
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Le dépistage de la fonctionnelle non-codant des variants génétiques utilisant mobilité électrophorétique Maj Assay (EMSA) et l'ADN-affinité Précipitations Assay (DAPA)
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
We present a strategic plan and protocol for identifying non-coding genetic variants affecting transcription factor (TF) DNA binding. A detailed experimental protocol is provided for electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genotype-dependent TF DNA binding.
Résumé
Population and family-based genetic studies typically result in the identification of genetic variants that are statistically associated with a clinical disease or phenotype. For many diseases and traits, most variants are non-coding, and are thus likely to act by impacting subtle, comparatively hard to predict mechanisms controlling gene expression. Here, we describe a general strategic approach to prioritize non-coding variants, and screen them for their function. This approach involves computational prioritization using functional genomic databases followed by experimental analysis of differential binding of transcription factors (TFs) to risk and non-risk alleles. For both electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genetic variants, a synthetic DNA oligonucleotide (oligo) is used to identify factors in the nuclear lysate of disease or phenotype-relevant cells. For EMSA, the oligonucleotides with or without bound nuclear factors (often TFs) are analyzed by non-denaturing electrophoresis on a tris-borate-EDTA (TBE) polyacrylamide gel. For DAPA, the oligonucleotides are bound to a magnetic column and the nuclear factors that specifically bind the DNA sequence are eluted and analyzed through mass spectrometry or with a reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blot analysis. This general approach can be widely used to study the function of non-coding genetic variants associated with any disease, trait, or phenotype.
Introduction
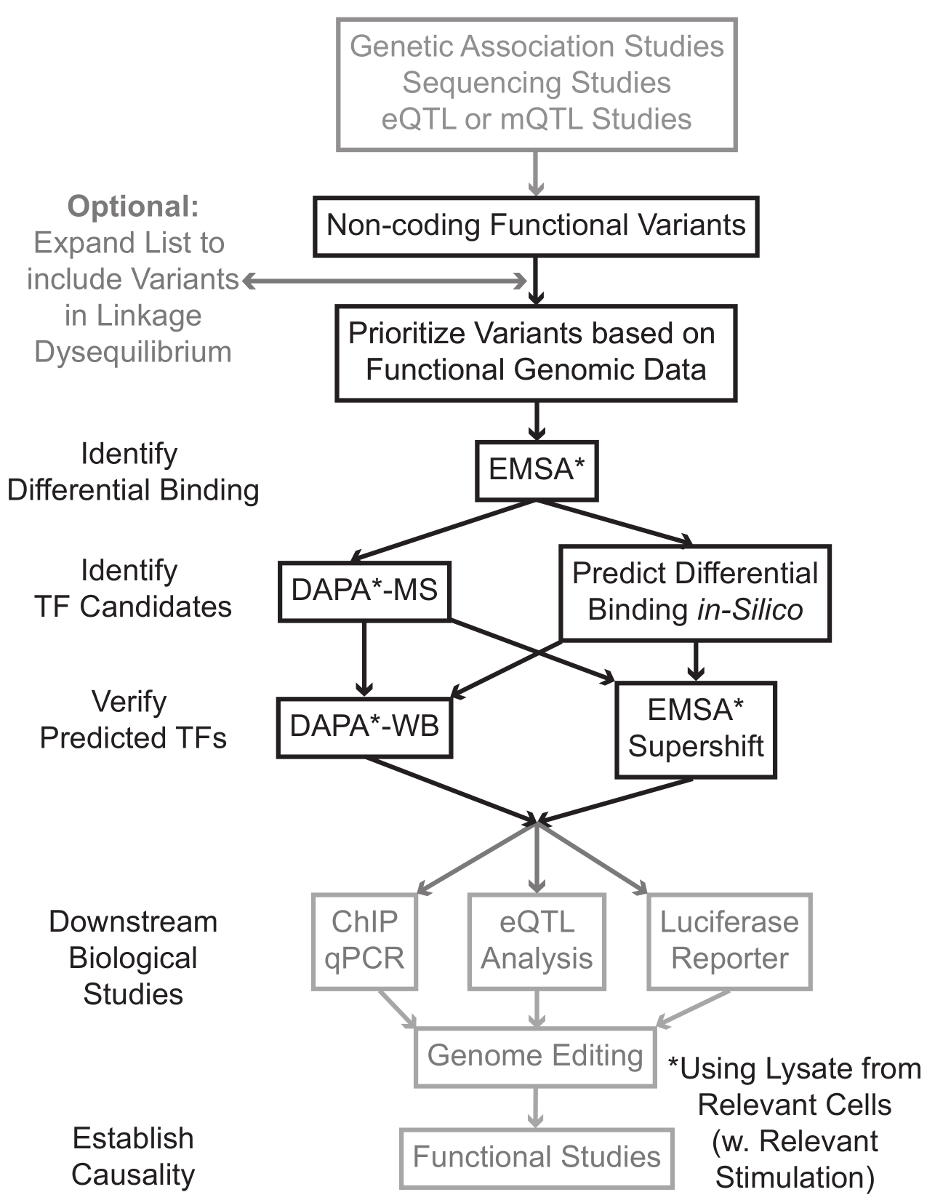
Des études de séquençage et de génotypage à base d'études, y compris l'ensemble du génome des études d'association (GWAS), des études de locus candidat, et profond de séquençage, ont identifié de nombreuses variantes génétiques qui sont statistiquement associés à une maladie, trait, ou d'un phénotype. Contrairement aux prévisions initiales, la plupart de ces variantes (85-93%) sont situées dans des régions non codantes et ne changent pas la séquence d'acides aminés des protéines 1,2. L' interprétation de la fonction de ces variantes non codantes et déterminer les mécanismes biologiques qui les relient à la maladie associée, trait ou un phénotype a révélé difficile 3-6. Nous avons développé une stratégie générale visant à identifier les mécanismes moléculaires qui lient les variantes à une importante phénotype intermédiaire - l'expression du gène. Ce pipeline est spécifiquement conçu pour identifier la modulation de la liaison par des variants génétiques TF. Cette stratégie combine les approches de calcul et des techniques de biologie moléculaire visant à prédireeffets biologiques des variants candidats in silico, et vérifier ces prédictions empiriquement (Figure 1).

Figure 1:.. Approche stratégique pour l'analyse des étapes non codantes variantes génétiques qui ne sont pas inclus dans le protocole détaillé associé à ce manuscrit sont en gris S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Dans de nombreux cas, il est important de commencer en élargissant la liste des variantes à inclure tous ceux en haute liaison-déséquilibre (LD) avec chaque variante statistiquement associée. LD est une mesure d'association non aléatoire des allèles à deux positions chromosomiques différentes, qui peut être mesurée par la r 2 statistique 7. r 2 est une mesure du linkage déséquilibre entre deux variantes, avec un r 2 = 1 désignant liaison parfaite entre deux variantes. Allèles en haute LD se trouvent à la co-ségrégation sur le chromosome à travers les populations ancestrales. tableaux de génotypage actuels ne comprennent pas toutes les variantes connues dans le génome humain. Au lieu de cela, ils exploitent la LD dans le génome humain et comprennent un sous - ensemble des variantes connues qui agissent comme substituts pour les autres variantes dans une région particulière de LD 8. Ainsi, une variante sans aucune conséquence biologique peut être associée à une maladie particulière, car il est en LD avec la variante-causal variante avec un effet biologique significatif. La procédure, il est recommandé de convertir la dernière version de 1000 génomes projeter 9 fichiers variante d'appel (VCF) dans des fichiers binaires compatibles avec PLINK 10,11, un outil open-source pour toute analyse d'association du génome. Par la suite, toutes les autres variantes génétiques avec LD r 2> 0,8 avec chacun va génétique d'entréeRiant peut être identifié en tant que candidats. Il est important d'utiliser la population de référence appropriée pour ce beau - par exemple, si un variant a été identifié chez les sujets d'origine européenne, des données provenant de sujets d'ascendance similaires devraient être utilisés pour l' expansion de LD.
l'expansion LD se traduit souvent par des dizaines de variantes de candidats, et il est probable que seule une petite fraction de ceux-ci contribuent au mécanisme de la maladie. Souvent, il est impossible d'étudier expérimentalement chacune de ces variantes individuellement. Il est donc utile de tirer parti des milliers de jeux de données accessibles au public de génomique fonctionnelle comme un filtre pour prioriser les variantes. Par exemple, le consortium ENCODE 12 a effectué des milliers d'expériences de ChIP-seq décrivant la liaison de TFs et co-facteurs, et les marques de histones dans un large éventail de contextes, ainsi que des données chromatine d'accessibilité des technologies telles que DNase-seq 13, ATAC -seq 14 et suivants 15-FAIRE. Databases et les serveurs Web tels que le navigateur UCSC Genome 16, Feuille de route Epigenomics 17, Blueprint Epigénome 18, cistrome 19 et ReMap 20 offrent un accès gratuit aux données produites par ces autres techniques expérimentales et à travers un large éventail de types et conditions cellulaires. Quand il y a trop de variantes pour examiner expérimentalement, ces données peuvent être utilisées pour établir des priorités de ceux situés dans les régions régulatrices probables dans les types de cellules et de tissus concernés. En outre, dans les cas où une variante est dans un pic de ChIP-seq pour une protéine spécifique, ces données peuvent fournir des pistes potentielles à la TF (s) spécifique ou co-facteurs dont la liaison pourrait affecter.
Ensuite, les variants résultants en priorité sont sélectionnés expérimentalement pour valider la liaison en utilisant EMSA 21,22 prédit une protéine dépendante du génotype. EMSA mesure la variation de la migration de l'oligonucléotide sur un gel TBE non réducteur. oligo- marqué par fluorescence est mis en incubation avec lelysat nucléaire, et la liaison des facteurs nucléaires retarder le mouvement de l'oligo sur le gel. De cette manière, oligo qui a lié des facteurs plus nucléaires présentera comme un signal fluorescent plus fort lors de la numérisation. Notamment, l'EMSA ne nécessite pas des prédictions sur les protéines spécifiques dont la liaison sera affectée.
Une fois que les variantes sont identifiées qui sont situés dans des régions régulatrices prévues et sont capables de facteurs nucléaires différentiellement contraignants, les méthodes de calcul sont utilisées pour prédire la TF spécifique (s) dont la liaison qu'ils pourraient affecter. Nous préférons utiliser CIS-BP 23,24, RegulomeDB 25, UNIProbe 26 et JASPAR 27. Une fois que le candidat TFs sont identifiés, ces prévisions peuvent être spécifiquement testées en utilisant des anticorps contre ces TFs (EMSA-supershifts et DAPA-Westerns). Une EMSA-supershift implique l'addition d'un anticorps spécifique TF au lysat nucléaire et oligo. Un résultat positif dans un EMSA-supershift est réédesented comme un nouveau changement dans la bande EMSA, ou une perte de la bande (examiné en référence 28). Dans le DAPA complémentaire, d'un duplex oligonucléotidique 5'-biotinylée contenant de la variante et la 20 paire de bases de nucléotides flanquantes sont mises en incubation avec un lysat nucléaire du type de cellule approprié (s) pour capturer des facteurs nucléaires se liant spécifiquement oligos. Le complexe facteur nucléaire duplex oligonucléotide est immobilisé par des microbilles de streptavidine dans une colonne magnétique. Les facteurs liés nucléaires sont collectés directement par élution 29,48. prédictions de liaison peuvent alors être évalués par un transfert de Western en utilisant des anticorps spécifiques de la protéine. Dans les cas où il n'y a pas de prédictions évidentes, ou trop de prédictions, les élutions de variantes pull-downs des expériences de DAPA peuvent être envoyés à un noyau de protéomique pour identifier les TFs candidats en utilisant la spectrométrie de masse, qui peuvent ensuite être validées en utilisant ces décrits précédemment méthodes.
Dans la suite de l'article, le protocole détaillé pour EMSA et DAPA analyse des variants génétiques est fourni.
Protocole
1. Préparation des solutions et réactifs
- Commandez des sondes ADN d'oligonucléotides personnalisés pour une utilisation dans EMSA et DAPA.
- Pour réduire la protéine liaison non spécifique, concevoir des oligos courts (entre 35-45 paires de bases (pb) de longueur) 30, et placer la variante d'intérêt dans le centre flanqué de sa séquence génomique endogène 17 pb. Pour oligos EMSA, ajoutez un 'fluorophore 5. Pour oligos DAPA, ajouter une balise 5 'de la biotine.
- Commander à la fois le brin sens et son complément inverse brin. Sinon, l'ordre duplex (pré-recuites) oligos. Lorsque vous nommez les oligos, fonder la nomenclature sur un génome de référence établie.
Note: Le «risque» et la désignation «non-risque» peut être la maladie et projet spécifique, tandis que «référence» et «non-référence" sont plus universellement pertinents. - À l'arrivée des oligos, centrifuger brièvement vers le bas le contenu et les remettre en suspension dans l'eau sans nucléase à une concentration finale de 100 μ; M. Magasin resuspendu stock à -20 ° C. Protéger oligos marqués avec un fluorophore de la lumière en enroulant une feuille d'aluminium.
| prénom | Séquence |
| rs76562819_REF_FOR | GTAATGCCTTAATGAGAGAG A GTTAGTCATCTTCTCACTTC |
| rs76562819_REF_REV | GAAGTGAGAAGATGACTAAC T CTCTCTCATTAAGGCATTAC |
| rs76562819_NONREF_FOR | GTAATGCCTTAATGAGAGAG G GTTAGTCATCTTCTCACTTC |
| rs76562819_NONREF_REV | GAAGTGAGAAGATGACTAAC C CTCTCTCATTAAGGCATTAC |
Tableau 1: Exemple EMSA / DAPA conception d'oligonucléotides pour tester un SNP pour bin différentielding. "REF" signifie l'allèle de référence, tandis que "NONREF" signifie l'allèle non-référence. "POUR" représente le brin avant, tandis que "REV" indique son complément. Le SNP est vu en rouge.
- Préparer l'extraction cytoplasmique (CE), un tampon avec une concentration finale de 10 mM de HEPES (pH 7,9), 10 mM de KCl et 0,1 mM d'EDTA dans de l'eau déminéralisée.
- Préparer l'extraction nucléaire (NE) du tampon à une concentration finale de 20 mM de HEPES (pH 7,9), 0,4 M de NaCl et 1 mM d'EDTA dans de l'eau déminéralisée.
- Préparer un tampon d'hybridation à une concentration finale de 10 mM de Tris (pH 7,5 à 8,0), 50 mM de NaCl et 1 mM d'EDTA dans de l'eau déminéralisée.
2. Préparation de lysat nucléaire à partir des cellules cultivées
Remarque: Le protocole expérimental a été optimisé en utilisant des lignées de cellules lymphoblastoïdes B, mais a été testée dans plusieurs autres lignées cellulaires adhérentes non apparentées / suspension et fonctionne de manière universelle.
- Culture des cellules B lymphoblastoïdes à Roswell Memorial Institute Park (RPMI) 1640 avec 2 mM de L-glutamine, du sérum de veau fœtal à 10%, et 1 x antibiotique antimycotique contenant 100 unités / ml de pénicilline, 100 ug / ml de streptomycine et 250 ng / ml d'amphotéricine B.
- Semences dans une gamme de 200,000-500,000 cellules viables / ml et on incube les flacons à 37 ° C avec 5% de dioxyde de carbone en position verticale avec des bouchons ventilés ou en vrac.
Remarque: La croissance des cellules B lymphoblastoïdes ralentit quand elles atteignent plus de 1.000.000 cellules / ml. Brisez explosions de cellules par pipetage de haut en bas plusieurs fois et revenir cellules 200,000-500,000 cellules / ml pour maintenir un taux de croissance rapide.
- Semences dans une gamme de 200,000-500,000 cellules viables / ml et on incube les flacons à 37 ° C avec 5% de dioxyde de carbone en position verticale avec des bouchons ventilés ou en vrac.
- Laver les cellules en culture deux fois avec 10 ml de glace froide phosphate saline tamponnée (PBS), centrifuger à 4 ° C, 300 g pendant 5 min et retirez PBS par aspiration.
- Compter les cellules en utilisant un hémocytomètre et remettre le culot en 1 ml PBS glacé par 10 7 cellules.
Remarque: Par exemple, si lyser 2 x 10 7 </ Sup> cellules, remettre en suspension dans 2 ml de PBS. - Aliquote de 1 ml à 1,5 ml microtubes de sorte que chaque tube contient 10 7 cellules dans du PBS. Centrifuger à 3300 xg pendant 2 min 4 ° C et Aspirer PBS.
- Avant utilisation, ajouter mM dithiothréitol 1 (DTT), inhibiteur de la phosphatase 1x et 1x inhibiteur de la protéase à un stock de travail de tampon de la CE. Resuspendre le culot de cellules avec 400 pi de tampon CE et laisser incuber sur de la glace pendant 15 minutes.
- Ajouter 25 ul de 10% de Nonidet P-40 et mélanger par pipetage. Centrifugeuse à 4 ° C, vitesse max pendant 3 min. Décanter et jeter le surnageant.
- Avant utilisation, ajouter mM DTT, inhibiteur de la phosphatase 1x et 1x inhibiteur 1 de la protéase à un stock de travail de tampon NE. Resuspendre le culot cellulaire avec 30 pi de tampon NE et mélanger par tourbillonnement.
- Incuber à 4 ° C dans un dispositif de rotation du tube ou sur la glace pendant 10 min. Centrifugeuse 3300 xg pendant 2 min 4 ° C.
- Recueillir le surnageant clair (lysat nucléaire) et aliquote avant de les stocker à -806; C afin d'éviter de multiples cycles de congélation-décongélation qui peuvent dégrader la protéine. Laisser un aliquote de 10 pi pour mesurer la concentration de protéines en utilisant le dosage de l' acide bichoninic (BCA) 31.
3. Mobilité électrophorétique Maj Assay (EMSA)
- Préparer Oligo Stock de travail et EMSA Gel.
- Si oligos ont été commandés en duplex, dégeler les 100 uM stock et diluer 1: 2000 dans un tampon de recuit pour obtenir un stock de travail de 50 nM.
- Si les oligos ont été commandés à simple brin, décongeler les stocks de 100 um et dilué 1:10 dans du tampon d'hybridation pour atteindre 100 valeurs de travail nm. Combiner 100 ul de la solution de complément de brin de 100 nm avec l'autre dans un microtube.
- Placez dans un bloc thermique à 95 ° C pendant 5 min. Désactiver le bloc thermique et permettent oligos refroidir lentement jusqu'à la température ambiante pendant au moins une heure avant utilisation.
- Pré-exécuter le Gel EMSA.
- Retirer la lame d'un pré-cast 6% TBE gel et rincer sous l'eau déminéralisée plusieurs fois pour éliminer tout tampon dans les puits. Préparer 1 litre de tampon 0,5 x TBE en ajoutant 50 ml de TBE 10x à 950 ml d'eau déminéralisée.
- Assembler l'appareil d'électrophorèse sur gel et vérifier les fuites en remplissant la chambre intérieure avec un tampon 0,5x TBE. Si aucun tampon fuit dans la chambre extérieure, remplir la chambre extérieure à peu près deux tiers du chemin.
- Pré-exécuter le gel à 100 V pendant 60 min.
- Rincer chaque puits avec 200 pi de tampon 0,5x TBE.
- Préparer Binding Buffer Master Mix.
- 10x préparer un tampon de liaison avec une concentration finale de 100 mM de Tris, 500 mM de KCl, 10 mM de DTT; pH 7,5 dans de l'eau déminéralisée.
- Dans un tube de microcentrifugeuse, créer un mélange maître constitué des réactifs communs à toutes les réactions (10 ul 10x tampon de liaison, 10 pi de DTT / polysorbate, 5 pi Poly d (IC), et d' ADN de sperme de saumon 2,5 pi; tableau 2). Préparer un montant supplémentaire de 10%pour tenir compte de la perte de volume due à pipetage.
| Réactif | Conc final. | Rxn n ° 1 | Rxn n ° 2 | Rxn n ° 3 | Rxn n ° 4 |
| eau ultrapure | 20 ul de vol. | 13,5 pi | 11.98 ul | 13.5μl | 11.98 ul |
| 10x Binding Buffer | 1 fois | 2 ul | 2 ul | 2 ul | 2 ul |
| DTT / TW-20 | 1 fois | 2 ul | 2 ul | 2 ul | 2 ul |
| ADN de sperme de saumon | 500 ng / ul | 0,5 ul | 0,5 ul | 0,5 ul | 0,5 ul |
| 1 pg / pl Poly d (IC) | 1 pg | 1 ul | 1 ul | 1 ul | 1 ul |
| Extrait nucléaire (5,26 ug / ul) | 8 pg | - | 1,52 pi | - | 1,52 pi |
| NE Buffer | 1,52 pi | - | 1,52 pi | - | |
| Référence allèle oligo | 50 fmol | 1 ul | 1 ul | - | - |
| Non-Référence allèle oligo | 50 fmol | - | - | 1 ul | 1 ul |
Tableau 2: Exemple EMSA réaction setup. Le tableau illustre un exemple EMSA pour tester l'hypothèse selon laquelle il est le génotype dépendant liaison de TFs à un SNP spécifique.
- Ajouter de l'eau sans nuclease à chaque microtube de telle sorte que le volume final après l'addition de tous les réactifs est de 20 ul.
- Ajouter la quantité appropriée (5,5 pi) de mélange maître à chaque tube de microcentrifugeuse.
- Ajouter 8 pg de lysat nucléaire aux microtubes appropriés. Inclure des tubes contenant l'oligo sans extrait nucléaire comme témoins négatifs (par exemple le tableau 2, Rxn # 1 et Rxn 3 #).
Note: La quantité optimale de lysat par réaction doit être déterminée expérimentalement par titrage. Généralement, titration une gamme de 2-10 pg de lysat est suffisante. - Ajouter 50 fmoles d'oligo aux microtubes appropriés. Flick pour mélanger et faire tourner brièvement le contenu au fond du tube. Incuber pendant 20 min à température ambiante.
Note: Si attempting un supershift, incuber le mélange de lysat avec l'anticorps pendant 20 minutes à température ambiante avant l'addition d'oligos. Il est recommandé d'utiliser 1 pg d'un anticorps ChIP de qualité pour de meilleurs résultats. - Ajouter 2 pi de 10x orange Loading Dye à chaque tube de microcentrifugeuse. Pipeter haut et en bas pour mélanger.
- Charger les échantillons dans le gel à 6% TBE pré-exécution par pipetage de haut en bas pour mélanger, puis expulser chaque échantillon dans un puits séparé. Exécutez le gel à 80 V jusqu'à ce que le colorant orange, a migré du 02.03 au 03.04 le chemin vers le bas du gel. Cela devrait prendre environ 60-75 min.
- Retirer le gel de la cassette en plastique en le soulevant avec un couteau de gel et placer le gel dans un récipient avec un tampon de TBE à 0,5% pour l'empêcher de se dessécher.
- Placer le gel sur la surface d'un système d'imagerie infrarouge et de chimioluminescence, en étant sûr d'éliminer toutes les bulles ou les contaminants qui perturbent l'image.
- Utilisation du logiciel de système de balayage, cliquez sur l'onglet "Acquisition"puis sélectionnez "Dessiner Nouveau" pour dessiner une zone autour de la zone correspondant à l'endroit où le gel est situé sur la surface du scanner.
- Dans la section "Chaînes" de l'onglet "Acquisition", sélectionnez le canal correspondant à la longueur d'onde de la balise fluorophore sur l'oligo. Dans la section "Scanner", cliquez sur "preview" pour obtenir un aperçu de la numérisation à faible qualité. Réglez la zone de numérisation en faisant glisser la boîte bleue qui entoure l'image de prévisualisation acquis jusqu'à la partie du gel à imager.
Remarque: Par exemple, si en utilisant les oligos marqués avec un fluorophore nm 700, assurez-vous que le canal "700 nm" est sélectionné avant la numérisation. - Dans la section "Commandes de numérisation", sélectionnez l'option "84 uM" résolution et l'option de qualité "moyenne". Définir le focus pour compenser la moitié de l'épaisseur du gel. Remarque: Par exemple, un gel de 1 mm serait d'utiliser un 0,5 mm de décalage de focalisation.
- Dans la section "Scanner", cliquez sur "Démarrer" à bEgin l'analyse.
Remarque: Lors de l'analyse, le régime luminosité, le contraste, et la couleur peut souvent être ajustée manuellement selon le fabricant du système de balayage. - Une fois l'analyse terminée, sélectionnez l'onglet "Image" et cliquez sur "Rotation ou Flip" dans la section "Créer" pour corriger l'orientation. Enregistrez le fichier d'image en cliquant sur "Exporter" dans le menu principal, puis sélectionnez "Image Single View."
4. ADN Purification par affinité de dosage (DAPA)
- Préparation de 5 uM Oligo travail Stock.
- Si oligos ont été commandés en duplex, dégeler les 100 uM stock et diluer 1:20 dans un tampon de recuit pour obtenir un stock de travail de 5 uM.
- Si les oligos ont été commandés à simple brin, décongeler les stocks de 100 um et dilué 1:10 dans du tampon d'hybridation pour obtenir des stocks 10 iM de travail. Combiner 10 pl de 10 pM brins complémentaires les uns avec les autres. Placez dans un bloc thermique à 95 ° C pendant5 min. Éteignez le bloc de chaleur et laisser les oligos refroidir lentement à la température ambiante avant utilisation.
- Avant de commencer, réchauffer le tampon de liaison, un tampon de lavage de faible stringence, un tampon de lavage à haute stringence, et un tampon d'élution à température ambiante.
Remarque: Une concentration finale de 50 ng / ml de poly d (IC) peut être ajouté au tampon de fixation, un tampon de lavage à faible stringence, et un tampon de lavage de stringence élevée pour réduire le potentiel de liaison non spécifique des protéines à oligos. - Préparer les mélanges de liaison pour chaque variante.
- Mélanger 1 volume de lysat nucléaire avec 2 volumes de tampon de liaison.
Remarque: La quantité requise de lysat doit être déterminée expérimentalement en raison de l'abondance des TFs variables. Utilisation entre 100-250 pg de lysat nucléaire par colonne est suffisante dans la plupart des cas. - Ajouter inhibiteur 1x phosphatase, un inhibiteur de protéase 1x et activateur de liaison 1x (en option) et mélanger en tapotant le tube plusieurs fois.
Note: 100x liaisonactivateur est constitué de 750 mM MgCl 2 et 300 mM ZnCl2. Ajouter activateur de liaison si la liaison de l'ADN TF dépend des cofacteurs ou des agents réducteurs. Si cette information est inconnue, ajouter l'activateur de liaison. - Ajouter 10 pi de 5 pM d'ADN de capture biotinylée (50 pmol) à chaque mélange de liaison respectif. Incuber pendant 20 min à température ambiante.
Nota: Le temps d'incubation et la température peut varier en fonction de la TF. Les valeurs optimales doivent être déterminées expérimentalement.
- Mélanger 1 volume de lysat nucléaire avec 2 volumes de tampon de liaison.
- Ajouter 100 pi de microbilles de streptavidine. Incuber pendant 10 min à température ambiante.
- Pour chaque sonde oligo testée, placer une colonne de liaison dans le séparateur magnétique. Placer un tube de microcentrifugation directement sous chaque colonne de liaison et d'appliquer 100 pi de tampon de liaison pour rincer la colonne.
- Pipeter le contenu de chaque mélange de liaison dans des colonnes séparées et de permettre au liquide de circuler complètement à travers la colonne dans la microcentrifugeusele tube avant de poursuivre. Assurez-vous d'étiqueter les colonnes avec la variante oligo qui a été utilisé dans le mélange de liaison. Etiqueter les échantillons accréditives et les remplacer par de nouveaux tubes à centrifuger pour collecter les lavages à faible stringence.
- Appliquer 100 pi de faible stringence tampon de lavage à la colonne; attendre jusqu'à ce que le réservoir de la colonne est vide. Répétez lavage 4x. Etiqueter les échantillons de lavage à faible stringence et les remplacer par de nouveaux tubes à centrifuger pour collecter les lavages à haute stringence.
- Appliquer 100 pi de haute stringence tampon de lavage à la colonne; attendre jusqu'à ce que le réservoir de la colonne est vide. Répétez lavage 4x. Etiqueter les échantillons de lavage à haute stringence et les remplacer par de nouveaux tubes à centrifuger pour recueillir le pré-élution.
- Ajouter 30 pi de tampon d'élution native à la colonne et laisser reposer pendant 5 min. Etiqueter les échantillons de pré-élution et les remplacer par de nouveaux tubes à centrifuger pour recueillir l'élution.
Remarque: Cela n'éluer pas la protéine liée; il lave le reste haute stringencele tampon de la colonne et on le remplace par un tampon d'élution pour optimiser l'efficacité de l'élution. - Ajouter un 50 ul élution native tampon supplémentaire pour éluer les TFs liés. Pour un rendement plus élevé, mais éluat moins concentré, ajouter un 50 pi supplémentaires de tampon d'élution natif et de recueillir l'écoulement.
Note: Analyser des échantillons d'élution par spectrométrie de masse pour déterminer l'identité du TFs lié 32. Par la suite, vérifier les résultats protéomiques par le biais dodécylsulfate de sodium électrophorèse sur gel de polyacrylamide au sulfite (SDS-PAGE) , suivie d'un transfert Western 33. Si la spectrométrie de masse ne sont pas disponibles, exécutez une tache d'argent en utilisant la technique standard au lieu d'un Western blot pour déterminer la taille de la protéine (s) représentant la liaison génotype-dépendante. Utilisez ces informations pour affiner la liste des TFs prédites des approches de calcul détaillées dans l'introduction.
Résultats
Dans cette section, les résultats représentatifs de ce qui les attend sont fournies lors de l'exécution d'un EMSA ou DAPA, et la variabilité en ce qui concerne la qualité de lysat est caractérisé. Par exemple, il a été suggéré que le gel et les échantillons de protéines de décongélation plusieurs fois peut entraîner une dénaturation. Afin d'explorer la reproductibilité de l'analyse EMSA dans le contexte de ces cycles "gel-dégel", deux 35 oligo...
Discussion
Bien que les progrès dans les technologies de séquençage et de génotypage ont grandement amélioré notre capacité à identifier les variants génétiques associés à la maladie, notre capacité à comprendre les mécanismes fonctionnels impactés par ces variantes est à la traîne. Une source importante du problème est que de nombreuses variantes associées à la maladie sont situés dans n sur le codage des régions du génome, qui affectent probablement plus difficiles à prédire-mécanismes qui contr?...
Déclarations de divulgation
Les auteurs n'ont rien à dévoiler.
Remerciements
We thank Erin Zoller, Jessica Bene, and Lindsey Hays for input and direction in protocol development. MTW was supported in part by NIH R21 HG008186 and a Trustee Award grant from the Cincinnati Children's Hospital Research Foundation. ZHP was supported in part by T32 GM063483-13.
matériels
| Name | Company | Catalog Number | Comments |
| Custom DNA Oligonucleotides | Integrated DNA Technologies | http://www.idtdna.com/site/order/oligoentry | |
| Potassium Chloride | Fisher Scientific | BP366-500 | KCl, for CE buffer |
| HEPES (1 M) | Fisher Scientific | 15630-080 | For CE and NE buffer |
| EDTA (0.5M), pH 8.0 | Life Technologies | R1021 | For CE, NE, and annealing buffer |
| Sodium Chloride | Fisher Scientific | BP358-1 | NaCl, for NE buffer |
| Tris-HCl (1M), pH 8.0 | Invitrogen | BP1756-100 | For annealing buffer |
| Phosphate Buffered Saline (1x) | Fisher Scientific | MT21040CM | PBS, for cell wash |
| DL-Dithiothreitol solution (1 M) | Sigma | 646563 | Reducing agent |
| Protease Inhibitor Cocktail | Thermo Scientific | 87786 | Prevents catabolism of TFs |
| Phosphatase Inhibitor Cocktail | Thermo Scientific | 78420 | Prevents dephosphorylation of TFs |
| Nonidet P-40 Substitute | IBI Scientific | IB01140 | NP-40, for nuclear extraction |
| BCA Protein Assay Kit | Thermo Scientific | 23225 | For measuring protein concentration |
| Odyssey EMSA Buffer Kit | Licor | 829-07910 | Contains all necessary EMSA buffers |
| TBE Gels, 6%, 12 Wells | Invitrogen | EC6265BOX | For EMSA |
| TBE Buffer (10x) | Thermo Scientific | B52 | For EMSA |
| FactorFinder Starting Kit | Miltenyi Biotec | 130-092-318 | Contains all necessary DAPA buffers |
| Licor Odyssey CLx | Licor | Recommended scanner for DAPA/EMSA | |
| Antibiotic-Antimycotic | Gibco | 15240-062 | Contains 10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of Fungizone® Antimycotic |
| Fetal Bovine Serum | Gibco | 26140-079 | FBS, for culture media |
| RPMI 1640 Medium | Gibco | 22400-071 | Contains L-glutamine and 25 mM HEPES |
Références
- Hindorff, L. A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 106 (23), 9362-9367 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Ward, L. D., Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 30 (11), 1095-1106 (2012).
- Paul, D. S., Soranzo, N., Beck, S. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays. 36 (2), 191-199 (2014).
- Zhang, F., Lupski, J. R. Non-coding genetic variants in human disease. Hum Mol Genet. , (2015).
- Lee, T. I., Young, R. A. Transcriptional regulation and its misregulation in disease. Cell. 152 (6), 1237-1251 (2013).
- Slatkin, M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 9 (6), 477-485 (2008).
- Bush, W. S., Moore, J. H. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 8 (12), e1002822 (2012).
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 491 (7422), 56-65 (2012).
- Chang, C. C., et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
- Purcell, S., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81 (3), 559-575 (2007).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16 (1), 123-131 (2006).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 10 (12), 1213-1218 (2013).
- Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R., Lieb, J. D. FAIRE Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17 (6), 877-885 (2007).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Martens, J. H., Stunnenberg, H. G. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 98 (10), 1487-1489 (2013).
- Liu, T., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12 (8), R83 (2011).
- Griffon, A., et al. Integrative analysis of public ChIP-seq experiments reveals a complex multi-cell regulatory landscape. Nucleic Acids Res. 43 (4), e27 (2015).
- Staudt, L. M., et al. A lymphoid-specific protein binding to the octamer motif of immunoglobulin genes. Nature. 323 (6089), 640-643 (1986).
- Singh, H., Sen, R., Baltimore, D., Sharp, P. A. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature. 319 (6049), 154-158 (1986).
- Weirauch, M. T., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 158 (6), 1431-1443 (2014).
- Ward, L. D., Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40 (Database issue), D930-D934 (2012).
- Boyle, A. P., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22 (9), 1790-1797 (2012).
- Hume, M. A., Barrera, L. A., Gisselbrecht, S. S., Bulyk, M. L. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 43 (Database issue), D117-D122 (2015).
- Mathelier, A., et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42 (Database issue), 142-147 (2014).
- Smith, M. F., Delbary-Gossart, S. Electrophoretic Mobility Shift Assay (EMSA). Methods Mol Med. 50, 249-257 (2001).
- Franza, B. R., Josephs, S. F., Gilman, M. Z., Ryan, W., Clarkson, B. Characterization of cellular proteins recognizing the HIV enhancer using a microscale DNA-affinity precipitation assay. Nature. 330 (6146), 391-395 (1987).
- . BCA Protein Assay Kit: User Guide Available from: https://tools.thermofisher.com/content/sfs/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf (2014)
- Wijeratne, A. B., et al. Phosphopeptide separation using radially aligned titania nanotubes on titanium wire. ACS Appl Mater Interfaces. 7 (21), 11155-11164 (2015).
- Silva, J. M., McMahon, M. The Fastest Western in Town: A Contemporary Twist on the Classic Western Blot Analysis. J. Vis. Exp. (84), (2014).
- Lu, X., et al. Lupus Risk Variant Increases pSTAT1 Binding and Decreases ETS1 Expression. Am J Hum Genet. 96 (5), 731-739 (2015).
- Ramana, C. V., Chatterjee-Kishore, M., Nguyen, H., Stark, G. R. Complex roles of Stat1 in regulating gene expression. Oncogene. 19 (21), 2619-2627 (2000).
- Fillebeen, C., Wilkinson, N., Pantopoulos, K. Electrophoretic Mobility Shift Assay (EMSA) for the Study of RNA-Protein Interactions: The IRE/IRP Example. J. Vis. Exp. (94), e52230 (2014).
- Heng, T. S., Painter, M. W. Immunological Genome Project, C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 9 (10), 1091-1094 (2008).
- Wu, C., et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10 (11), R130 (2009).
- Wu, C., Macleod, I., Su, A. I. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 41 (Database issue), D561-D565 (2013).
- Wang, J., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22 (9), 1798-1812 (2012).
- Holden, N. S., Tacon, C. E. Principles and problems of the electrophoretic mobility shift assay. J Pharmacol Toxicol Methods. 63 (1), 7-14 (2011).
- Xu, J., Liu, H., Park, J. S., Lan, Y., Jiang, R. Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development. 141 (7), 1442-1452 (2014).
- Yang, T. -. P., et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 26 (19), 2474-2476 (2010).
- Fort, A., et al. A liver enhancer in the fibrinogen gene cluster. Blood. 117 (1), 276-282 (2011).
- Solberg, N., Krauss, S. Luciferase assay to study the activity of a cloned promoter DNA fragment. Methods Mol Biol. 977, 65-78 (2013).
- Rahman, M., et al. A repressor element in the 5'-untranslated region of human Pax5 exon 1A. Gene. 263 (1-2), 59-66 (2001).
- Mali, P., et al. RNA-Guided Human Genome Engineering via Cas9. Science. 339 (6121), 823-826 (2013).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.