
Method Article
Microdisseção laser industrializada e guiada por inteligência artificial para análise proteômica microescalada do Microambiente tumoral
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
Este protocolo descreve um fluxo de trabalho de alto rendimento para segmentação de regiões de interesse confirmadas pela patologia de imagens de seção de tecidos manchados e finos para enriquecimento de populações celulares resolvidas por histologia usando microdisseção a laser. Esta estratégia inclui um novo algoritmo que permite a transferência de demarcações denotando populações celulares de interesse diretamente para microscópios a laser.
Resumo
O microambiente tumoral (TME) representa um ecossistema complexo composto por dezenas de tipos de células distintas, incluindo populações de tumores, estroma e células imunes. Para caracterizar a variação do nível de proteome e a heterogeneidade tumoral em escala, métodos de alto rendimento são necessários para isolar seletivamente populações celulares discretas em malignidades tumorais sólidas. Este protocolo descreve um fluxo de trabalho de alto rendimento, habilitado pela inteligência artificial (IA), que segmenta imagens de hematoxilina e eosina (H&E) manchadas de tecidos finos em regiões de interesse confirmadas pela patologia para a colheita seletiva de populações celulares resolvidas por histologia usando microdisseção a laser (LMD). Essa estratégia inclui um novo algoritmo que permite a transferência de regiões denotando populações celulares de interesse, anotadas usando software de imagem digital, diretamente para microscópios a laser, permitindo assim coleções mais fáceis. Foi realizada a implementação bem-sucedida deste fluxo de trabalho, demonstrando a utilidade deste método harmonizado para colher seletivamente populações de células tumorais do TME para análise proteômica quantitativa e multiplexada por espectrometria de massa de alta resolução. Essa estratégia integra-se plenamente à revisão histopatologia de rotina, aproveitando a análise de imagens digitais para apoiar o enriquecimento das populações celulares de interesse e é totalmente generalizável, possibilitando colheitas harmonizadas de populações celulares do TME para análises multiômicas.
Introdução
O TME representa um ecossistema complexo povoado por uma variedade altamente diversificada de tipos celulares, como células tumorais, células estromais, células imunes, células endoteliais, outros tipos de células mesenquimais e adipócitos, juntamente com uma complexa matriz extracelular1. Esse ecossistema celular varia dentro e em diferentes sítios de órgãos de doença, resultando em heterogeneidade tumoral complexa 2,3. Estudos recentes têm demonstrado que tumores heterogêneos e tumores com baixa celularidade tumoral (baixa pureza) muitas vezes se correlacionam com o prognóstico da doença ruim 2,3.
Para entender a interação molecular entre populações de tumores e células não tumorais dentro do TME em escala, estratégias padronizadas e de alto rendimento são necessárias para colher seletivamente populações celulares distintas de interesse para análise multiômica a jusante. A proteômica quantitativa representa uma técnica em rápida evolução e cada vez mais importante para promover a compreensão da biologia do câncer. Até o momento, a preponderância dos estudos que empregam proteômica tem feito isso com proteínas extraídas de preparações inteiras de tecido tumoral (por exemplo, criopulverizadas), levando a uma escassez na compreensão da heterogeneidade de nível proteome no TME 4,5,6.
O desenvolvimento de estratégias de coleta de amostras que se integrem perfeitamente e aproveitem informações a partir de fluxos de trabalho de patologia clínica permitirão uma nova geração de proteômicas resolvidas em histologia que sejam altamente complementares aos fluxos de trabalho de patologia diagnóstica padrão-ouro. A LMD permite a coleta direta e seletiva de subpopulações celulares ou regiões de interesse (ROIs) através de inspeção microscópica das seções finas de tecido histologicamentemanchados 7. Os recentes grandes avanços na patologia digital e na análise habilitada para IA demonstraram a capacidade de identificar características composicionais únicas e ROIs dentro do TME de forma automatizada, muitas das quais se correlacionam com alterações moleculares e características clínicas da doença, como resistência à terapia e prognóstico dedoenças 8.
O fluxo de trabalho descrito no protocolo aqui apresentado aproveita soluções de software comercial para anotar seletivamente ROIs tumorais dentro de imagens de histopatologia digital, e utiliza ferramentas de software desenvolvidas internamente para transferir esses ROIs tumorais para microscópios laser para coleta automatizada de populações celulares discretas de interesse que se integra perfeitamente com fluxos de trabalho de análise multiômica a jusante. Esta estratégia integrada diminui significativamente o tempo do operador de LMD e minimiza a duração para a qual os tecidos são necessários para estar em temperatura ambiente. A integração da seleção automatizada de recursos e da colheita de LMD com proteômica quantitativa de alto rendimento é demonstrada através de uma análise diferencial do TME de dois subtipos histológicos representativos do câncer de ovário epitelial, câncer de ovário soros de alto grau (HGSOC) e carcinoma de células claras ovarianas (OCCC).
Protocolo
Todos os protocolos de estudo foram aprovados para uso sob um protocolo aprovado pelo IRB ocidental "Uma Análise Molecular Integrada do Câncer Endometrial e Ovariano para identificar e validar biomarcadores clinicamente informativos" considerados isentos sob a regulamentação federal dos EUA 45 CFR 46.102(f). Todos os protocolos experimentais envolvendo dados humanos neste estudo estavam de acordo com a Declaração de Helsinque. O consentimento informado foi obtido de todos os sujeitos envolvidos no estudo.
ATENÇÃO: Os seguintes reagentes utilizados ao longo do protocolo são conhecidos ou suspeitos de cancerígenos e/ou contêm materiais perigosos: etanol, água DEPC, solução de Hematoxilina de Mayer, solução Eosin Y, metanol, acetonitrila e ácido fórmico. O manuseio adequado, conforme descrito nas respectivas fichas de segurança (SDS), e o uso de equipamentos de proteção individual (EPI) adequados, são obrigatórios.
1. Gerando o arquivo de dados de lista de forma padrão (.sld) contendo fiduciais calibradores
NOTA: As etapas de protocolo descritas nesta seção são específicas para uso com um microscópio laser invertido e o software associado (ver a Tabela de Materiais). A criação de um arquivo .sld padrão só é necessária uma vez por microscópio a laser. O arquivo resultante pode ser usado para cortar fiduciários em todos os slides PEN usados posteriormente. Tempo aproximado: 5 min (apenas uma vez).
- Abra o software LMD e carregue a membrana de naftato de polietileno (PEN) no estágio LMD de frente para baixo, com a etiqueta mais próxima do usuário. Desmarque a caixa de linha fechada no lado direito da janela do programa.
- Use a função PtoP (ponto a ponto) sob alta ampliação (63x) para desenhar três setas "V" para servir como fiduciários de calibração. Começando em um ponto externo no V, desenhe uma linha para o ponto médio do V e clique único. Em seguida, desenhe uma segunda linha do ponto central do V até o final do segundo ponto V externo, e clique duas vezes para criar uma forma V única e sem fechamento das duas linhas.
NOTA: Estes fiduciários de calibração devem ser colocados em três cantos do slide: frente direita, direita traseira, esquerda traseira. - Selecione a opção AF (foco automático) antes da opção de corte . Corte o slide na posição 1, mova-o para cada uma das posições restantes do slide e rastreie precisamente sobre os cortes de calibração.
- Salve o arquivo .sld e selecione a opção Salvar sem calibração da caixa de diálogo pop-up para evitar cortar os fiduciários de calibração na membrana.
NOTA: Um arquivo .sld representativo contendo fiduciais calibradores padrão para quatro posições de slides é fornecido no Arquivo Suplementar 1.
2. Preparação de slides LMD
NOTA: As etapas de protocolo descritas nesta seção são específicas para uso com um microscópio laser invertido e o software associado (ver a Tabela de Materiais). Tempo aproximado: 5 min.
- Certifique-se de que o slide está completamente seco antes de cortar os fiduciários de calibração de referência. Abra o software LMD e abra o arquivo padrão de calibração .sld na opção Formas de importação .
- Selecione a opção AF (foco automático) antes da opção de corte . Carregue o slide(s) com o tecido voltado para baixo e a etiqueta deslize mais perto do operador para o suporte de slides no estágio LMD.
- Usando o microscópio laser e o arquivo de calibração padrão .sld, corte fiduciais de calibração na membrana PEN.
- OPCIONAL: Corte os fiduciários de calibração na membrana PEN antes ou depois que a seção de tecido é/são colocadas na lâmina. Se os fiduciais de calibração forem cortados antes da colocação do tecido, certifique-se de que o tecido e/ou fixador não se sobreponha aos calibradores quando o tecido for colocado na lâmina na Etapa 2.5. Se os fiduciais de calibração forem cortados após a colocação do tecido, pare após a conclusão da Etapa 2.4 e prossiga para a seção 3.
- Revise todos os calibradores individualmente para garantir que cada corte esteja completo e visível.
NOTA: Use o recurso Mover e Cortar para direcionar manualmente o laser sobre quaisquer fiduciários de calibração que não cortaram totalmente a membrana PEN. - Coloque a seção de tecido congelado ou fixo de parafina (FFPE) na lâmina contendo os fiduciários de calibração.
3. Coloração de tecido
NOTA: Tempo aproximado: 30 min.
- Fixar os slides de tecido LMD congelados em 70% de etanol (EtOH) contendo reagentes inibidores de fosfattase por 5 min.
- Lave os slides em água de pirocarboco dietil (DEPC) contendo reagentes inibidores de fosfatase por 1 min.
- Lave os slides em água DEPC por 1 min.
- Incubar os slides na solução de Hematoxilina de Mayer por 3 minutos.
- Enxágüe os slides em água DEPC por 3 minutos.
- Enxágüe os slides em uma nova troca de água DEPC por 1 min.
- Incubar os slides em solução Aquosa Eosin Y para 1 s.
- Enxágüe os slides 2 x 5 s em 95% EtOH.
- Enxágüe os slides 3 x 10 s em 100% EtOH.
- Limpe o excesso de EtOH da parte de trás dos slides e deixe os slides secarem ao ar.
- Armazene os slides a -80 °C se a LMD não for executada imediatamente.
4. Imagem de slides
NOTA: As etapas de protocolo descritas nesta seção são específicas para slides digitalizados (ver a tabela de materiais) e as imagens resultantes salvas como arquivos .svs. Use qualquer scanner e seu software associado que gerem arquivos de imagem em um formato que o software de análise de imagem (veja a Tabela de Materiais) possa abrir. Os tipos de arquivos que usam tiffs piramida que são suportados incluem JPG, TIF, MRXS, QPTIFF, componente TIFF, SVS, AFI, SCN, LIF, DCM, OME. TIFF, ND2, VSI, NDPI, NDPIS, CZI, BIF, KFB e ISYNTAX. Tempo aproximado: 5 min.
- Ligue o scanner e abra o software do scanner de slides. Carregue a lâmina com o tecido voltado para o único estágio de deslizamento no scanner. Certifique-se de que a lâmina está completamente seca e coloque delicadamente uma mancha sobre o tecido. Não use etanol ou óleo de imersão sob a tampa.
- Capture a imagem do micrográfico usando configurações calibradas para ajustar para a membrana PEN em vez de fundo de vidro e ignorar a coloração da membrana de fundo, de acordo com as instruções do fabricante.
- Ajuste a área de imagem arrastando e redimensionando o perímetro verde interno para capturar toda a área da membrana PEN, conforme necessário. Adicione quatro pontos de foco no tecido clicando duas vezes na imagem de visão geral do snapshot e três pontos de foco na membrana perto dos fiduciais de calibração (um ponto de foco por cada um dos três fiduciários de calibração).
NOTA: Os quatro pontos de foco podem ser colocados em quase qualquer lugar na seção tecidual, embora a colocação no tecido que está muito manchado e pareça preto pode causar a falha da varredura. - No menu Exibir , selecione Monitor de vídeo. Ajuste manualmente o foco usando o controle deslizante de foco fino e/ou macro, conforme necessário, para cada ponto ao redor do tecido LMD. Capture a varredura de imagem sob alta ampliação (20x). Confirme se todas as fiduciais de calibração são visíveis e claras na imagem salva.
5. Seleção automatizada de recursos usando software de análise de imagens
- Para coleções inteiras de tumores (tempo aproximado: 5 min; dependente de caso):
- Abra o software de análise de imagens (veja a Tabela de Materiais). Selecione Abrir Imagens e na janela pop-up, selecione o arquivo de imagem .svs gerado a partir da digitalização do slide no scanner AT2.
NOTA: Um arquivo de imagem .svs representativo é fornecido no Arquivo Suplementar 2. - Navegue até a guia Anotações . Selecione a ferramenta Caneta na barra de ferramentas de anotação e desenhe uma forma ao redor do tecido.
- Selecione a forma e clique com o botão direito do mouse na imagem. No menu suspenso Avançado , selecione Partição (Tiled). Defina o tamanho e o espaço entre 500 e 40, respectivamente, e selecione OK para gerar as telhas. Selecione e exclua a forma do perímetro usado para gerar as telhas na etapa 5.1.2.
- Selecione o menu suspenso de ações de camada | Exporte para salvar as anotações de ladrilhos como um arquivo .anotação.
NOTA: Um arquivo de anotação .representativo para toda uma coleta de tecido tumoral é fornecido no Arquivo Suplementar 3. - Crie uma pasta para a sessão ou projeto e salve o arquivo .anotação dentro de uma subpasta rotulada com o identificador exclusivo para o slide.
- Navegue até a guia Anotações . Selecione a | suspensa de ações de camada Exclua todas as camadas para remover todas as anotações da imagem. Selecione a ferramenta de caneta e desenhe uma linha curta da ponta interna da ponta da seta para cada fiduciário de calibração. Desenhe as linhas das marcas na seguinte ordem: superior esquerdo, superior direito, inferior direito.
- Selecione o menu suspenso de ações de camada | Exportar para salvar as anotações da linha como um arquivo .anotação. Adicione _calib ao nome do arquivo e coloque o arquivo na subpasto que contém as coordenadas para as formas de ladrilhos.
NOTA: Um arquivo representativo _calib.anotação é fornecido no Arquivo Suplementar 4. - Copie o endereço para a pasta principal do projeto ou sessão. Abra o script gerador de importação XML, "Malleator" (disponível via https://github.com/GYNCOE/Mitchell.et.al.2022), usando o ambiente de desenvolvimento integrado IDLE e cole o endereço da pasta do projeto entre as cotações na parte inferior do script.
- Selecione o menu suspenso run | Execute o Módulo para executar o script.
NOTA: O arquivo de importação LMD .xml será gerado dentro da subpasta criada para a imagem/slide. Um arquivo .xml representativo é fornecido no Arquivo Suplementar 5.
- Abra o software de análise de imagens (veja a Tabela de Materiais). Selecione Abrir Imagens e na janela pop-up, selecione o arquivo de imagem .svs gerado a partir da digitalização do slide no scanner AT2.
- Apenas para coleções enriquecidas com LMD (tempo aproximado: 15 min; dependente de caso):
- Abra o software de análise de imagens (veja a Tabela de Materiais). Selecione Abrir imagens e, na janela pop-up, selecione o arquivo de imagem .svs gerado a partir da digitalização do slide.
- Navegue até a guia Anotações . Selecione e use a ferramenta de anotação Retângulo para desenhar uma caixa ao redor do tecido.
- Selecione a anotação da caixa e clique com o botão direito do mouse na imagem. Selecione o menu suspenso avançado | Opção de particionamento (Tiled). Defina o tamanho e o espaço entre 500 e 40, respectivamente, e selecione OK para gerar as telhas. Selecione e exclua a anotação da caixa do perímetro usada para gerar as telhas na etapa 5.2.2.
- Selecione o menu suspenso de ações de camada | Exporte para salvar as anotações de ladrilhos como um arquivo .anotação.
- Coloque uma cópia salva do algoritmo Python "Dapě" (disponível via https://github.com/GYNCOE/Mitchell.et.al.2022), desenvolvido para mesclar as camadas de anotação classificadas por IA, na mesma pasta do arquivo de anotações de ladrilhos.
- Copie o nome do arquivo de anotação de ladrilhos. Abra o programa Python usando o ambiente de desenvolvimento integrado IDLE e cole o nome do arquivo de anotação de ladrilhos entre as cotações na parte inferior do programa.
- Selecione o menu suspenso run | Módulo de execução. Aguarde que um novo arquivo seja gerado que tenha todas as anotações de ladrilhos mescladas sob uma única camada.
- Abra o software de análise de imagens e navegue até a guia Anotações . Selecione o menu suspenso de ações de camada | Exclua todas as camadas para remover todas as anotações da imagem.
- Selecione o menu suspenso de ações de camada | Importe arquivo de anotação local. Na janela pop-up, selecione o arquivo de anotação .', que foi gerado pelo script. Certifique-se de que todas as telhas importadas estão sob a mesma camada de anotação.
- Navegue até a guia Classificador e siga as instruções do fabricante para gerar formas para os ROIs. Antes de executar o classificador, selecione as camadas de anotação desejadas (ou seja, a camada tumor ou estroma) verificando a caixa de ROI ou caixas na guia Anotações , sob as Opções de Classificador Avançado. Use a opção Camada de Anotação do menu Ações de Classificação para executar o classificador.
- Uma vez concluída a análise do Classificador, navegue até a guia Anotações e selecione a camada de anotação gerada a partir da análise. Selecione o menu suspenso de ações de camada | Exclua todas as camadas mas atual para remover todas as outras camadas de anotação da imagem.
- Selecione o menu suspenso de ações de camada | Exporte para salvar as anotações como um arquivo .anotação. Crie uma pasta para a sessão ou projeto e salve o arquivo .anotação dentro de uma subpasta rotulada com o identificador exclusivo para o slide.
NOTA: Um arquivo de anotação .um representante para uma coleta de tecido enriquecido LMD classificado é fornecido no Arquivo Suplementar 6. - Navegue até a guia Anotações, selecione a lista suspensa de ações de camada | Exclua todas as camadas para remover todas as anotações da imagem. Selecione a ferramenta caneta e desenhe uma linha curta de cada fiduciário de calibração. Desenhe linhas das marcas na seguinte ordem: superior esquerdo, superior direito, inferior direito.
- Selecione o menu suspenso de ações de camada | Exportar para salvar as anotações da linha como um arquivo .anotação. Adicione _calib ao nome do arquivo e coloque o arquivo na subpasto que contém as coordenadas para as formas de ladrilhos.
- Copie o endereço para a pasta principal do projeto ou sessão. Abra o script gerador de importação XML, "Malleator" (disponível via https://github.com/GYNCOE/Mitchell.et.al.2022), usando o ambiente de desenvolvimento integrado IDLE e cole o endereço da pasta do projeto entre as cotações na parte inferior do script.
- Selecione o menu suspenso run | Execute o Módulo para executar o script.
NOTA: O arquivo de importação LMD .xml será gerado dentro da subpasta criada para a imagem/slide.
6. Microdisseção a laser
NOTA: As etapas de protocolo descritas nesta seção são específicas para uso com um microscópio laser invertido e o software associado (ver a Tabela de Materiais). Tempo aproximado: 2 h; caso dependente.
- Carregue o slide de membrana marcado (contendo fiduciais de calibração) com o tecido voltado para baixo e o lado da etiqueta mais próximo do operador no suporte de slides no estágio do microscópio laser.
- Selecione Formas de importação no menu suspenso do arquivo . Selecione o arquivo de importação LMD .xml gerado para o slide. Selecione Não na janela pop-up para evitar carregar pontos de referência do arquivo e não na segunda janela pop-up para evitar o uso de quaisquer pontos de referência armazenados anteriormente para calibração.
- Siga as instruções da aplicação LMD e alinhe a cruz de calibração para cada uma das três fiduciárias de calibração no slide. Procure por fiduciais de calibração que aparecem no canto superior esquerdo, superior direito e inferior direito da imagem de slides no software de análise de imagem que corresponderá a pontos de referência nos cantos dianteiros, traseiros e esquerdo traseiros, respectivamente, do slide LMD invertido no estágio do microscópio. Alternar entre usar a lente objetiva de 5x para localizar e o objetivo de 63x para alinhar cada fiduciário de calibração. Selecione Não na janela pop-up para evitar salvar os pontos de referência para arquivar e OK na segunda janela pop-up para confirmar se o slide está inserido.
- Mova a lente objetiva de 5x para a posição e selecione Sim na janela pop-up para usar a ampliação real. Uma vez que as formas importadas aparecem, concentre a câmera no tecido.
- Destaque e selecione todas as formas na janela Lista de formas , arraste-as para o lugar usando uma ou duas anotações dentro do campo de visão como referências e alinhe o eixo z vertical para cortar com o laser.
- Revise as formas importadas e atribua-as à posição apropriada do tubo para coleta. Pressione o Start Cut para iniciar o laser.
NOTA: As formas importadas no arquivo .xml serão automaticamente atribuídas à posição "sem tampa" na janela Lista de formas . Para colher tecido, as formas importadas devem ser remanejadas para uma posição contendo um tubo carregado.
7. Digestão de proteínas pela tecnologia de ciclo de pressão (PCT)
NOTA: Tempo aproximado: 4 h (3h sem tempo de secagem da centrífuga de vácuo).
- Coloque tubos de 0,5 mL contendo os MicroTubos PCT tampados contendo tecido colhido LMD em 20 μL de 100 mM TEAB/10% acetonitrilo em um termociclador e aqueça a 99 °C por 30 min, depois esfrie a 50 °C por 10 minutos.
- Gire os tubos por 30 s a 4.000 × g e, em seguida, remova os MicroTubes dos tubos de 0,5 mL. Usando a ferramenta MicroCap, remova e descarte as MicroCaps dos MicroTutos pct. Adicione trippsina (veja a tabela de materiais) a uma razão de 1 μg por tecido de 30 mm2 e insira uma MicroPestle no MicroTube usando a ferramenta MicroCap.
- Transfira os MicroTubes para um cartucho barocicr e monte o cartucho completo. Coloque o cartucho na câmara de pressão do barocicr e fixe a tampa. Barociclo a 45.000 psi para 50 s e pressão atmosférica para 10 s a 50 °C para 60 ciclos.
- Uma vez que a barocíclação esteja completa, transfira os microtubos para um tubo de microcentrífugo de 0,5 mL e centrífuga por 2 min a 4.000 × g.
- Remova o MicroTube do tubo de microcentrifuuge de 0,5 mL usando a ferramenta de tampa. Remova cuidadosamente o pilão usando a ferramenta da tampa e enxágue a metade inferior do pilão com 20 μL de espectrometria de massa cromatografia líquida (LC-MS) e colete a lavagem em um tubo de microcentrifusagem de 0,5 mL limpo.
- Toque suavemente no MicroTube na parte superior do banco para mover o líquido para a parte inferior e transfira toda a solução do MicroTube para o tubo de microcentrifuuge de 0,5 mL.
- Adicione 20 μL de água de grau LC-MS ao MicroTube e toque-a suavemente na parte superior do banco. Transfira a solução de lavagem para o tubo de 0,5 mL e repita esta etapa de lavagem mais uma vez.
- Centrífuga de vácuo para secar as amostras até ~2 μL e adicionar 100 μL de 100 mM TEAB, pH 8.0.
- Determinar a concentração de peptídeos usando um ensaio de ensaio colorimétrico (ácido bicinchonínico (BCA) ; consulte a Tabela de Materiais) de acordo com o protocolo do fabricante.
8. Rotulagem de etiqueta de massa tandem (TMT) e limpeza easypep
NOTA: Tempo aproximado: 7 h 20 min (2 h 20 min sem tempo de secagem de centrífugas de vácuo).
- Leve os reagentes isobáricos de rotulagem TMT à temperatura ambiente antes de abrir. Adicione 500 μL de acetonitrilo 100% a cada frasco TMT (5 mgs). Incubar por 10 minutos com vórtice ocasional.
- Dissolva 5 μg da amostra de peptídeo em 100 μL de 100 mM TEAB, pH 8.0 e adicione 10 μL de um determinado reagente TMT. Construa e inclua pools de referência representando cada amostra individual no experimento em cada conjunto multiplex TMT de amostras para facilitar a quantificação de amostras em múltiplos multiplexes TMT9. Incubar reações por 1h à temperatura ambiente com agitação/toque ocasional.
- Sacie a reação de rotulagem TMT adicionando 10 μL de hidroxilamina de 5% e incubar por 30 minutos em temperatura ambiente com toques ocasionais. Após a extinção, misture as amostras rotuladas por TMT em um tubo e seque a aproximadamente 200 μL.
- Adicione 1.800 μL de ácido fórmico de 0,1%. Verifique o pH com papel pH: se pH ~3, adicione 1 mL de ácido fórmico de 0,1%; se o pH >3, adicione 10-20 μL de ácido fórmico de 5% até pH ~3. Adicione 0,1% de ácido fórmico para trazer a um volume final de 3 mL.
- Remova a guia na parte inferior da Coluna de Limpeza do Peptídeo, remova a tampa e coloque-a em um tubo cônico de 15 mL. Transfira a amostra com etiqueta TMT para a coluna e proceda com a limpeza de acordo com o protocolo do fabricante.
- Centrífuga de vácuo para secar os peptídeos elucidos para ~20 μL, transfira para um frasco de LC usando bicarbonato de amônio de 25 mM para um volume final de 80 μL, e prossiga para fracionamento off-line.
9. Fracionamento e agrupamento da amostra multiplex TMT
NOTA: Tempo aproximado: 3 h 30 min (1 h 30 min sem tempo de secagem de centrífugas de vácuo).
- Fracionar os multiplexes de peptídeos rotulados por TMT por cromatografia básica de fase invertida em 96 frações, desenvolvendo um gradiente linear crescente (0,69% min-1) da fase móvel B (acetonitrila) na fase móvel A (10 mM NH4HCO3, pH 8.0).
- Gerar 36 frações concatenadas por meio da junção de poços amostrais. Centrifuugar a vácuo para secar frações a ~2 μL e resuspend em 25 mM de bicarbonato de amônio (concentração final 1,5 μg/10 μL), centrífuga a 15.000 × g por 10-15 min, e transferência para frascos de LC para análise de MS.
10. Espectrometria de massa de cromatografia líquida (LC-MS/MS)
NOTA: Tempo aproximado: Método de instrumento e design experimental dependente.
- Calibrar o espectrômetro de massa de acordo com as instruções/protocolos do fabricante.
- Prepare novas fases e padrões móveis e realize preparações apropriadas de pré-execução de LC (incluindo, mas não se limitando a purgar solventes, ar de descarga e scripts de teste de vazamento para o instrumento referenciado [ver a Tabela de Materiais]). Equilibre colunas pré e analíticas e loop de amostra antes das análises iniciais.
- Antes e entre as análises de multiplex TMT seriais, valide que o sistema LC-MS atende métricas de desempenho previamente benchmarked usando digestores de peptídeos rotulados por TMT de garantia de qualidade (QA/QC) e (por exemplo, MSPE (ver a Tabela de Materiais), HeLa).
- Carregue frascos de feixe automático nas posições apropriadas no autosampler LC. Analisar frações individuais com um método gradiente/Esclerose múltipla apropriado. Intercalar uma corrida de "lavagem" com padrão peptídeo (por exemplo, calibração do tempo de retenção de peptídeos [PRTC]) aproximadamente uma vez por dia para avaliar o desempenho espectral cromatográfico e em massa. Após a análise de cada série de frações de amostra multiplex TMT, execute os padrões de benchmark QA/QC TMT para avaliar o desempenho do sistema.
- Execute rotinas de avaliação de espectrômetros de massa após os padrões de benchmark QA/QC TMT para avaliar o desempenho pós-amostra e, em seguida, calibrar o sistema como na etapa 10.1 para o próximo conjunto amostral.
11. Análise de dados bioinformáticas
NOTA: Tempo aproximado: Dependente de design experimental.
- Transfira todos os dados de amostra (por exemplo, .raw arquivos) para uma unidade de armazenamento/computador de rede apropriada.
- Pesquise todas as frações em conjunto usando o aplicativo de análise de dados desejado (por exemplo, Proteome Discover, Mascot) usando parâmetros apropriados9 contra um banco de dados de referência proteica específico da espécie para gerar correspondências espectrais de peptídeos (PSMs) e extrair intensidades de sinal de íons de repórter TMT. Filtrar PSMs baseados em métricas adequadas de controle de qualidade e agregar abundâncias de proporção de íons de repórter tmt deregistro mediano de 2 transformações em abundâncias globais de nível de proteína, como descrito anteriormente 3,9.
- Compare alterações proteicas em condições de interesse usando o software de análise diferencial desejado.
Resultados
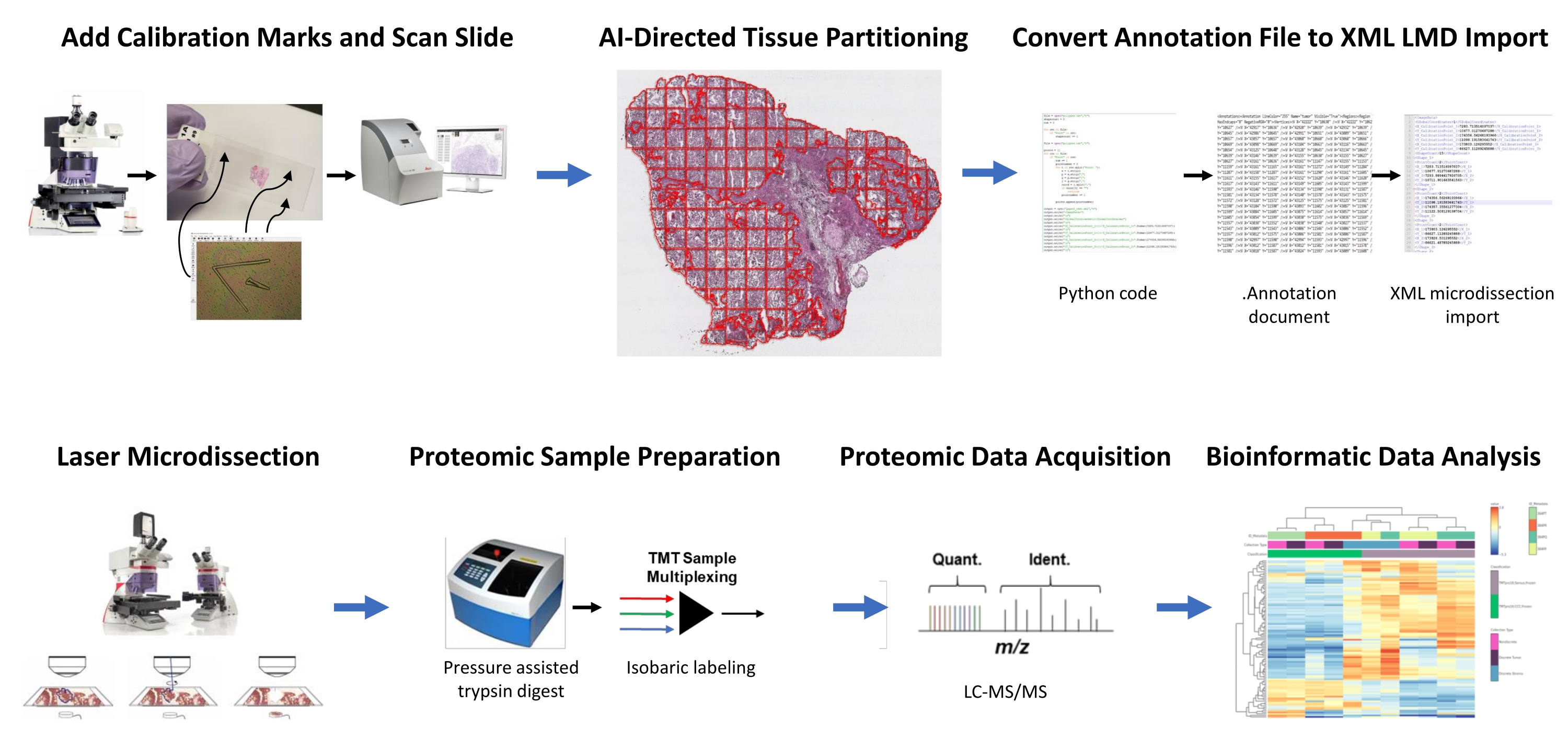
Seções finas de tecido congelado fresco de dois pacientes com HGSOC e dois OCCC foram analisadas utilizando-se esta identificação integrada de ROI de tecido, segmentação, LMD e fluxo de trabalho de análise proteômica quantitativa (Figura 1). As seções representativas de tecido sátmica H&E para cada tumor foram revisadas por um patologista certificado pelo conselho; a celularidade tumoral variou de 70% a 99%. Os tecidos foram seccionados finos em lâminas de membrana PEN (Arquivo Suplementar 2) e pré-cortados com fiduciais calibradores (Arquivo Suplementar 1), permitindo a integração dos dados de orientação posicional a partir de anotações geradas no software de análise de imagem (ver a Tabela de Materiais) com orientação coordenada cartesiana no software LMD. Após a coloração da H&E, foram capturadas imagens de alta resolução (20x) dos slides PEN contendo o tecido mais calibradores.
As populações de tumores e células estromes nos micrografos foram segmentadas por meio de software de análise de imagem (ver a Tabela de Materiais) para colheita seletiva por LMD, juntamente com colheitas representando toda a seção fina do tecido (por exemplo, tecido tumoral inteiro) (Figura 1). Anotações não discriminantes para coleções inteiras de tecido tumoral foram geradas particionando toda a seção tecidual com telhas de 500 μm2, deixando uma lacuna de 40 μm entre as telhas para manter a integridade da membrana PEN e evitar que a membrana se enrole durante a LMD. Em slides para enriquecimento de LMD resolvido em histologia, o classificador de IA no software de análise de imagem (ver a Tabela de Materiais) foi treinado para discriminar entre tumor e células estromicanas, juntamente com o fundo de deslizamento de vidro em branco. As regiões representativas de tumor, estroma e vidro em branco foram destacadas manualmente, e a ferramenta de classificação foi utilizada para segmentar esses ROIs em toda a seção tecidual. As camadas segmentadas que representam tecido inteiro, epitélio tumor e estroma foram salvas separadamente como arquivos individuais de anotação .(Arquivo Suplementar 3 e Arquivo Suplementar 6). Em uma cópia separada do arquivo de imagem (sem as anotações particionadas do ROI), uma linha curta da ponta central de cada um dos três calibradores fiduciais foi anotada e salva como um arquivo .anotação usando o mesmo nome de arquivo que cada um dos arquivos de camada de anotação LMD, mas anexada com o sufixo "_calib" (Arquivo Suplementar 4). Estas linhas foram utilizadas para co-registrar a posição dos calibradores de membrana PEN com os dados da lista de forma de anotação desenhados no software de análise de imagem.
O presente estudo fornece dois algoritmos, "Malleator" e "Dapě" em Python para suportar este fluxo de trabalho LMD orientado por IA, que estão disponíveis em https://github.com/GYNCOE/Mitchell.et.al.2022. O algoritmo Malleator extrai as coordenadas cartesianas específicas para todas as anotações individuais (ROI de tecido e calibradores) dos arquivos de anotação .emparelhados e os mescla em um único arquivo de importação de Linguagem de Marcação Extensível (XML) (Arquivo Suplementar 5). Especificamente, o algoritmo Malleator usa o nome do diretório de uma pasta pai como entrada para pesquisar todas as pastas subdireciontárias e gera .xml arquivos para quaisquer subpastas que ainda não possuem um arquivo .xml mesclado. O algoritmo Malleator mescla todas as camadas de anotação no software de análise de imagens (ver a tabela de materiais) em uma única camada e converte os dados da lista de forma gerada por IA, que é salvo como tipo de arquivo proprietário .anotação, em formato .xml compatível com o software LMD. Depois de fundir os arquivos de anotação e calibrador, o arquivo .xml gerado por algoritmo é salvo e importado para o software LMD. Pequenos ajustes são necessários para ajustar manualmente o alinhamento das anotações, que também serve para registrar a posição vertical (z-plane) do estágio de slides no microscópio laser. O algoritmo Dapě é usado especificamente para coleções enriquecidas com LMD. As telhas particionadas são automaticamente atribuídas a camadas de anotação individuais pelo software de análise de imagens. O algoritmo Dapě mescla todas as telhas particionadas em uma única camada de anotação antes do uso da ferramenta Classificador, reduzindo assim o tempo de execução da análise do Classificador para coleções enriquecidas com LMD.
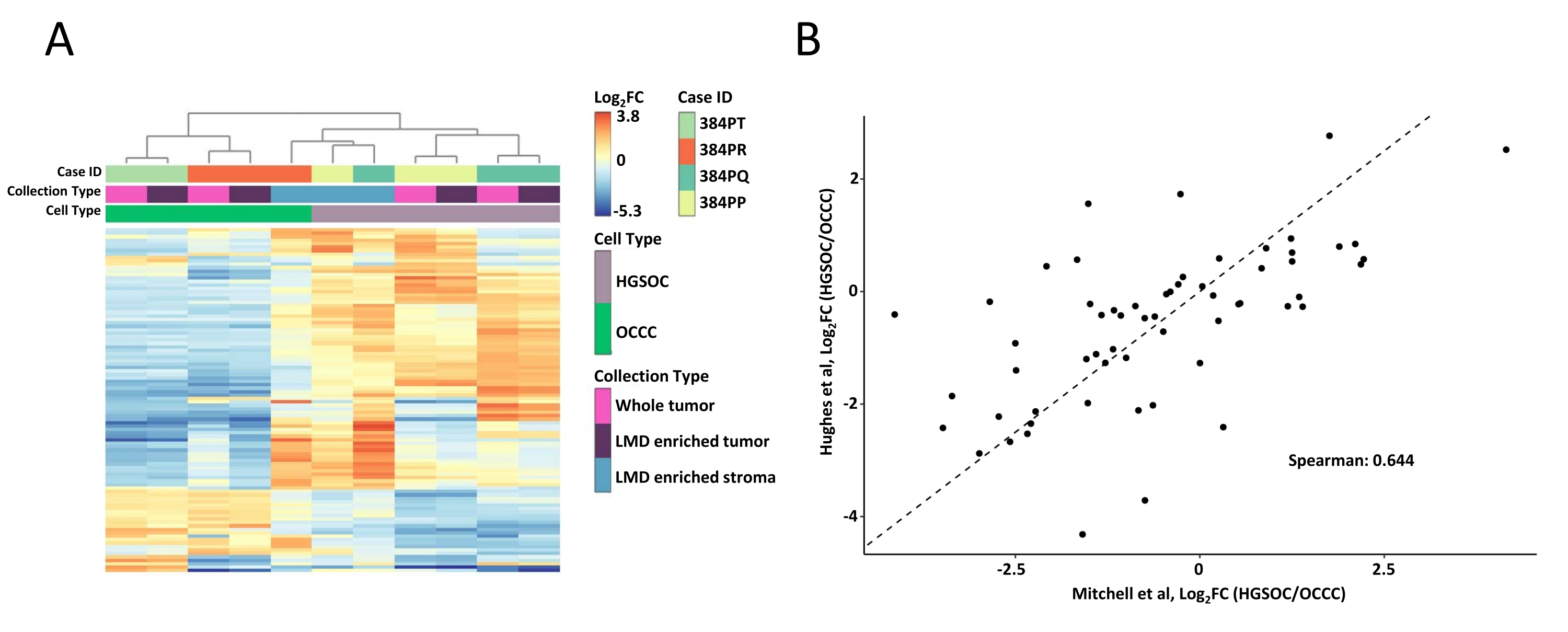
Foram digeridas amostras de tecido enriquecido com tumor e LMD, rotuladas com reagentes TMT, multiplexadas, fracionadas offline e analisadas via proteômica quantitativa baseada em MS, conforme descrito anteriormente9. O rendimento médio do peptídeo (43-60 μg) e a recuperação (0,46-0,59 μg/mm2) para amostras colhidas usando este fluxo de trabalho orientado por IA foram comparáveis com os relatórios anteriores 9,10. Um total de 5.971 proteínas foram co-quantificadas em todas as amostras (Tabela Suplementar S1). O agrupamento hierárquico não supervisionado utilizando as 100 proteínas mais variáveis resultou na segregação dos histotipos HGSOC e OCCC das amostras de tumores enriquecidos com LMD e inteiros (Figura 2A), semelhantes aos descritos anteriormente11. Em contraste, as amostras de estroma enriquecidas com LMD tanto do HGSOC quanto do OCCC agruparam-se e independentemente do tumor enriquecido com LMD e amostras de tumores inteiros. Das 5.971 proteínas quantificadas, 215 foram significativamente alteradas (LIMMA adj. p < 0,05) entre coleções inteiras de tumores de amostras de HGSOC e OCCC (Tabela Suplementar S2). Essas proteínas alteradas foram comparadas com aquelas identificadas para diferenciar o tecido tumoral HGSOC e OCCC por Hughes et al.11. Das 76 proteínas de assinatura quantificadas por Hughes et al., 57 foram co-quantificadas neste conjunto de dados e foram altamente correlacionadas (Spearman Rho = 0,644, p < 0,001) (Figura 2B).

Figura 1: Resumo do fluxo de trabalho integrado para seleção automatizada de tecido de interesse para microdisseção a laser para proteômica quantitativa a jusante. Fiduciais de calibração são cortados em lâminas de membrana PEN para co-registrar dados de orientação posicional de segmentos derivados de IA do ROI de tecido no software de análise de imagem, HALO, com posicionamento horizontal no microscópio LMD. O algoritmo Malleator é usado para mesclar os dados de segmentação anotados em todas as camadas de anotação para um slide com o arquivo de referência _calib e para convertê-los em um arquivo .xml compatível com o software LMD. O tecido colhido por LMD para análise proteômica é digerido e analisado por proteômica quantitativa de alto rendimento, como descrito anteriormente9. Abreviaturas: LMD = microdisseção a laser; ROI = região de interesse; TMT = tag de massa tandem; Quant. = quantificação; Ident. = identificação; LC-MS/MS = espectrometria de massa cromatografia líquida-tandem. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Análise das proteínas em amostras de tumores enriquecidos com LMD e inteiros. (A) Análise hierárquica não supervisionada das 100 proteínas mais variadamente abundantes em amostras de 100 tumores enriquecidos e tumores integrais do HGSOC e OCCC. (B) Correlação de abundâncias de proteínas de2 vezes de alteração de tronco entre as colheitas de tumores inteiros do HGSOC e OCCC no presente estudo (Mitchell et al., x-axis) e um estudo semelhante por Hughes et al. (y-axis)11. Abreviaturas: LMD = microdisseção a laser; HGSOC = câncer de ovário soroso de alto grau; OCCC = carcinoma de células claras ovarianas; log2FC = log2-transformado abundância proteômica. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Tabela Suplementar S1: Abundâncias de 5.971 proteínas co-quantificadas em todas as amostras de tumores enriquecidos e inteiros de LMD de amostras de tecido HGSOC e OCCC. Abreviaturas: LMD = microdisseção a laser; HGSOC = câncer de ovário soroso de alto grau; OCCC = carcinoma de células claras ovarianas. Clique aqui para baixar esta Tabela.
Tabela Suplementar S2: Proteínas expressas diferencialmente (215) em coleções inteiras de tumores de HGSOC vs OCCC (LIMMA adj. p < 0,05). Abreviaturas: HGSOC = câncer de ovário soroso de alto grau; OCCC = carcinoma de células claras ovarianas. Clique aqui para baixar esta Tabela.
Arquivo suplementar 1: Arquivo de lista de forma representativa (.sld) contendo fiduciais calibradores padrão para quatro posições de slides. O arquivo pode ser importado para o software LMD. Clique aqui para baixar este Arquivo.
Arquivo Suplementar 2: Arquivo de imagem .svs representativo de uma seção de tecido de alta resolução (20x) manchada de H&E. O arquivo pode ser aberto e visualizado usando software de análise de imagem ou software LMD. Abreviação: H&E = hematoxilina e eosina; LMD = microdisseção a laser. Clique aqui para baixar este Arquivo.
Arquivo Suplementar 3: Arquivo de anotação .representativo de segmentos de tumores inteiros particionados. O arquivo pode ser importado para o software de análise de imagens. Clique aqui para baixar este Arquivo.
Arquivo suplementar 4: Arquivo representativo _calib.anotação dos segmentos fiduciais calibradores. As informações de coordenadas representam o posicionamento oriental das linhas de calibrador curtas extraídas de cada fiduciário da ponta da seta. O arquivo pode ser importado para o software de análise de imagens. Clique aqui para baixar este Arquivo.
Arquivo suplementar 5: linguagem de marcação extensível representativa (.xml) arquivo gerado pelo algoritmo Malleator. O arquivo pode ser importado para o software de microdisseção a laser. Clique aqui para baixar este Arquivo.
Arquivo Suplementar 6: Arquivo de anotação .representativo de segmentos classificados por IA particionados para coleções enriquecidas com LMD. O arquivo pode ser importado para o software de análise de imagens. Abreviaturas: IA = inteligência artificial; LMD = microdisseção a laser. Clique aqui para baixar este Arquivo.
Discussão
Embora existam múltiplos precedentes de estudo destinados a desenvolver e/ou melhorar os fluxos de trabalho para enriquecimento de subpopulações celulares-alvo de tecidos e metodologias congeladas e frescas para a manutenção da qualidade da amostra durante o processamentode 9,12,13,14,15, existe uma necessidade substancial de desenvolver estratégias automatizadas para a preparação de amostras de tecido clínico para análises moleculares para diminuir a variabilidade e aumentar a reprodutibilidade. Este fluxo de trabalho descreve um protocolo padronizado e semautomado que integra ferramentas de software de análise de imagem existentes (ver a Tabela de Materiais) para a colheita de populações de células discretas por LMD de amostras de tecido clínico.
O enriquecimento de LMD espacialmente resolvido de ROIs capturando populações de células discretas representa uma etapa de processamento de tecidos de próxima geração antes de análises multiômicas para melhorar a caracterização e identificação molecular e facilitar a descoberta de biomarcadores seletivos celulares. Este protocolo melhora as metodologias existentes, reduzindo a exposição muitas vezes longa de seções teciduais ao ambiente que está associada à segmentação manual do ROI por um histólogo (que pode levar >1-2 h antes da coleta de LMD). Este fluxo de trabalho permite que o ROI seja pré-identificado pela classificação e segmentação guiadas por IA. Limitar o tempo de moradia tecidual diminuirá variações espúrias nas avaliações de alvos moleculares altamente labiais, como fosforpeptídeos e mRNA, ou para técnicas analíticas baseadas em anticorpos que dependem de uma proteína-alvo estar em sua conformação nativa para detecção.
O corte de fiduciais de calibrador puro no slide da membrana PEN que são claramente visíveis na imagem de slide digitalizada é um dos componentes-chave que permitem a integração do software de análise de imagem (ver a Tabela de Materiais) com o fluxo de trabalho LMD. Garantir que os calibradores tenham um ponto preciso ("limpo") na parte inferior da forma "V" permite a seleção de um ponto preciso no software de análise de imagem para que as linhas calibradoras sejam desenhadas, conforme descrito nas etapas 5.1.6 e 5.2.13. O alinhamento desses pontos durante a importação para o software LMD é fundamental para sobrepor adequadamente as anotações (facilitadas através da geração de um arquivo .xml compatível usando os algoritmos "Malleator" e/ou "Dapě") no ROI de tecido relevante no slide LMD físico. É necessário destacar todas as formas e coletivamente "arrastar e soltar" coletivamente no lugar mesmo quando o alinhamento é preciso ao importar para o software LMD para registrar a posição vertical (z-plane) do estágio de slides no microscópio laser. Pequenos ajustes no posicionamento das anotações sobre o ROI tecidual também podem ser feitos durante esta etapa, se necessário.
Uma limitação da versão atual do algoritmo Malleator é que ele não é compatível com as ferramentas de forma de anotação predefinidas fornecidas pelo software de análise de imagem (ver a Tabela de Materiais), embora futuras atualizações/versões do algoritmo visarão melhorar essa compatibilidade. O arquivo .anotação para formas desenhadas usando essas ferramentas contém apenas dois conjuntos de coordenadas emparelhadas x e y para cada anotação, sem a orientação espacial completa em torno desses pontos. O uso atual dessas ferramentas resulta na conversão das anotações em linhas retas definidas por apenas dois pontos durante o processo de importação. A definição manual dos segmentos de ROI de tecido é necessária para conversão bem-sucedida para formato XML e importação de LMD. Isso pode ser realizado definindo manualmente cada ROI com anotações poligonais individuais à mão livre específicas da área alvo ou aplicando uma anotação circular ou retangular aproximada em todos os segmentos de ROI de tecido, se desejar, e será compatível com este fluxo de trabalho.
Embora o fluxo de trabalho aqui apresentado tenha sido demonstrado para análise proteômica de amostras de tecido de câncer humano congelado fresco, este fluxo de trabalho LMD orientado por IA pode ser usado equivalentemente com tecidos FFPE, tipos de tecidos não cancerígenos, e aqueles de fontes não humanas. Ele também pode suportar outros fluxos de trabalho de perfil molecular a jusante, incluindo análises transcriômicas, genômicas ou fosfoproteômicas. Este fluxo de trabalho também pode aproveitar outros usos do software de análise de imagens (ver a Tabela de Materiais), inclusive com recursos associados à contagem de células ou outros módulos analíticos, incluindo o módulo "Multiplex IHC" ou o Add-on "Tissue Microarray (TMA)." Aplicações futuras deste fluxo de trabalho também podem se beneficiar de predefinir o número de células por segmento de ROI, garantindo entradas celulares equivalentes em várias coleções, ou usando métodos alternativos para definir ROIs celulares de interesse, como por imunohistoquímica ou sociologia celular.
Divulgações
T.P.C. é membro da ThermoFisher Scientific, Inc SAB e recebe financiamento de pesquisa da AbbVie.
Agradecimentos
O financiamento para este projeto foi fornecido em parte pelo Programa de Saúde de Defesa (HU0001-16-2-0006 e HU0001-16-2-00014) para a Universidade de Serviços Uniformizados para o Centro de Excelência do Câncer Ginecológico. Os patrocinadores não tiveram papel no projeto, execução, interpretação ou redação do estudo. Disclaimer: As opiniões aqui expressas são dos autores e não refletem a política oficial do Departamento do Exército/Marinha/Força Aérea, Departamento de Defesa ou Governo dos EUA.
Materiais
| Name | Company | Catalog Number | Comments |
| 1260 Infinity II System | Agilent Technologies Inc | Offline LC system | |
| 96 MicroCaps (150uL) in bulk | Pressure Biosciences Inc | MC150-96 | |
| 96 MicroPestles in bulk | Pressure Biosciences Inc | MP-96 | |
| 96 MicroTubes in bulk (no caps) | Pressure Biosciences Inc | MT-96 | |
| 9mm MS Certified Clear Screw Thread Kits | Fisher Scientific | 03-060-058 | Sample vial for offline LC frationation and mass spectrometry |
| Acetonitrile, Optima LC/MS Grade | Fisher Chemical | A995-4 | Mobile phase solvent |
| Aperio AT2 | Leica Microsystems | 23AT2100 | Slide scanner |
| Axygen PCR Tubes with 0.5 mL Flat Cap | Fisher Scientific | 14-222-292 | Sample tubes; size fits PCT tubes and thermocycler |
| Barocycler 2320EXT | Pressure Biosciences Inc | 2320-EXT | Barocycler |
| BCA Protein Assay Kit | Fisher Scientific | P123225 | |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 | |
| Easy-nLC 1200 | Thermo Fisher Scientific | Liquid Chromatography | |
| EasyPep Maxi Sample Prep Kit | Thermo Fisher Scientific | NCI5734 | Post-label sample clean up column |
| EASY-SPRAY C18 2UM 50CM X 75 | Fisher Scientific | ES903 | Analytical column |
| Eosin Y Solution Aqueous | Sigma Aldrich | HT110216 | |
| Formic Acid, 99+ % | Thermo Fisher Scientific | 28905 | Mobile phase additive |
| ggplot2 version 3.3.5 | CRAN | https://cran.r-project.org/web/packages/ggplot2/ | |
| HALO | Indica Labs | Image analysis software | |
| IDLE (Integrated Development and Learning Environment) | Python Software Foundation | ||
| iheatmapr version 0.5.1 | CRAN | https://cran.r-project.org/web/packages/iheatmapr/ | |
| iRT Kit | Biognosys | Ki-3002-1 | LC-MS QAQC Standard |
| limma version 3.42.2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html | |
| LMD Scanning stage Ultra LMT350 | Leica Microsystems | 11888453 | LMD stage model outfitted with PCT tube holder |
| LMD7 (software version 8.2.3.7603) | Leica Microsystems | LMD apparatus (microscope, laser, camera, PC, tablet) | |
| Mascot Server | Matrix Science | Data analysis software | |
| Mass Spec-Compatible Human Protein Extract, Digest | Promega | V6951 | LC-MS QAQC Standard |
| Mayer’s Hematoxylin Solution | Sigma Aldrich | MHS32 | |
| PEN Membrane Glass Slides | Leica Microsystems | 11532918 | |
| Peptide Retention Time Calibration Mixture | Thermo Fisher Scientific | 88321 | LC-MS QAQC Standard |
| Phosphatase Inhibitor Cocktail 2 | Sigma Aldrich | P5726 | |
| Phosphatase Inhibitor Cocktail 3 | Sigma Aldrich | P0044 | |
| Pierce LTQ Velos ESI Positive Ion Calibration Solution | Thermo Fisher Scientific | 88323 | Instrument calibration solution |
| PM100 C18 3UM 75UMX20MM NV 2PK | Fisher Scientific | 164535 | Pre-column |
| Proteome Discoverer | Thermo Fisher Scientific | OPTON-31040 | Data analysis software |
| Python | Python Software Foundation | ||
| Q Exactive HF-X | Thermo Fisher Scientific | Mass spectrometer | |
| R version 3.6.0 | CRAN | https://cran-archive.r-project.org/bin/windows/base/old/2.6.2/ | |
| RColorBrewer version 1.1-2 | CRAN | https://cran.r-project.org/web/packages/RColorBrewer/ | |
| Soluble Smart Digest Kit | Thermo Fisher Scientific | 3251711 | Digestion reagent |
| TMTpro 16plex Label Reagent Set | Thermo Fisher Scientific | A44520 | isobaric TMT labeling reagents |
| Veriti 60 well thermal cycler | Applied Biosystems | 4384638 | Thermocycler |
| Water, Optima LC/MS Grade | Fisher Chemical | W6-4 | Mobile phase solvent |
| ZORBAX Extend 300 C18, 2.1 x 12.5 mm, 5 µm, guard cartridge (ZGC) | Agilent Technologies Inc | 821125-932 | Offline LC trap column |
| ZORBAX Extend 300 C18, 2.1 x 150 mm, 3.5 µm | Agilent Technologies Inc | 763750-902 | Offline LC analytical column |
Referências
- Motohara, T., et al. An evolving story of the metastatic voyage of ovarian cancer cells: cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene. 38 (16), 2885-2898 (2019).
- Aran, D., Sirota, M., Butte, A. J. Systematic pan-cancer analysis of tumour purity. Nature Communications. 6, 8971 (2015).
- Hunt, A. L., et al. Extensive three-dimensional intratumor proteomic heterogeneity revealed by multiregion sampling in high-grade serous ovarian tumor specimens. iScience. 24 (7), 102757 (2021).
- Dou, Y., et al. Proteogenomic characterization of endometrial carcinoma. Cell. 180 (4), 729-748 (2020).
- Zhang, H., et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 166 (3), 755-765 (2016).
- Gillette, M. A., et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell. 182 (1), 200-225 (2020).
- Silvestri, A., et al. Protein pathway biomarker analysis of human cancer reveals requirement for upfront cellular-enrichment processing. Laboratory Investigation. 90 (5), 787-796 (2010).
- Echle, A., et al. Deep learning in cancer pathology: a new generation of clinical biomarkers. British Journal of Cancer. 124 (4), 686-696 (2021).
- Lee, S., et al. Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Reports. 31 (2), 107502 (2020).
- Xuan, Y., et al. Standardization and harmonization of distributed multi-center proteotype analysis supporting precision medicine studies. Nature Communications. 11 (1), 5248 (2020).
- Hughes, C. S., et al. Quantitative profiling of single formalin fixed tumour sections: proteomics for translational research. Scientific Reports. 6 (1), 34949 (2016).
- Espina, V., et al. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Molecular & Cellular Proteomics. 7 (10), 1998-2018 (2008).
- Espina, V., Heiby, M., Pierobon, M., Liotta, L. A. Laser capture microdissection technology. Expert Review of Molecular Diagnostics. 7 (5), 647-657 (2007).
- Havnar, C. A., et al. Automated dissection protocol for tumor enrichment in low tumor content tissues. Journal of Visualized Experiments. (169), e62394 (2021).
- Mueller, C., et al. One-step preservation of phosphoproteins and tissue morphology at room temperature for diagnostic and research specimens. PLoS One. 6 (8), (2011).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados