
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
機能電気泳動移動度シフトアッセイ(EMSA)を用いた非コーディング遺伝的変異体およびDNA親和性沈殿アッセイ(DAPA)のスクリーニング
* これらの著者は同等に貢献しました
要約
We present a strategic plan and protocol for identifying non-coding genetic variants affecting transcription factor (TF) DNA binding. A detailed experimental protocol is provided for electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genotype-dependent TF DNA binding.
要約
Population and family-based genetic studies typically result in the identification of genetic variants that are statistically associated with a clinical disease or phenotype. For many diseases and traits, most variants are non-coding, and are thus likely to act by impacting subtle, comparatively hard to predict mechanisms controlling gene expression. Here, we describe a general strategic approach to prioritize non-coding variants, and screen them for their function. This approach involves computational prioritization using functional genomic databases followed by experimental analysis of differential binding of transcription factors (TFs) to risk and non-risk alleles. For both electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genetic variants, a synthetic DNA oligonucleotide (oligo) is used to identify factors in the nuclear lysate of disease or phenotype-relevant cells. For EMSA, the oligonucleotides with or without bound nuclear factors (often TFs) are analyzed by non-denaturing electrophoresis on a tris-borate-EDTA (TBE) polyacrylamide gel. For DAPA, the oligonucleotides are bound to a magnetic column and the nuclear factors that specifically bind the DNA sequence are eluted and analyzed through mass spectrometry or with a reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blot analysis. This general approach can be widely used to study the function of non-coding genetic variants associated with any disease, trait, or phenotype.
概要
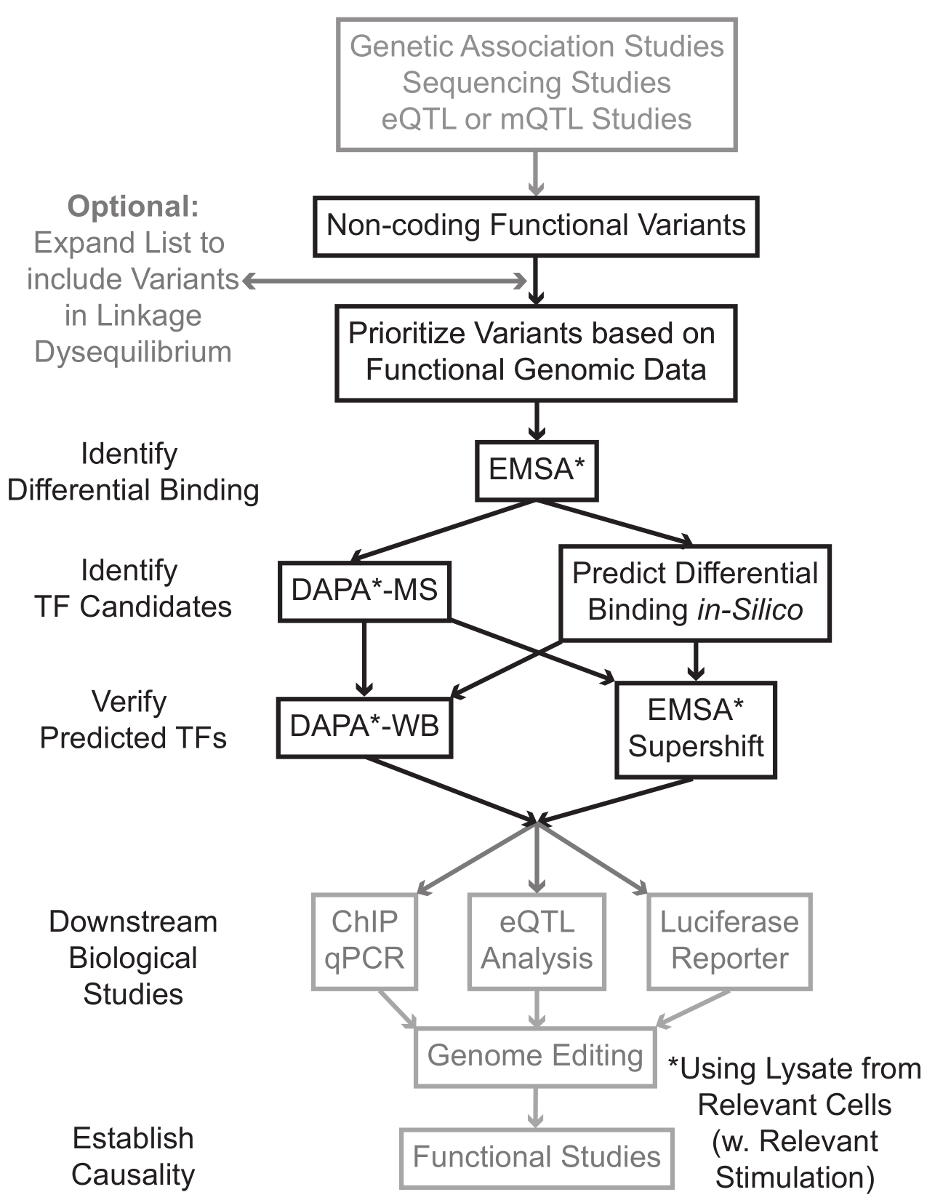
ゲノムワイド関連研究(GWAS)を含むシーケンシングおよびジェノタイピングベースの研究、候補遺伝子座の研究、およびディープシーケンシング研究は、統計学的疾患、形質または表現型と関連している多くの遺伝的変異を同定しました。初期の予測に反して、これらの変異体(85-93%)の大部分は非コード領域に位置しており、タンパク質1,2のアミノ酸配列を変更しません。これらの非コード変異体の機能の解釈と関連する疾患、形質または表現型にそれらを接続する生物学的メカニズムを決定することは3-6に挑戦証明されています。遺伝子発現 - 私たちは、重要な中間表現型にバリアントをリンクする分子メカニズムを識別するための一般的な戦略を開発しました。このパイプラインは、特に遺伝的変異による結合TFの調節を同定するために設計されています。この戦略を予測することを目的とした計算手法および分子生物学技術を組み合わせインシリコ候補変異体の生物学的効果、および( 図1)、経験的に、これらの予測を検証します。

図1: 非コード遺伝子変異体の解析のための戦略的アプローチ 、この原稿に関連付けられた詳細なプロトコールに含まれていない手順は灰色で網掛けされている。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
多くの場合、それぞれの統計的に関連する変異体と高い連鎖、不平衡(LD)にあるすべてのものを含むように変異体のリストを拡張することによって開始することが重要です。 LDは、R 2統計7によって測定することができる2つの異なる染色体位置における対立遺伝子の非ランダムな関連の尺度です。 r 2は林の尺度でありますR 2の変種間の完全な結合を示す= 1 2を持つ2つの変異体、間の影の不均衡。高LDにおける対立遺伝子は祖先集団全体の染色体上に同時分離することが判明しています。現在のジェノタイピングアレイは、ヒトゲノム中のすべての既知の亜種が含まれていません。その代わりに、彼らは人間のゲノム内のLDを利用し、LD 8の特定の領域内の他の変異体のためのプロキシとして機能する知られている変異体のサブセットを含みます。それは意味のある生物学的効果との因果バリアントバリアントとLDにあるため、このように、任意の生物学的影響のない変異体は、特定の疾患に関連することができます。手続きは、PLINK 10,11、全ゲノム関連解析のためのオープンソースのツールと互換性のあるバイナリファイルに千ゲノムプロジェクト9バリアントコールファイル(VCF)の最新リリースを変換することをお勧めします。続いて、各入力遺伝VAとLDのR 2> 0.8と他のすべての遺伝的変異ほほ笑むは候補として同定することができます。変異体がヨーロッパ系の科目で同定された場合、同様の祖先の被験者からのデータは、LDの拡張のために使用されるべきで、STEP- 例えばこのための適切な参照集団を使用することが重要です。
LDの拡張はしばしば、候補変異体の数十をもたらし、これらのうちのごく一部が、疾患機構に寄与する可能性があります。多くの場合、実験的に個別にこれらの変異体のそれぞれを調べることは実行不可能です。バリアントの優先順位を決定するためのフィルタとして一般に公開機能ゲノムデータセットの数千人を活用することが有効です。例えば、ENCODEコンソーシアム12は 、DNアーゼ-seqの13、ATACなどの技術からクロマチンアクセシビリティデータと共に、文脈の広い範囲でのTFおよび補因子の結合を記述したChIP-seqの実験で何千もの、およびヒストンマークを行いました-seq 14、およびFAIRE-seqの15。 DATABこのようなUCSCゲノムブラウザ16、ロードマップエピゲノミクス17、青写真エピゲノム18、Cistrome 19、及び再マップ20としてのASおよびWebサーバは、細胞型および条件の広い範囲にわたって、これらおよび他の実験技術によって生成されたデータへの無料アクセスを提供します。実験的に調べるためにあまりにも多くの変異体が存在する場合、これらのデータは、関連する細胞および組織型における可能性の調節領域内に位置するものを優先順位付けするために使用することができます。さらに、変異体は、特定のタンパク質のためのChIP-seqのピークの範囲内にある場合には、これらのデータは、特定のTF(複数可)またはその影響を与える可能性があります結合補因子に関して潜在的なリードを提供することができます。
次に、優先順位を付け、得られた変異体は、EMSA 21,22を使用して予測結合遺伝子型依存性タンパク質を検証するために実験的にスクリーニングされます。 EMSAは、非還元TBEゲル上でオリゴの移行の変化を測定します。蛍光標識オリゴとともにインキュベートします核溶解液、および核因子の結合は、ゲル上のオリゴの動きを遅らせるます。このように、より多くの核因子を結合したオリゴは、スキャン時に強い蛍光シグナルとして提示します。特に、EMSAは、バインディング影響される特定のタンパク質についての予測を必要としません。
変異体は、予測調節領域内に位置し、示差的結合核因子することができるされていることが確認されると、計算方法は、その結合それらが影響する可能性のある特定のTF(複数可)を予測するために使用されます。私たちは、CIS-BP 23,24、RegulomeDB 25、UniProbe 26、およびJASPAR 27を使用することを好みます。候補者のTFが識別されると、これらの予測は、具体的には、これらのTF(EMSA-スーパーシフトとDAPA-西部劇)に対する抗体を用いて試験することができます。 EMSA-スーパーシフトは、核溶解液とオリゴにTF特異的抗体の添加を含みます。 EMSA-スーパーシフトで陽性の結果がのreprですEMSAバンドのさらなるシフト、またはバンドの損失としてesented(参照28に概説されています)。相補DAPAにおいて、変異体および20塩基対の隣接ヌクレオチドを含む5 'ビオチン化オリゴ二本鎖を特異的オリゴを結合、核因子を捕捉するために、関連する細胞型(複数可)からの核溶解物と共にインキュベートします。オリゴ二重核因子複合体を磁気カラムにビーズをストレプトアビジンにより固定されています。バインドされた核要因が溶出29,48を介して直接収集されます。結合予測は、その後、タンパク質に特異的な抗体を用いたウェスタンブロットによって評価することができます。明らかな予測、またはあまりにも多くの予測が存在しない場合には、DAPA実験の変異プルダウンからの溶出は、質量分析を使用して候補のTFを同定するためにプロテオミクスコアに送信することができ、続いて前述のこれらを使用して検証することができますメソッド。
articlの残りの部分でE、EMSA及び遺伝的変異のDAPA分析のための詳細なプロトコルが提供されます。
プロトコル
ソリューションおよび試薬の調製
- EMSAとDAPAで使用するためのカスタムDNAオリゴヌクレオチドプローブを注文してください。
- 、非特異的タンパク質結合を減少させる(長さ35から45塩基対(bp)の間の)短いオリゴ30を設計し、その17 bpの内在性ゲノム配列によって挟ま中心部に関心の変形を配置します。 EMSAオリゴについては、5 '蛍光団を追加します。 DAPAオリゴについては、5 'ビオチンタグを追加します。
- センス鎖およびその逆相補鎖の両方を注文。また、二重(予備アニール)オリゴを注文。オリゴに名前を付けるときは、確立された基準ゲノム上の命名法の基礎としています。
注:「リスク」と「基準」と「非参照」は、より普遍的に関連している一方で、「非危険」指定は、病気やプロジェクト固有することができます。 - オリゴの到着すると、簡単に100μの最終濃度にヌクレアーゼフリー水に内容を再懸濁をスピンダウン; M。ストアは、-20℃でストックを再懸濁しました。アルミホイルで包むことにより光からフォアでタグ付けされたオリゴを保護します。
| 名 | シーケンス |
| rs76562819_REF_FOR | GTAATGCCTTAATGAGAGAG A GTTAGTCATCTTCTCACTTC |
| rs76562819_REF_REV | GAAGTGAGAAGATGACTAAC T CTCTCTCATTAAGGCATTAC |
| rs76562819_NONREF_FOR | GTAATGCCTTAATGAGAGAG G GTTAGTCATCTTCTCACTTC |
| rs76562819_NONREF_REV | GAAGTGAGAAGATGACTAAC C CTCTCTCATTAAGGCATTAC |
表1:差動ビンのSNPをテストする例EMSA / DAPAオリゴヌクレオチドの設計「NONREFは「非参照対立遺伝子を表しながら、鼎。「REF」は、参照対立遺伝子を意味します。 「FOR」「REV」は、その補数を示しながら、フォワード鎖を意味します。 SNPは赤で見られます。
- 最終10mMのHEPES(pHは7.9)の濃度が、10のKCl、および脱イオン水に0.1 mMのEDTAで細胞質抽出(CE)のバッファを準備します。
- 脱イオン水中20mMのHEPES(pHは7.9)、0.4 MのNaCl、および1mMのEDTAの最終濃度でバッファ核抽出(NE)を準備します。
- 脱イオン水中の10 mMトリス(pHは7.5〜8.0)、50mMのNaCl、および1mMのEDTAの最終濃度でアニーリングバッファーを準備します。
培養細胞からの核溶解物の調製
注:この実験プロトコールは、Bリンパ芽球様細胞株を用いて最適化したが、いくつかの他の無関係な付着/懸濁細胞系において試験されており、普遍的に動作します。
- Cultu2mMのL-グルタミン、10%ウシ胎児血清、およびペニシリンの100単位/ ml、ストレプトマイシン100μg/ mlを含む1×抗生物質 - 抗真菌、および250 ngのロズウェルパーク記念研究所(RPMI)1640中のBリンパ芽球様細胞の再アンホテリシンB / mlの
- 200,000-500,000生存細胞/ mlの範囲で種子及び通気又はルーズキャップを直立位置に5%二酸化炭素、37℃でフラスコをインキュベートします。
注:彼らは1,000,000 cells / mlに達したときに、Bリンパ芽球様細胞の成長が遅くなります。ピペッティングにより細胞ブラストを破壊し、上下に数回と成長の急速な速度を維持するために、200,000-500,000細胞/ mlに細胞を返します。
- 200,000-500,000生存細胞/ mlの範囲で種子及び通気又はルーズキャップを直立位置に5%二酸化炭素、37℃でフラスコをインキュベートします。
- 5分間、4℃で300×gでスピンダウンし、吸引を介してPBSを除去し、二回10ミリリットルの氷冷リン酸緩衝生理食塩水(PBS)で培養した細胞を洗浄。
- 10 7個の細胞あたり1ミリリットルの氷冷PBSとして、血球計数器を再懸濁ペレットを用いて細胞を数えます。
注:例えば、2×10 7を溶解した場合</ SUP>細胞、2mlのPBSで再懸濁。 - アリコート1.5ミリリットルのマイクロチューブに1ミリリットル各チューブは、PBS中の10 7個の細胞を含むように。 PBSオフ2分4°Cと吸引用の3300×gで遠心分離します。
- 使用CEバッファのワーキングストックを、1mMのジチオスレイトール(DTT)、1×ホスファターゼ阻害剤、および1×プロテアーゼ阻害剤を添加する前に。 CEバッファー400μlので再懸濁細胞ペレットを、15分間氷上でインキュベートします。
- 10%のNonidet P-40の25μl加え、ピペッティングにより混合します。 4°C、3分間、最大速度で遠心分離します。デカントして上清を捨てます。
- 使用前に、NEバッファのワーキングストックに1 mMのDTT、1×ホスファターゼ阻害剤、および1×プロテアーゼ阻害剤を追加します。 NE緩衝液30μlで再懸濁細胞ペレットを、ボルテックスで混和します。
- チューブローテーターにおいてまたは氷上で10分間4℃でインキュベートします。 2分4°Cのための遠心分離機3300×gで。
- -80で保存する前に透明な上清(核溶解液)、アリコートを収集6; Cは、タンパク質を分解することができる複数の凍結融解サイクルを回避します。 bichoninic酸アッセイ(BCA)31を用いてタンパク質濃度を測定するために、10μlのアリコートを残します。
3.電気泳動移動度シフトアッセイ(EMSA)
- オリゴワーキングストックとEMSAゲルを準備します。
- オリゴが二重に発注された場合は、100μMのストックを解凍し、1を希釈:2000をアニーリング緩衝液中で50 nMの作業ストックを達成するために。
- オリゴは、一本鎖発注された場合は、100μMの株式を解凍し、100 nMの作業の株式を達成するために、アニーリング緩衝液で1:10に希釈します。マイクロ遠心チューブ内で相互に100 nMの相補鎖溶液100μlを組み合わせます。
- 95℃で5分間加熱ブロックに置き。ヒートブロックの電源をオフにして、オリゴはゆっくりと、使用前に少なくとも1時間、室温まで冷却することができます。
- EMSAゲルを事前に実行します。
- プレキャスト6%Tからスライドを削除しますゲルBEと井戸から任意の緩衝液を除去するために脱イオン水の下で数回すすぎます。脱イオン水950ミリリットルに10倍TBEの50ミリリットルを追加することにより、0.5×TBE緩衝液1リットルを準備します。
- ゲル電気泳動装置を組み立て、0.5×TBE緩衝液を用いて、内部チャンバを充填することによって漏れをチェックします。何のバッファが外側室に漏れるない場合は、おおよその方法の三分の二を外側室を埋めます。
- 60分間、100Vでゲルを事前に実行します。
- 0.5×TBE緩衝液200μlで各ウェルを洗浄します。
- 結合バッファーマスターミックスを調製します。
- 100 mMトリス、500 mMの塩化カリウム、10mMのDTTの最終濃度で10×結合緩衝液を準備します。脱イオン水のpH 7.5。
- マイクロ遠心チューブでは、すべての反応(; 表210μlの10×結合緩衝液、10μlのDTT /ポリソルベート、5μlのポリD(IC)、および2.5μlのサケ精子DNA)に共通の試薬 からなるマスターミックスを作成します。追加の10%を準備ピペッティングによる体積損失を考慮します。
| 試薬 | 最終濃度。 | #1 RXN | #2 RXN | #3 RXN | #4 RXN |
| 超純水 | 20μlの容量に。 | 13.5μlの | 11.98μlの | 13.5μl | 11.98μlの |
| 10×結合緩衝液 | 1倍 | 2μlの | 2μlの | 2μlの | 2μlの |
| DTT / TW-20 | 1倍 | 2μlの | 2μlの | 2μlの | 2μlの |
| サケ精子DNA | 500 ngの/μlの | 0.5μlの | 0.5μlの | 0.5μlの | 0.5μlの |
| 1μgの/μlのポリD(IC) | 1μgの | 1μlの | 1μlの | 1μlの | 1μlの |
| 核抽出(5.26 UG /μl)を | 8μgの | - | 1.52μlの | - | 1.52μlの |
| NEバッファー | 1.52μlの | - | 1.52μlの | - | |
| 参考アレルオリゴ | 50 fmolで | 1μlの | 1μlの | - | - |
| 非参照アレルオリゴ | 50 fmolで | - | - | 1μlの | 1μlの |
表2:例EMSA反応setu頁の表は、EMSAは、遺伝子型に依存する特定のSNPへのTFの結合が存在するという仮説をテストする例を示しています。
- 全ての試薬を添加した後の最終容量は20μlとなるような各マイクロチューブにヌクレアーゼフリー水を追加します。
- 各マイクロチューブにマスターミックスの適切な量(5.5μl)を追加します。
- 適切なマイクロ遠心チューブに核溶解物の8μgのを追加します。核陰性対照として抽出物( 例えば、 表2、RXN#1とRXN#3)なしでオリゴを含むチューブを含めます。
注:反応あたりの溶解物の最適量は滴定により実験的に決定されなければなりません。一般的に、溶解物の2-10μgの範囲を滴定することで十分です。 - 適切なマイクロ遠心チューブにオリゴの50 fmolでを追加します。混ぜると簡単にチューブの底に内容を回転させるフリック。室温で20分間インキュベートします。
注:attempti場合スーパーシフトngを、オリゴの添加前に室温で20分間、抗体と溶解物混合物をインキュベートします。最良の結果を得るためのChIPグレード抗体を1μgを使用することをお勧めします。 - 各マイクロチューブに10倍オレンジローディング色素の2μLを加えます。アップピペットとミックスダウンします。
- ピペッティング混合すると、その後別々のウェルに各サンプルを排出することにより、プレラン6%TBEゲルにサンプルをロードします。オレンジ色の色素がゲルダウン道の3/4に2/3を移動するまで80 Vでゲルを実行します。これは、約60-75分を取る必要があります。
- ゲルナイフでそれをこじ開けによりプラスチックカセットからゲルを外し、乾燥からそれを維持するために0.5%TBE緩衝液を用いて容器にゲルを置きます。
- 画像を混乱させるだろう任意の気泡や汚染物質を排除することを確認され、赤外線および化学発光イメージングシステムの表面上にゲルを置きます。
- 走査システムソフトウェアを使用して、「獲得」タブをクリックしますそしてその後、ゲルをスキャナの表面上に配置されている場所に対応する領域の周りにボックスを描画するために「新を描く」を選択します。
- 「獲得」タブの「チャネル」セクションでは、オリゴ上のフルオロフォアタグの波長に対応するチャネルを選択します。 「スキャナ」セクションでは、低品質のプレビュースキャンを取得するには、「プレビュー」をクリックします。撮像すべきゲルの部分まで取得したプレビュー画像を囲む青いボックスをドラッグしてスキャン領域を調整します。
注:たとえば、700 nmの蛍光団で標識されたオリゴを使用している場合は、「700 nmの "チャンネルがスキャン前に選択されていることを確認。 - 「スキャンコントロール」セクションで、「84μM」解決オプションと「中」品質オプションを選択します。ゲルの半分の厚さに相殺するためにフォーカスを設定します。注:たとえば、1ミリメートルゲルは、オフセット0.5ミリメートルフォーカスを使用します。
- 「スキャナ」セクションでは、Bに「スタート」をクリックスキャンをegin。
注:スキャン中、明るさ、コントラスト、カラースキームは、多くの場合、走査システムの製造者によって手動で調整することができます。 - スキャンが終了した後、「イメージ」タブを選択し、向きを修正するために、「作成」セクションの「回転または反転」をクリックします。メインメニューの「エクスポート」をクリックすることで、画像ファイルを保存し、選択し、 "シングルイメージビューを。」
4. DNAアフィニティー精製アッセイ(DAPA)
- 5μMオリゴワーキングストックの調製。
- オリゴが二重に発注された場合は、100μMのストックを解凍し、5μMワーキングストックを達成するために、アニーリングバッファー中で1:20希釈します。
- オリゴは、一本鎖発注された場合は、100μMの株式を解凍し、株式を作業、10μMを達成するために、アニーリング緩衝液で1:10に希釈します。互いに10μMの相補鎖の10μLを兼ね備えています。 95℃のヒートブロックに配置します5分。ヒートブロックの電源をオフにして、オリゴはゆっくりと使用前に室温まで冷却することができます。
- 開始する前に、室温に結合緩衝液、低ストリンジェンシー洗浄緩衝液、高ストリンジェンシー洗浄バッファー、および溶出バッファーを温めます。
注:50ngの最終濃度/ mLのポリD(IC)は、オリゴへのタンパク質の潜在的な非特異的結合を減少させるために、結合緩衝液、低ストリンジェンシー洗浄緩衝液、および高ストリンジェンシー洗浄緩衝液に添加することができます。 - 各変異体の結合混合物を準備します。
- 結合緩衝液の2倍量の核溶解物の1ボリュームを混ぜます。
注:溶解液の必要量が原因のTFの存在量を変化させることに実験的に決定されなければなりません。列ごとに、核ライセートμgの100から250の間を使うことで、ほとんどの場合は十分です。 - 1×ホスファターゼ阻害剤、1×プロテアーゼ阻害剤、および1×結合エンハンサー(オプション)を追加し、チューブを数回フリックすることにより混合します。
注:100X結合エンハンサーは、750のMgCl 2、300mMのZnCl 2を構成されています。 DNAへのTFの結合は、補因子または還元剤に依存している場合、結合エンハンサーを追加します。この情報は知られていない場合は、結合促進剤を加えます。 - それぞれの結合混合物に5μMビオチン化された捕捉DNA(50ピコモル)の10μLを加えます。室温で20分間インキュベートします。
注意:インキュベーションの時間および温度は、TFに応じて変化し得ます。最適値は、実験的に決定される必要があります。
- 結合緩衝液の2倍量の核溶解物の1ボリュームを混ぜます。
- ストレプトアビジンマイクロビーズ100μlのを追加します。室温で10分間インキュベートします。
- 各オリゴプローブは、テストされているために、磁気分離器に結合列を配置します。各結合列の下に直接マイクロチューブを置き、カラムを洗浄するために結合緩衝液100μlを適用します。
- 別々の列に各結合混合物の内容をピペットで、液体が微量にカラムを通って完全に流れることを可能にします先に進む前にチューブ。結合混合物中で使用された変異型オリゴ列にラベルを付けていることを確認してください。フロースルーサンプルにラベルを付け、低ストリンジェンシー洗浄を収集するために、新しいマイクロ遠心チューブと交換してください。
- カラムへの低ストリンジェンシー洗浄バッファー100μlを適用します。列のリザーバが空になるまで待ちます。洗浄4倍を繰り返します。低ストリンジェンシー洗浄のサンプルにラベルを付け、高ストリンジェンシー洗浄を収集するために、新しいマイクロ遠心チューブと交換してください。
- カラムに高ストリンジェンシー洗浄バッファー100μlを適用します。列のリザーバが空になるまで待ちます。洗浄4倍を繰り返します。高ストリンジェンシーの洗浄サンプルにラベルを付け、プリ溶出を収集するために、新しいマイクロ遠心チューブと交換してください。
- 列にネイティブの溶出緩衝液30μlを加え、5分間放置しました。プレ溶出サンプルにラベルを付け、溶出を収集するために、新しいマイクロ遠心チューブと交換してください。
注:これは、結合したタンパク質を溶出しません。それは残りの高ストリンジェンシーを洗いますカラムから緩衝液および溶出の効率を最大にするために、溶出緩衝液に置き換え。 - バインドされたTFを溶出するために追加の50μlのネイティブの溶出バッファーを追加します。より高い収率が、あまり集中した溶出液については、ネイティブの溶出バッファーの追加の50μlを添加し、フロースルーを収集します。
注:バインドされたTF 32の同一性を決定するために質量分析を介して、溶出サンプルを分析します。その後、ウエスタンブロット33に続くドデシルナトリウム亜硫酸ポリアクリルアミドゲル電気泳動(SDS-PAGE)を介してプロテオミクスの結果を検証します。質量分析を使用できない場合は、遺伝子型依存性結合を示すタンパク質のサイズを決定する代わりに、ウエスタンブロットを、標準的な技術を用いて銀染色を実行します。導入に詳述した計算のアプローチからの予測TFの一覧を絞り込むために、この情報を使用してください。
結果
このセクションでは、EMSAまたはDAPAを実行するときに何を期待するの代表的な結果が提供され、そして溶解物の品質に関して変動性が特徴です。例えば、複数回の凍結融解のタンパク質試料は変性をもたらし得ることが示唆されています。これらの「凍結融解」サイクルの文脈においてEMSA分析の再現性を調査するために、1遺伝子変異体で異なる2 35 bpのオリゴが指示?...
ディスカッション
シーケンシングおよびジェノタイピング技術の進歩は著しく、疾患に関連する遺伝子変異体を同定するために我々の能力を強化しているが、これらの変異体によって影響を受ける機能のメカニズムを理解するために我々の能力が遅れています。問題の主な原因は、多くの疾患関連変異体は、上のコード可能性が高い遺伝子発現を制御困難ツー予測メカニズムに影響を与えるゲノムの領域?...
開示事項
著者らは、開示することは何もありません。
謝辞
We thank Erin Zoller, Jessica Bene, and Lindsey Hays for input and direction in protocol development. MTW was supported in part by NIH R21 HG008186 and a Trustee Award grant from the Cincinnati Children's Hospital Research Foundation. ZHP was supported in part by T32 GM063483-13.
資料
| Name | Company | Catalog Number | Comments |
| Custom DNA Oligonucleotides | Integrated DNA Technologies | http://www.idtdna.com/site/order/oligoentry | |
| Potassium Chloride | Fisher Scientific | BP366-500 | KCl, for CE buffer |
| HEPES (1 M) | Fisher Scientific | 15630-080 | For CE and NE buffer |
| EDTA (0.5M), pH 8.0 | Life Technologies | R1021 | For CE, NE, and annealing buffer |
| Sodium Chloride | Fisher Scientific | BP358-1 | NaCl, for NE buffer |
| Tris-HCl (1M), pH 8.0 | Invitrogen | BP1756-100 | For annealing buffer |
| Phosphate Buffered Saline (1x) | Fisher Scientific | MT21040CM | PBS, for cell wash |
| DL-Dithiothreitol solution (1 M) | Sigma | 646563 | Reducing agent |
| Protease Inhibitor Cocktail | Thermo Scientific | 87786 | Prevents catabolism of TFs |
| Phosphatase Inhibitor Cocktail | Thermo Scientific | 78420 | Prevents dephosphorylation of TFs |
| Nonidet P-40 Substitute | IBI Scientific | IB01140 | NP-40, for nuclear extraction |
| BCA Protein Assay Kit | Thermo Scientific | 23225 | For measuring protein concentration |
| Odyssey EMSA Buffer Kit | Licor | 829-07910 | Contains all necessary EMSA buffers |
| TBE Gels, 6%, 12 Wells | Invitrogen | EC6265BOX | For EMSA |
| TBE Buffer (10x) | Thermo Scientific | B52 | For EMSA |
| FactorFinder Starting Kit | Miltenyi Biotec | 130-092-318 | Contains all necessary DAPA buffers |
| Licor Odyssey CLx | Licor | Recommended scanner for DAPA/EMSA | |
| Antibiotic-Antimycotic | Gibco | 15240-062 | Contains 10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of Fungizone® Antimycotic |
| Fetal Bovine Serum | Gibco | 26140-079 | FBS, for culture media |
| RPMI 1640 Medium | Gibco | 22400-071 | Contains L-glutamine and 25 mM HEPES |
参考文献
- Hindorff, L. A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 106 (23), 9362-9367 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Ward, L. D., Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 30 (11), 1095-1106 (2012).
- Paul, D. S., Soranzo, N., Beck, S. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays. 36 (2), 191-199 (2014).
- Zhang, F., Lupski, J. R. Non-coding genetic variants in human disease. Hum Mol Genet. , (2015).
- Lee, T. I., Young, R. A. Transcriptional regulation and its misregulation in disease. Cell. 152 (6), 1237-1251 (2013).
- Slatkin, M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 9 (6), 477-485 (2008).
- Bush, W. S., Moore, J. H. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 8 (12), e1002822 (2012).
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 491 (7422), 56-65 (2012).
- Chang, C. C., et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
- Purcell, S., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81 (3), 559-575 (2007).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16 (1), 123-131 (2006).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 10 (12), 1213-1218 (2013).
- Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R., Lieb, J. D. FAIRE Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17 (6), 877-885 (2007).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Martens, J. H., Stunnenberg, H. G. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 98 (10), 1487-1489 (2013).
- Liu, T., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12 (8), R83 (2011).
- Griffon, A., et al. Integrative analysis of public ChIP-seq experiments reveals a complex multi-cell regulatory landscape. Nucleic Acids Res. 43 (4), e27 (2015).
- Staudt, L. M., et al. A lymphoid-specific protein binding to the octamer motif of immunoglobulin genes. Nature. 323 (6089), 640-643 (1986).
- Singh, H., Sen, R., Baltimore, D., Sharp, P. A. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature. 319 (6049), 154-158 (1986).
- Weirauch, M. T., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 158 (6), 1431-1443 (2014).
- Ward, L. D., Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40 (Database issue), D930-D934 (2012).
- Boyle, A. P., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22 (9), 1790-1797 (2012).
- Hume, M. A., Barrera, L. A., Gisselbrecht, S. S., Bulyk, M. L. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 43 (Database issue), D117-D122 (2015).
- Mathelier, A., et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42 (Database issue), 142-147 (2014).
- Smith, M. F., Delbary-Gossart, S. Electrophoretic Mobility Shift Assay (EMSA). Methods Mol Med. 50, 249-257 (2001).
- Franza, B. R., Josephs, S. F., Gilman, M. Z., Ryan, W., Clarkson, B. Characterization of cellular proteins recognizing the HIV enhancer using a microscale DNA-affinity precipitation assay. Nature. 330 (6146), 391-395 (1987).
- . BCA Protein Assay Kit: User Guide Available from: https://tools.thermofisher.com/content/sfs/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf (2014)
- Wijeratne, A. B., et al. Phosphopeptide separation using radially aligned titania nanotubes on titanium wire. ACS Appl Mater Interfaces. 7 (21), 11155-11164 (2015).
- Silva, J. M., McMahon, M. The Fastest Western in Town: A Contemporary Twist on the Classic Western Blot Analysis. J. Vis. Exp. (84), (2014).
- Lu, X., et al. Lupus Risk Variant Increases pSTAT1 Binding and Decreases ETS1 Expression. Am J Hum Genet. 96 (5), 731-739 (2015).
- Ramana, C. V., Chatterjee-Kishore, M., Nguyen, H., Stark, G. R. Complex roles of Stat1 in regulating gene expression. Oncogene. 19 (21), 2619-2627 (2000).
- Fillebeen, C., Wilkinson, N., Pantopoulos, K. Electrophoretic Mobility Shift Assay (EMSA) for the Study of RNA-Protein Interactions: The IRE/IRP Example. J. Vis. Exp. (94), e52230 (2014).
- Heng, T. S., Painter, M. W. Immunological Genome Project, C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 9 (10), 1091-1094 (2008).
- Wu, C., et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10 (11), R130 (2009).
- Wu, C., Macleod, I., Su, A. I. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 41 (Database issue), D561-D565 (2013).
- Wang, J., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22 (9), 1798-1812 (2012).
- Holden, N. S., Tacon, C. E. Principles and problems of the electrophoretic mobility shift assay. J Pharmacol Toxicol Methods. 63 (1), 7-14 (2011).
- Xu, J., Liu, H., Park, J. S., Lan, Y., Jiang, R. Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development. 141 (7), 1442-1452 (2014).
- Yang, T. -. P., et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 26 (19), 2474-2476 (2010).
- Fort, A., et al. A liver enhancer in the fibrinogen gene cluster. Blood. 117 (1), 276-282 (2011).
- Solberg, N., Krauss, S. Luciferase assay to study the activity of a cloned promoter DNA fragment. Methods Mol Biol. 977, 65-78 (2013).
- Rahman, M., et al. A repressor element in the 5'-untranslated region of human Pax5 exon 1A. Gene. 263 (1-2), 59-66 (2001).
- Mali, P., et al. RNA-Guided Human Genome Engineering via Cas9. Science. 339 (6121), 823-826 (2013).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved