi regolazioni per regolare manualmente l'allineamento delle annotazioni, che serve anche a registrare la posizione verticale (piano z) dello stadio del vetrino sul microscopio laser. L'algoritmo Dapọ viene utilizzato specificamente per le collezioni arricchite di LMD. I riquadri partizionati vengono assegnati automaticamente ai singoli livelli di annotazione dal software di analisi delle immagini. L'algoritmo Dapọ unisce tutti i riquadri partizionati in un unico livello di annotazione prima dell'utilizzo dello strumento Classificatore, riducendo così il tempo di esecuzione dell'analisi del classificatore per le raccolte arricchite di LMD.
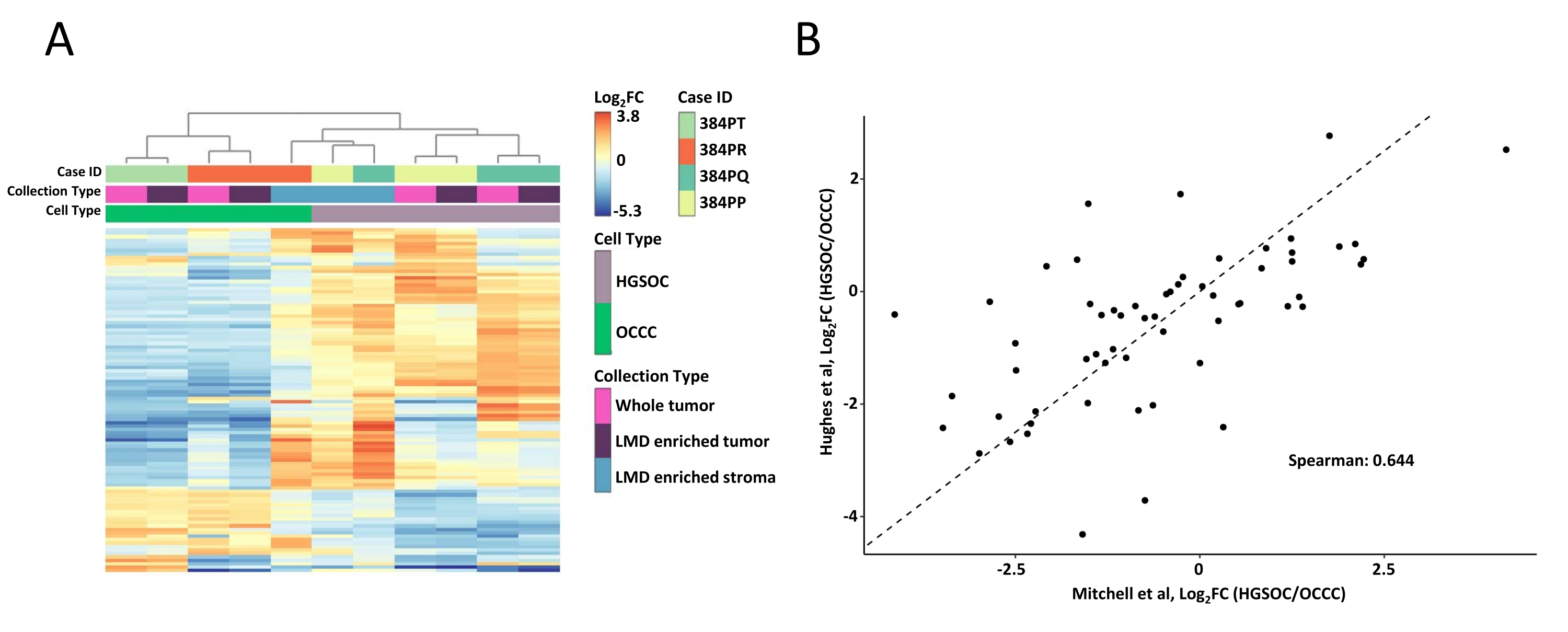
L'intero tumore e i campioni di tessuto arricchito con LMD sono stati digeriti, etichettati con reagenti TMT, multiplexati, frazionati offline e analizzati tramite proteomica quantitativa basata sulla SM come descritto in precedenza9. La resa peptidica media (43-60 μg) e il recupero (0,46-0,59 μg/mm2) per i campioni raccolti utilizzando questo flusso di lavoro basato sull'intelligenza artificiale erano comparabili con i rapporti precedenti 9,10. Un totale di 5.971 proteine sono state co-quantificate in tutti i campioni (Tabella supplementare S1). Il clustering gerarchico non supervisionato utilizzando le 100 proteine più variabili ha portato alla segregazione degli istotipi HGSOC e OCCC dai campioni tumorali completi e arricchiti con LMD (Figura 2A), simile a quello precedentemente descritto11. Al contrario, i campioni di stroma arricchiti con LMD provenienti da HGSOC e OCCC si sono raggruppati insieme e indipendentemente dai campioni tumorali arricchiti con LMD e da campioni di tumore intero. Tra le 5.971 proteine quantificate, 215 sono state significativamente alterate (LIMMA adj. p < 0,05) tra intere raccolte tumorali da campioni HGSOC e OCCC (Tabella supplementare S2). Queste proteine alterate sono state confrontate con quelle identificate per differenziare il tessuto tumorale HGSOC e OCCC da Hughes et al.11. Delle 76 proteine di firma quantificate da Hughes et al., 57 erano co-quantificate in questo set di dati ed erano altamente correlate (Spearman Rho = 0,644, p < 0,001) (Figura 2B).

Figura 1: Riepilogo del flusso di lavoro integrato per la selezione automatizzata delle regioni tissutali di interesse per la microdissezione laser per la proteomica quantitativa a valle. I fiduciali di calibrazione vengono tagliati su vetrini a membrana PEN per co-registrare i dati di orientamento posizionale da segmenti derivati dall'IA del ROI tissutale nel software di analisi delle immagini, HALO, con posizionamento orizzontale sul microscopio LMD. L'algoritmo Malleator viene utilizzato per unire i dati di segmentazione annotati su tutti i livelli di annotazione per una diapositiva con il file di riferimento _calib e per convertirlo in un file .xml compatibile con il software LMD. Il tessuto raccolto da LMD per l'analisi proteomica viene digerito e analizzato mediante proteomica quantitativa ad alto rendimento come descritto in precedenza9. Abbreviazioni: LMD = microdissezione laser; ROI = regione di interesse; TMT = tag di massa tandem; Quant. = quantificazione; Ident. = identificazione; LC-MS/MS = cromatografia liquida-spettrometria di massa tandem. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Analisi delle proteine in campioni tumorali interi e arricchiti con LMD. (A) Analisi di cluster gerarchici non supervisionati delle 100 proteine più variabilmente abbondanti in campioni tumorali arricchiti con HGSOC e OCCC LMD e interi. (B) Correlazione delle abbondanze proteiche log2 fold-change tra HGSOC e OCCC raccolti di tumori interi nel presente studio (Mitchell et al., asse x) e uno studio simile di Hughes et al. (asse y)11. Abbreviazioni: LMD = microdissezione laser; HGSOC = carcinoma ovarico sieroso di alto grado; OCCC = carcinoma ovarico a cellule chiare; log2FC = log2-trasformata abbondanza proteomica. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Tabella supplementare S1: Abbondanza di 5.971 proteine co-quantificate tra tutti i campioni di tumore arricchiti con LMD e interi da campioni di tessuto HGSOC e OCCC. Abbreviazioni: LMD = microdissezione laser; HGSOC = carcinoma ovarico sieroso di alto grado; OCCC = carcinoma ovarico a cellule chiare. Fare clic qui per scaricare questa tabella.
Tabella supplementare S2: Proteine differenzialmente espresse (215) in intere raccolte tumorali da HGSOC vs OCCC (LIMMA adj. p < 0,05). Abbreviazioni: HGSOC = carcinoma ovarico sieroso di alto grado; OCCC = carcinoma ovarico a cellule chiare. Fare clic qui per scaricare questa tabella.
File supplementare 1: file di dati dell'elenco di forme rappresentative (.sld) contenente fiduciali di calibratori standard per quattro posizioni di scorrimento. Il file può essere importato nel software LMD. Fare clic qui per scaricare questo file.
File supplementare 2: file immagine .svs rappresentativo di una sezione di tessuto ad alta risoluzione (20x) macchiata di H&E. Il file può essere aperto e visualizzato utilizzando un software di analisi delle immagini o un software LMD. Abbreviazione: H&E = ematossilina ed eosina; LMD = microdissezione laser. Fare clic qui per scaricare questo file.
File supplementare 3: file di annotazione rappresentativo di interi segmenti tumorali partizionati. Il file può essere importato in un software di analisi delle immagini. Fare clic qui per scaricare questo file.
File supplementare 4: File _calib.annotation rappresentativo dei segmenti fiduciari del calibratore. Le informazioni sulle coordinate rappresentano il posizionamento orientale delle brevi linee di calibrazione disegnate da ciascuna punta di freccia fiduciale. Il file può essere importato in un software di analisi delle immagini. Fare clic qui per scaricare questo file.
File supplementare 5: file rappresentativo del linguaggio di markup estensibile (.xml) generato dall'algoritmo Malleator. Il file può essere importato nel software di microdissezione laser. Fare clic qui per scaricare questo file.
File supplementare 6: file di annotazione rappresentativo di segmenti classificati AI partizionati per raccolte arricchite con LMD. Il file può essere importato in un software di analisi delle immagini. Abbreviazioni: AI = intelligenza artificiale; LMD = microdissezione laser. Fare clic qui per scaricare questo file.
Discussione
Sebbene vi siano stati diversi precedenti di studio volti a sviluppare e/o migliorare i flussi di lavoro per l'arricchimento di sottopopolazioni cellulari bersaglio da FFPE e/o tessuti freschi congelati e metodologie per mantenere la qualità del campione durante l'elaborazione 9,12,13,14,15, esiste una sostanziale necessità di sviluppare strategie automatizzate per la preparazione di campioni di tessuto clinico per analisi molecolari per ridurre la variabilità e aumentare la riproducibilità. Questo flusso di lavoro descrive un protocollo standardizzato e semiautomatico che integra gli strumenti software di analisi delle immagini esistenti (vedere la Tabella dei materiali) per la raccolta risolta in istologia di popolazioni cellulari discrete da LMD da campioni di tessuto clinico.
L'arricchimento LMD spazialmente risolto di ROI che catturano popolazioni cellulari discrete rappresenta una fase di elaborazione tissutale di prossima generazione prima delle analisi multiomiche per migliorare la caratterizzazione e l'identificazione molecolare e facilitare la scoperta di biomarcatori selettivi delle cellule. Questo protocollo migliora le metodologie esistenti riducendo l'esposizione spesso lunga delle sezioni di tessuto all'ambiente ambientale associata alla segmentazione manuale del ROI da parte di un istologo (che può richiedere >1-2 ore prima della raccolta LMD). Questo flusso di lavoro consente invece di preidentificare il ROI dalla classificazione e dalla segmentazione guidate dall'intelligenza artificiale. Limitare il tempo di permanenza nei tessuti ridurrà le variazioni spurie nelle valutazioni di bersagli molecolari altamente labili, come fosfopeptidi e mRNA, o per tecniche analitiche basate su anticorpi che si basano su una proteina bersaglio nella sua conformazione nativa per il rilevamento.
Il taglio di fiduciali di calibrazione puliti sul vetrino a membrana PEN che sono chiaramente visibili nell'immagine della diapositiva scansionata è uno dei componenti chiave che consentono l'integrazione del software di analisi delle immagini (vedere la Tabella dei materiali) con il flusso di lavoro LMD. Garantire che i calibratori abbiano un punto preciso ("pulito") nella parte inferiore della forma a "V" consente di selezionare un punto preciso nel software di analisi delle immagini per le linee del calibratore da cui disegnare, come descritto nei passaggi 5.1.6 e 5.2.13. L'allineamento di questi punti durante l'importazione nel software LMD è fondamentale per sovrapporre correttamente le annotazioni (facilitato attraverso la generazione di un file .xml compatibile utilizzando gli algoritmi "Malleator" e/o "Dapọ") sul ROI del tessuto pertinente sulla diapositiva LMD fisica. È necessario evidenziare tutte le forme e collettivamente "trascinare e rilasciare" in posizione anche quando l'allineamento è preciso al momento dell'importazione nel software LMD per registrare la posizione verticale (piano z) dello stadio del vetrino sul microscopio laser. Piccoli aggiustamenti al posizionamento delle annotazioni sul ROI del tessuto possono anche essere effettuati durante questa fase, se necessario.
Una limitazione della versione corrente dell'algoritmo Malleator è che non è compatibile con gli strumenti di forma di annotazione predefiniti forniti dal software di analisi delle immagini (vedere la Tabella dei materiali), sebbene i futuri aggiornamenti / versioni dell'algoritmo mireranno a migliorare questa compatibilità. Il file .annotation per le forme disegnate utilizzando questi strumenti contiene solo due set di coordinate x e y accoppiate per ogni annotazione, senza l'orientamento spaziale completo attorno a tali punti. L'uso corrente di questi strumenti comporta la conversione delle annotazioni in linee rette definite da soli due punti durante il processo di importazione. La definizione manuale dei segmenti roi dei tissue è necessaria per una conversione di successo in formato XML e l'importazione LMD. Questo può essere eseguito definendo manualmente ogni ROI con singole annotazioni poligonali a mano libera specifiche per l'area di destinazione o applicando un'annotazione circolare o rettangolare approssimata su tutti i segmenti di ROI del tessuto, se lo si desidera, e sarà compatibile con questo flusso di lavoro.
Mentre il flusso di lavoro qui presentato è stato dimostrato per l'analisi proteomica di campioni di tessuto tumorale umano appena congelati, questo flusso di lavoro LMD basato sull'intelligenza artificiale può essere utilizzato in modo equivalente con tessuti FFPE, tipi di tessuto non canceroso e quelli provenienti da fonti non umane. Può anche supportare altri flussi di lavoro di profilazione molecolare a valle, tra cui analisi trascrittomiche, genomiche o fosfoproteomiche. Questo flusso di lavoro può anche sfruttare altri usi del software di analisi delle immagini (vedere la Tabella dei materiali), anche con funzionalità associate al conteggio delle cellule o ad altri moduli analitici, tra cui il modulo "Multiplex IHC" o il "Tissue Microarray (TMA) Add-on". Le applicazioni future di questo flusso di lavoro possono anche trarre vantaggio dalla predefinizione del numero di cellule per segmento ROI, garantendo così input cellulari equivalenti su più raccolte o utilizzando metodi alternativi per definire i ROI cellulari di interesse, come l'immunoistochimica o la sociologia cellulare.
Divulgazioni
T.P.C. è un membro SAB di ThermoFisher Scientific, Inc e riceve finanziamenti per la ricerca da AbbVie.
Riconoscimenti
Il finanziamento per questo progetto è stato fornito in parte dal Defense Health Program (HU0001-16-2-0006 e HU0001-16-2-00014) alla Uniformed Services University per il Gynecologic Cancer Center of Excellence. Gli sponsor non hanno avuto alcun ruolo nella progettazione, esecuzione, interpretazione o scrittura dello studio. Disconoscimento: Le opinioni qui espresse sono quelle degli autori e non riflettono la politica ufficiale del Dipartimento dell'Esercito/Marina/Aeronautica, del Dipartimento della Difesa o del Governo degli Stati Uniti.
Materiali
| Name | Company | Catalog Number | Comments |
| 1260 Infinity II System | Agilent Technologies Inc | Offline LC system | |
| 96 MicroCaps (150uL) in bulk | Pressure Biosciences Inc | MC150-96 | |
| 96 MicroPestles in bulk | Pressure Biosciences Inc | MP-96 | |
| 96 MicroTubes in bulk (no caps) | Pressure Biosciences Inc | MT-96 | |
| 9mm MS Certified Clear Screw Thread Kits | Fisher Scientific | 03-060-058 | Sample vial for offline LC frationation and mass spectrometry |
| Acetonitrile, Optima LC/MS Grade | Fisher Chemical | A995-4 | Mobile phase solvent |
| Aperio AT2 | Leica Microsystems | 23AT2100 | Slide scanner |
| Axygen PCR Tubes with 0.5 mL Flat Cap | Fisher Scientific | 14-222-292 | Sample tubes; size fits PCT tubes and thermocycler |
| Barocycler 2320EXT | Pressure Biosciences Inc | 2320-EXT | Barocycler |
| BCA Protein Assay Kit | Fisher Scientific | P123225 | |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 | |
| Easy-nLC 1200 | Thermo Fisher Scientific | Liquid Chromatography | |
| EasyPep Maxi Sample Prep Kit | Thermo Fisher Scientific | NCI5734 | Post-label sample clean up column |
| EASY-SPRAY C18 2UM 50CM X 75 | Fisher Scientific | ES903 | Analytical column |
| Eosin Y Solution Aqueous | Sigma Aldrich | HT110216 | |
| Formic Acid, 99+ % | Thermo Fisher Scientific | 28905 | Mobile phase additive |
| ggplot2 version 3.3.5 | CRAN | https://cran.r-project.org/web/packages/ggplot2/ | |
| HALO | Indica Labs | Image analysis software | |
| IDLE (Integrated Development and Learning Environment) | Python Software Foundation | ||
| iheatmapr version 0.5.1 | CRAN | https://cran.r-project.org/web/packages/iheatmapr/ | |
| iRT Kit | Biognosys | Ki-3002-1 | LC-MS QAQC Standard |
| limma version 3.42.2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html | |
| LMD Scanning stage Ultra LMT350 | Leica Microsystems | 11888453 | LMD stage model outfitted with PCT tube holder |
| LMD7 (software version 8.2.3.7603) | Leica Microsystems | LMD apparatus (microscope, laser, camera, PC, tablet) | |
| Mascot Server | Matrix Science | Data analysis software | |
| Mass Spec-Compatible Human Protein Extract, Digest | Promega | V6951 | LC-MS QAQC Standard |
| Mayer’s Hematoxylin Solution | Sigma Aldrich | MHS32 | |
| PEN Membrane Glass Slides | Leica Microsystems | 11532918 | |
| Peptide Retention Time Calibration Mixture | Thermo Fisher Scientific | 88321 | LC-MS QAQC Standard |
| Phosphatase Inhibitor Cocktail 2 | Sigma Aldrich | P5726 | |
| Phosphatase Inhibitor Cocktail 3 | Sigma Aldrich | P0044 | |
| Pierce LTQ Velos ESI Positive Ion Calibration Solution | Thermo Fisher Scientific | 88323 | Instrument calibration solution |
| PM100 C18 3UM 75UMX20MM NV 2PK | Fisher Scientific | 164535 | Pre-column |
| Proteome Discoverer | Thermo Fisher Scientific | OPTON-31040 | Data analysis software |
| Python | Python Software Foundation | ||
| Q Exactive HF-X | Thermo Fisher Scientific | Mass spectrometer | |
| R version 3.6.0 | CRAN | https://cran-archive.r-project.org/bin/windows/base/old/2.6.2/ | |
| RColorBrewer version 1.1-2 | CRAN | https://cran.r-project.org/web/packages/RColorBrewer/ | |
| Soluble Smart Digest Kit | Thermo Fisher Scientific | 3251711 | Digestion reagent |
| TMTpro 16plex Label Reagent Set | Thermo Fisher Scientific | A44520 | isobaric TMT labeling reagents |
| Veriti 60 well thermal cycler | Applied Biosystems | 4384638 | Thermocycler |
| Water, Optima LC/MS Grade | Fisher Chemical | W6-4 | Mobile phase solvent |
| ZORBAX Extend 300 C18, 2.1 x 12.5 mm, 5 µm, guard cartridge (ZGC) | Agilent Technologies Inc | 821125-932 | Offline LC trap column |
| ZORBAX Extend 300 C18, 2.1 x 150 mm, 3.5 µm | Agilent Technologies Inc | 763750-902 | Offline LC analytical column |
Riferimenti
- Motohara, T., et al. An evolving story of the metastatic voyage of ovarian cancer cells: cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene. 38 (16), 2885-2898 (2019).
- Aran, D., Sirota, M., Butte, A. J. Systematic pan-cancer analysis of tumour purity. Nature Communications. 6, 8971 (2015).
- Hunt, A. L., et al. Extensive three-dimensional intratumor proteomic heterogeneity revealed by multiregion sampling in high-grade serous ovarian tumor specimens. iScience. 24 (7), 102757 (2021).
- Dou, Y., et al. Proteogenomic characterization of endometrial carcinoma. Cell. 180 (4), 729-748 (2020).
- Zhang, H., et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 166 (3), 755-765 (2016).
- Gillette, M. A., et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell. 182 (1), 200-225 (2020).
- Silvestri, A., et al. Protein pathway biomarker analysis of human cancer reveals requirement for upfront cellular-enrichment processing. Laboratory Investigation. 90 (5), 787-796 (2010).
- Echle, A., et al. Deep learning in cancer pathology: a new generation of clinical biomarkers. British Journal of Cancer. 124 (4), 686-696 (2021).
- Lee, S., et al. Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Reports. 31 (2), 107502 (2020).
- Xuan, Y., et al. Standardization and harmonization of distributed multi-center proteotype analysis supporting precision medicine studies. Nature Communications. 11 (1), 5248 (2020).
- Hughes, C. S., et al. Quantitative profiling of single formalin fixed tumour sections: proteomics for translational research. Scientific Reports. 6 (1), 34949 (2016).
- Espina, V., et al. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Molecular & Cellular Proteomics. 7 (10), 1998-2018 (2008).
- Espina, V., Heiby, M., Pierobon, M., Liotta, L. A. Laser capture microdissection technology. Expert Review of Molecular Diagnostics. 7 (5), 647-657 (2007).
- Havnar, C. A., et al. Automated dissection protocol for tumor enrichment in low tumor content tissues. Journal of Visualized Experiments. (169), e62394 (2021).
- Mueller, C., et al. One-step preservation of phosphoproteins and tissue morphology at room temperature for diagnostic and research specimens. PLoS One. 6 (8), (2011).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati