
JoVE 비디오를 활용하시려면 도서관을 통한 기관 구독이 필요합니다. 전체 비디오를 보시려면 로그인하거나 무료 트라이얼을 시작하세요.
Method Article
전기 영동 이동성 분석 (EMSA) 및 Shift 키를 사용하여 기능 비 코딩 유전자 변형에 대한 심사 DNA 화성 강수 분석 (DAPA)
* 이 저자들은 동등하게 기여했습니다
요약
We present a strategic plan and protocol for identifying non-coding genetic variants affecting transcription factor (TF) DNA binding. A detailed experimental protocol is provided for electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genotype-dependent TF DNA binding.
초록
Population and family-based genetic studies typically result in the identification of genetic variants that are statistically associated with a clinical disease or phenotype. For many diseases and traits, most variants are non-coding, and are thus likely to act by impacting subtle, comparatively hard to predict mechanisms controlling gene expression. Here, we describe a general strategic approach to prioritize non-coding variants, and screen them for their function. This approach involves computational prioritization using functional genomic databases followed by experimental analysis of differential binding of transcription factors (TFs) to risk and non-risk alleles. For both electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genetic variants, a synthetic DNA oligonucleotide (oligo) is used to identify factors in the nuclear lysate of disease or phenotype-relevant cells. For EMSA, the oligonucleotides with or without bound nuclear factors (often TFs) are analyzed by non-denaturing electrophoresis on a tris-borate-EDTA (TBE) polyacrylamide gel. For DAPA, the oligonucleotides are bound to a magnetic column and the nuclear factors that specifically bind the DNA sequence are eluted and analyzed through mass spectrometry or with a reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blot analysis. This general approach can be widely used to study the function of non-coding genetic variants associated with any disease, trait, or phenotype.
서문
시퀀싱 및 게놈 넓은 협회 연구 (GWAS), 후보의 궤적 연구를 포함하여 유전자형을 기반으로 연구, 그리고 깊은 시퀀싱 연구는 통계적으로 질병, 특성, 또는 표현형과 관련된 많은 유전자 변형을 확인했다. 초기 예측과는 반대로, 이러한 변형 (85-93%)의 대부분은 비 - 코딩 영역에 위치하는 단백질 1,2의 아미노산 서열을 변경하지 않는다. 이들 비 - 코딩 변이체의 기능을 해석하고 관련된 질환에 접속 생물학적 메커니즘을 결정하는 단계, 특성, 또는 표현형 3-6 도전 입증되었다. 유전자 발현 - 우리는 중요한 중간 표현형 변이를 연결하는 분자 메커니즘을 식별하기위한 일반적인 전략을 개발했다. 이 파이프 라인은 특히 유전 적 변이에 의해 결합 TF의 변조를 식별 할 수 있도록 설계되었습니다. 이 전략은 예측하기위한 계산 방법과 분자 생물학 기술을 결합생물학적 인 실리코 후보 변종의 효과 및 확인이 예측 경험적으로 (그림 1).

그림 1 :.. 회색 음영이 원고와 관련된 상세한 프로토콜에 포함되지 않은 비 코딩 유전자 변형 단계의 분석을위한 전략적 접근 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
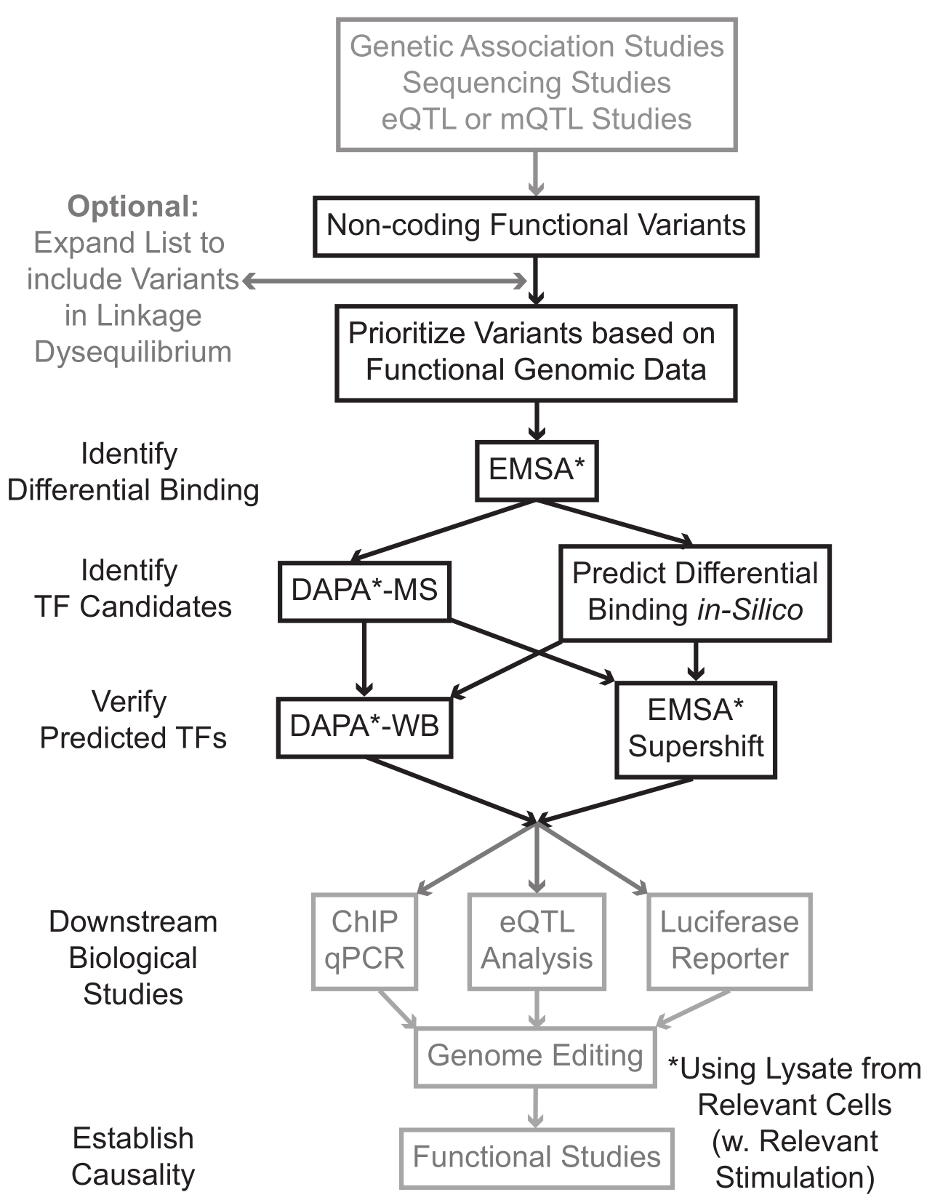{kind=link}
많은 경우, 각각 통계 학적으로 관련된 변이체 높은 링크 - 불평형 (LD)의 모든 것을 포함하는 변이체의 목록을 확대함으로써 시작하는 것이 중요하다. LD는 R 7이 통계에 의해 측정 될 수있는 두 개의 다른 대립 유전자의 염색체 위치에서의 비 - 무작위 교제의 척도이다. R 2는 린의 측정두 가지 변종 사이의 R 2 = 1 나타내는 완벽한 결합으로 두 가지 변형 사이의 케이지는 불균형. 높은 LD의 대립 유전자는 조상의 인구에서 염색체에 공동으로 분리하는 찾을 수 있습니다. 현재 유전자형 배열은 인간 게놈의 모든 알려진 변종을 포함하지 않는다. 대신, 그들은 인간 게놈 내에서 LD을 악용 LD (8)의 특정 지역 내에서 다른 변형에 대한 프록시 역할 알려진 변종의 서브 세트를 포함한다. 이 의미 생물학적 효과와 인과 변형 - 변형와 LD에 있기 때문에, 생물학적 결과없이 변이체는 특정 질환과 연관 될 수있다. 절차 적, 1000 게놈의 최신 버전은 가용 스루풋 10, 11, 전체 게놈 협회 분석을위한 오픈 소스 도구와 호환 바이너리 파일에 9 변형 호출 파일 (VCF)를 프로젝트로 변환하는 것이 좋습니다. 그 후, 각 입력 유전 버지니아와 LD의 R 2> 0.8 다른 모든 유전자 변형쾌활한 후보로서 식별 될 수있다. 변형이 유럽 가계의 주제, 비슷한 조상의 과목에서 데이터가 LD 확장을 사용해야에서 확인 된 경우는,이 스텝 - 예에 대한 적절한 기준 인구를 사용하는 것이 중요합니다.
LD 확장들은 후보 변형 수십 결과 및 이들의 단지 작은 분획이 질병기구에 기여할 것으로 예상된다. 종종, 실험적 개별적으로 각 변형을 조사하기 불가능하다. 변형의 우선 순위를 필터로 공개적으로 사용 가능한 기능 게놈 데이터 세트의 수천을 활용하는 것이 유용하다. 예를 들어, 인 코드 컨소시엄 (12)는의 DNase-SEQ 13 ATAC 같은 기술에서 염색질 접근성 데이터와 함께 콘텍스트의 넓은 범위에서 칩 배열하여 TFS의 결합을 설명하는 실험 및 공동 인자 및 히스톤 마크 수천을 수행 -seq 14 및 서열 FAIRE-15. Datab같은 UCSC 게놈 브라우저 (16), 로드맵 후성 유전체학 (17), 청사진 후성 유전체 (18), Cistrome (19), 및 매핑 20 ASE에 웹 서버는 세포 유형 및 조건의 넓은 범위에 걸쳐 이들과 다른 실험 기법에 의해 생성 된 데이터에 대한 무료 액세스를 제공합니다. 실험적 검토하기에 너무 많은 변형이있을 때, 이러한 데이터는 관련 세포 및 조직 유형의 것으로 조절 영역 내에 위치하는 우선 순위를 사용할 수있다. 또한, 변형 특정 단백질 칩 서열 피크 내에있는 경우,이 데이터는 특정 TF (들) 또는 그의 영향을 줄 수 있습니다 결합 공동 요인으로 잠재적 인 단서를 제공 할 수 있습니다.
다음으로, 우선 순위 결과 변종은 EMSA (21, 22)를 사용하여 결합 예측 유전자형에 의존하는 단백질을 확인하는 실험적으로 상영된다. EMSA은 비 환원성 TBE 겔상에서 올리고의 이동 변화를 측정한다. 형광 표지 올리고는 함께 배양핵 해물, 핵 인자의 결합은 올리고 겔상에서의 이동을 지연시킬 것이다. 이러한 방식으로, 스캔시 강한 형광 신호로 발표 할 예정보다 핵 요소를 결합했다 올리고있다. 특히, EMSA 바인딩 영향을받는 특정 단백질에 대한 예측을 필요로하지 않습니다.
변형이 예측 규제 지역 내에 위치하며, 차동 결합 핵 인자 할 수있는있다가 발견되면, 계산 방법은 누구가에 영향을 줄 수있는 바인딩 특정 TF (들)을 예측하기 위해 사용된다. 우리는 CIS-BP 23, 24, RegulomeDB 25 UniProbe (26), 및이 JasPAR (27)를 사용하는 것을 선호합니다. 후보 TF가 식별되면,이 예측은 구체적으로 다음과 TF (EMSA-supershifts 및 DAPA-서부)에 대한 항체를 사용하여 시험 할 수있다. EMSA-supershift 핵 분해물 및 올리고 TF에 특이 항체의 첨가를 포함한다. EMSA-supershift에서 긍정적 인 결과를 repr입니다EMSA 대역에서 추가의 이동 또는 대역의 손실로 esented (레퍼런스 28에서 검토). 상보 DAPA에서, 변형 및 뉴클레오티드의 측면에 20 염기쌍을 함유하는 5'- 비오틴 올리고 양면은 특히 올리고 결합 핵 인자 캡처 관련된 세포 유형 (들)로부터 핵 파쇄 액과 함께 배양된다. 올리고 이중 핵 인자 복합체는 자기 칼럼에서 마이크로 비드를 스트렙 타비 딘에 의해 고정된다. 결합 된 핵 인자 용출 29,48 통해 직접 수집됩니다. 바인딩 예측이어서 단백질에 특이적인 항체를 사용하여 웨스턴 블롯에 의해 평가 될 수있다. 후속하여 검증 할 수있는 명백한 예측 또는 너무 많은 예측, 질량 분석법을 이용하여 후보의 TF를 식별하기 위해 프로테오믹스 코어로 전송 될 수 DAPA 실험 변이체 풀 - 다운에서 용리가없는 경우에서, 이들은 전술 행동 양식.
articl의 나머지 부분에서즉, 유전자 변형 및 DAPA EMSA 분석 상세한 프로토콜이 제공된다.
프로토콜
솔루션 및 시약 1. 준비
- EMSA 및 방위 사업 청에 사용하기위한 사용자 정의 DNA 올리고 뉴클레오티드 프로브를 주문하십시오.
- 짧은 올리고 디자인, 바인딩이 아닌 특정 단백질을 감소시키기 위해 30, 및 17 bp의 내인성 게놈 서열에 의해 측면 중앙에 직접 관심의 변형을 배치 (35 ~ 45 염기의 길이 쌍 (BP) 사이). EMSA 올리고 들어, 5 '형광을 추가합니다. 방위 사업 청 올리고 들어, 5 '비오틴 태그를 추가합니다.
- 센스 가닥과 역 보완 가닥 모두를 주문하십시오. 또한, 주문 듀플렉스 (프리 어닐링) 올리고. 올리고 이름을 지정할 때 설립 참조 게놈의 명칭을 기반으로.
참고 : "위험"과 질병 및 프로젝트 특정 할 수있다 "비 위험"지정 "참조"와 "비 참조"더 보편적으로 관련이있다. - 올리고 도착하면, 간단히 100 μ의 최종 농도 클레아없는 물에서 재현 탁 내용과 스핀 다운;엠. 스토어는 -20 ° C에서 주식을 재현 탁. 알루미늄 호일로 포장하여 빛에서 형광 태그 올리고를 보호합니다.
| 이름 | 순서 |
| rs76562819_REF_FOR | GTAATGCCTTAATGAGAGAG GTTAGTCATCTTCTCACTTC |
| rs76562819_REF_REV | GAAGTGAGAAGATGACTAAC T CTCTCTCATTAAGGCATTAC |
| rs76562819_NONREF_FOR | GTAATGCCTTAATGAGAGAG G GTTAGTCATCTTCTCACTTC |
| rs76562819_NONREF_REV | GAAGTGAGAAGATGACTAAC C CTCTCTCATTAAGGCATTAC |
표 1 : 예 EMSA / 방위 사업 청 올리고 뉴클레오티드 디자인은 차동 빈의 SNP를 테스트"NONREF"은 비 기준 대립 약자 동안 딩. "REF"는 기준 대립 유전자를 나타낸다. "에 대한" "REV는"그 보완을 표시하면서, 앞으로 가닥을 의미합니다. SNP는 빨간색으로 볼 수있다.
- 최종 10 mM의 HEPES (pH를 7.9)의 농도, 10 밀리미터의 KCl, 탈 이온수 0.1 mM의 EDTA와 세포질 추출 (CE) 버퍼를 준비합니다.
- 핵 추출 (NE) 최종 20 mM의 HEPES (pH를 7.9)의 농도, 0.4 M의 NaCl, 및 탈 이온수에 1 mM의 EDTA와 버퍼를 준비합니다.
- 최종 10 mM 트리스 (PH 7.5-8.0)의 농도, 50 mM의 염화나트륨, 탈 이온수에 1 mM의 EDTA와 어닐링 버퍼를 준비합니다.
배양 된 세포에서 핵 해물 2. 준비
참고 :이 실험 프로토콜은 B-임파 세포 라인을 사용하여 최적화되었지만, 여러 가지 다른 관련이없는 접착 / 서스펜션 세포주에서 테스트를 거쳤으며 보편적으로 작동합니다.
- Cultu2 mM L- 글루타민, 10 % 소 태아 혈청, 페니실린 100 유니트 / ㎖, 스트렙토 마이신 100 ㎍ / ml를 함유 1X 항생제 - 항진균제 250 NG와 로스웰 파크 메모리얼 연구소 (RPMI) 1640 B-임파 세포 재 암포 테리 신 B의 / ㎖
- 200,000-500,000 생존 세포 / ml의 범위에서 시드와 통하게되거나 느슨한 캡 세워서 5 % 이산화탄소, 37 ℃에서 플라스크 배양한다.
참고 :이 1,000,000 이상 세포 / ml에 도달했을 때 B-임파 세포의 성장이 느려집니다. 최대 피펫 팅에 의해 여러 번 아래로 셀 폭발을 휴식과 성장의 빠른 속도를 유지하기 위해 200,000-500,000 세포 / ml로 세포를 반환합니다.
- 200,000-500,000 생존 세포 / ml의 범위에서 시드와 통하게되거나 느슨한 캡 세워서 5 % 이산화탄소, 37 ℃에서 플라스크 배양한다.
- 5 분, 4 ° C에서 300 XG에 스핀 다운 및 흡입을 통해 PBS를 제거, 두 번 10 ml의 차가운 얼음 인산염 완충 식염수 (PBS)로 배양 된 세포를 씻으십시오.
- 10 7 세포 당 차가운 PBS 1 ml의 얼음 같은 혈구와에 resuspend 펠렛을 사용하여 세포를 계산합니다.
참고 : 예를 들어, 2 × 10 7 용균 경우 </ SUP> 셀, 2 ml의 PBS에 재현 탁. - 나누어지는 1.5 ml의 마이크로 원심 튜브에 1 ㎖의 각 튜브는 PBS에서 10 7 세포를 포함하도록. PBS 오프 2 분 4 ° C 및 흡인을위한 3,300 XG에 원심 분리기.
- 사용 CE 버퍼의 작업 스톡 1 mM 디티 오 트레이 톨 (DTT), 1X 포스파타제 억제제 및 1X 프로테아제 억제제를 추가하기 전에. 재현 탁 셀 CE 버퍼 400 μL로 펠릿 15 분 동안 얼음 위에서 배양한다.
- 10 % Nonidet P-40의 25 μl를 추가하고 피펫으로 혼합한다. 4 ° C, 3 분 동안 최대 속도로 원심 분리기. 가만히 따르다 및 상층 액을 버린다.
- 사용 NE 버퍼의 작업 재고 1 mM의 DTT, 1X 포스파타제 억제제, 및 1X 프로테아제 억제제를 추가하기 전에. 재현 탁 세포 NE 버퍼의 30 μL와 펠렛 및 텍싱하여 혼합한다.
- 튜브 회전에서 10 분 동안 얼음에 4 ° C에서 품어. 원심 분리기 2 분 4 ° C를위한 3,300 XG.
- 에 저장하기 전에 맑은 상층 액 (핵 해물) 및 분취를 수집 -806; C는 단백질을 분해 할 수있다 여러 동결 - 해동 사이클을 방지 할 수 있습니다. bichoninic 산 분석 (BCA) (31)를 사용하여 단백질 농도를 측정하기 위해 10 μL 나누어지는 둡니다.
3. 전기 영동 이동성 이동 분석 (EMSA)
- 올리고 작업 주식 및 EMSA 젤을 준비합니다.
- 올리고가 양면에 정렬 된 경우, 100 μM 스톡을 해동 한 희석 : 2000 어닐링 완충액에 50 nM의 작업 스톡을 달성했다.
- 올리고는 단일 가닥 주문 된 경우, 100 μM 주식을 해동 및 100 nm의 작업 주식을 달성하기 위해 어닐링 버퍼에 1:10 희석. 미세 원심 분리 관에서 서로 100 nM의 상보 가닥 용액 100 ㎕를 결합.
- 5 분 동안 95 ℃의 열 블록에 놓는다. 열 블록을 끄고 올리고 천천히 사용 전 적어도 한 시간 동안 실온까지 냉각 할 수있다.
- EMSA 젤 사전 실행합니다.
- 프리 캐스트 6 % T에서 슬라이드를 제거젤을하고 우물에서 모든 버퍼를 제거하는 탈 이온수에서 여러 번 헹군다. 탈 이온수 950 ml의 10 배 TBE 50 ㎖를 추가하여 0.5 배의 TBE 버퍼 1 L를 준비합니다.
- 겔 전기 영동 장치를 조립하고 0.5 배의 TBE 버퍼와 내부 챔버를 작성하여 누출이 있는지 확인합니다. 어떤 버퍼가 외부 챔버로 누출되지 않으면 외부 챔버에게 길의 약 2/3 채운다.
- 60 분 동안 100 V에서 젤을 사전 실행합니다.
- 0.5 배의 TBE 버퍼 200 μL 잘 각을 플래시합니다.
- 버퍼 마스터 믹스 바인딩 준비합니다.
- 100 mM 트리스, 500 밀리미터의 KCl, 10 mM의 DTT의 최종 농도로 버퍼를 바인딩 10 배를 준비; 탈 이온수의 pH가 7.5.
- 미세 원심 분리 관에, 모든 반응 (표 2 내지 10 μL의 10 배의 결합 완충액 10 μL DTT / 소르 베이트, 5 μL 폴리 D (IC), 2.5 ㎕의 연어 정자 DNA)에 공통 시약 이루어진 마스터 믹스를 생성한다. 추가로 10 %를 준비피펫에 의한 볼륨 손실을 설명합니다.
| 시약 | 최종 용액에 농축. | # 1 RXN | # 2 RXN | # 3 RXN | # 4 RXN |
| 초순수 | 20 μL의 부피합니다. | 13.5 μL | 11.98 μL | 13.5μl | 11.98 μL |
| 10 배 버퍼 바인딩 | 1 배 | 2 μL | 2 μL | 2 μL | 2 μL |
| DTT / TW-20 | 1 배 | 2 μL | 2 μL | 2 μL | 2 μL |
| 연어 정자 DNA | 500 NG / μL | 0.5 μL | 0.5 μL | 0.5 μL | 0.5 μL |
| 1μg / μL 폴리 D (IC) | 1 μg의 | 1 μL | 1 μL | 1 μL | 1 μL |
| 핵 추출물 (5.26 UG / μL) | 8 μg의 | - | 1.52 μL | - | 1.52 μL |
| NE 버퍼 | 1.52 μL | - | 1.52 μL | - | |
| 참조 대립 유전자 올리고 | 50 fmol | 1 μL | 1 μL | - | - |
| 비 참조 대립 유전자 올리고 | 50 fmol | - | - | 1 μL | 1 μL |
표 2 : 예 EMSA 반응 세투P. 표는 유전자형 - 의존성 특정 SNP TF들로의 결합이 있다는 가설을 테스트하기 위해 예 EMSA를 나타낸다.
- 이러한 모든 시약을 첨가 한 다음 최종 부피가 20 μL 될 것으로 각 microcentrifuge 관에 뉴 클레아없는 물을 추가합니다.
- 각 microcentrifuge 관에 마스터 믹스의 적당량 (5.5 μL)를 추가합니다.
- 적절한의 microcentrifuge 튜브에 핵 해물의 8 μg의 추가. 핵 부정적인 컨트롤 등의 추출물 (예를 들어, 표 2, RXN # 1과 RXN # 3)없이 올리고을 포함하는 튜브를 포함합니다.
주의 : 반응 당 분해물의 최적 량은 적정에 의해 실험적으로 결정되어야한다. 일반적으로, 해물의 2-10 μg의 범위를 적정하면 충분하다. - 적절한의 microcentrifuge 튜브에 올리고 50 fmol를 추가합니다. 톡 믹스 앤 간단히 튜브의 바닥에 내용을 회전합니다. 실온에서 20 분 동안 인큐베이션.
참고 : 만약 attemptisupershift 겨, 올리고 첨가하기 전에 실온에서 20 분 동안 항체 해물 혼합물을 배양한다. 최상의 결과를위한 칩 학년 항체의 1 μg의 사용을 권장합니다. - 각 microcentrifuge 관에 10 배 오렌지로드 염료의 2 μl를 추가합니다. 최대 피펫 아래로 혼합.
- 별도의 우물에 각 샘플을 추방 한 후로 pipetting 아래로 혼합에 의해 사전 실행 6 % TBE 겔에 샘플을로드합니다. 주황색 염료가 젤 내리막 길의 3/4에 2/3를 마이그레이션 할 때까지 80 V에서 젤을 실행합니다. 이 약 60-75 분 정도 소요됩니다.
- 겔 칼이 열려 들리 플라스틱 카세트에서 젤을 제거하고 마르지을 유지하기 위해 0.5 % TBE 버퍼와 용기에 젤을 배치합니다.
- 이미지를 방해 할 어떤 거품이나 오염 물질을 제거해야되고, 적외선 및 화학 발광 영상 시스템의 표면에 젤을 놓습니다.
- 스캐닝 시스템 소프트웨어를 사용하면은 "획득"탭을 클릭합니다다음 겔 스캐너의 표면에있는 위치에 대응하는 영역 주위에 상자를 그립니다 "새 그리기"를 선택합니다.
- 은 "획득"탭의 "채널"섹션에서 올리고에 형광 태그의 파장에 해당하는 채널을 선택합니다. 은 "스캐너"섹션에서 낮은 품질의 미리보기 스캔을 얻기 위해 "미리보기"를 클릭합니다. 겔의 부분에 아래로 취득 미리보기 이미지를 둘러싼 파란색 상자를 드래그하여 스캔 영역을 조정하는 이미징합니다.
참고 : 예를 들어, 700 나노 형광으로 표지 올리고를 사용하는 경우 "700 nm의"채널 스캔하기 전에 선택되어 있는지 확인합니다. - 은 "스캔 제어"섹션에서 "84 μM"해상도 옵션과 "중간"품질 옵션을 선택합니다. 겔의 절반 두께 오프셋에 포커스를 설정한다. 참고 : 예를 들어, 1 mm 겔 오프셋 0.5 mm의 초점을 사용한다.
- 은 "스캐너"섹션에서 b에 "시작"버튼을 클릭스캔을 egin.
주 : 스캔하는 동안, 휘도, 명암 및 색상이 종종 주사 시스템의 제조사에 따라 수동으로 조정될 수있다. - 검사가 완료되면, "영상"탭을 선택하고 방향을 수정하려면 "만들기"절에서 "회전 또는 뒤집기"를 클릭합니다. 메인 메뉴에서 "내보내기"를 클릭하여 이미지 파일을 저장 한 다음 선택 "단일 이미지보기를."
4. DNA 선호도 정화 분석 (DAPA)
- 5 μM 올리고 작업 주식의 준비.
- 올리고는 양면 인쇄로 주문 된 경우, 100 μM의 주식을 해동하고 5 μm의 작업 주식을 달성하기 위해 어닐링 버퍼에 1:20 희석.
- 올리고는 단일 가닥 주문 된 경우, 100 μM 주식을 해동과 주식을 작업 10 μM을 달성하기 위해 어닐링 버퍼에 1:10 희석. 서로 10 μM 상보 가닥의 10 μl를 결합한다. 95 ℃에서 가열 블럭에서 발생5 분. 열 블록을 끄고 올리고 천천히 사용하기 전에 실온까지 냉각 할 수 있습니다.
- 시작하기 전에 실온으로 결합 완충액 낮은 엄격 세척 완충액, 고 엄격 세척 완충액 및 용출 완충액을 따뜻하게.
참고 : 50 ng의 최종 농도 / ㎖ 폴리 D (IC)를 올리고 발 단백질의 잠재적 비 - 특이 적 결합을 감소시키기 위해 결합 완충액 낮은 엄격 세척 완충액, 및 고 엄격 세척 완충액에 첨가 할 수있다. - 각 변형에 대한 바인딩 혼합물을 준비합니다.
- 버퍼를 바인딩의 2 권으로 핵 해물의 1 볼륨을 섞는다.
참고 : 해물의 필요한 양으로 인해 TF가 풍부한 변화를 실험적으로 결정되어야한다. 열 당 핵 해물의 μg의 100 ~ 250 사이를 사용하면 대부분의 경우에 충분하다. - 1 배 포스 파타 아제 억제제, 1X 프로테아제 억제제 및 1 배 결합 증강 (옵션)을 추가하고 튜브를 여러 번 쓸어 넘겨 섞는다.
참고 : 100 배 바인딩증강은 750 밀리미터의 MgCl 2, 300MM ZnCl 2로 구성되어 있습니다. DNA에 TF의 결합은 보조 인자 또는 환원제에 의존하는 경우 바인딩 인핸서를 추가합니다. 이 정보가 알려져 있지 않은 경우, 상기 바인딩 인핸서를 추가한다. - 각각의 결합 혼합물에 5 μm의 바이오틴 캡처 DNA (50 pmol의) 10 μl를 추가합니다. 실온에서 20 분 동안 인큐베이션.
참고 : 배양 시간 및 온도는 TF에 따라 다를 수 있습니다. 최적의 값은 실험적으로 결정되어야한다.
- 버퍼를 바인딩의 2 권으로 핵 해물의 1 볼륨을 섞는다.
- 스트렙 타비 딘 마이크로 비드의 100 μl를 추가합니다. 실온에서 10 분 동안 인큐베이션.
- 각 올리고 프로브를 테스트중인 경우, 자기 분리기에 바인딩 열을 배치합니다. 각 바인딩 열 아래 직접 microcentrifuge 관을 삽입하고 열을 씻어 바인딩 버퍼 100 μl를 적용합니다.
- 분리 컬럼에 각각 결합 혼합물의 내용을 피펫 및 액체가 미세 원심으로 칼럼을 통해 완전히 흐를 수 있도록진행하기 전에 관. 바인딩 혼합물에 사용 된 변형 올리고과 열 레이블을해야합니다. 플로우를 통해 샘플을 레이블과 낮은 엄격 세척를 수집하는 새로운 마이크로 원심 튜브로 교체합니다.
- 열을 둘러 엄격 세척 버퍼 100 μl를 적용; 열 저수지가 비어 때까지 기다립니다. 세척 4 배를 반복합니다. 저 엄격 세척 샘플 라벨과 높은 엄격 세척를 수집하는 새로운 마이크로 원심 튜브로 교체합니다.
- 칼럼에 높은 엄격 세척 버퍼 100 μl를 적용; 열 저수지가 비어 때까지 기다립니다. 세척 4 배를 반복합니다. 높은 엄격 세척 샘플 라벨 및 사전 용출를 수집하는 새로운 마이크로 원심 튜브로 교체합니다.
- 열 네이티브 용출 버퍼의 30 μl를 추가하고 5 분 동안 서 보자. 사전 용출 샘플 라벨 및 용출를 수집하는 새로운 마이크로 원심 튜브로 교체합니다.
주 :이 결합 된 단백질을 용출하지 않는다; 그것은 나머지 고 엄격 세척컬럼에서 용출 완충액의 효율을 극대화하는 용출 버퍼로 대체. - 바인딩과 TF를 용출하는 추가 50 μl의 기본 용출 버퍼를 추가합니다. 높은 수율 미만 농축 용출액 들어 천연 용출 버퍼의 추가 50 μL를 추가하고 관류를 수집한다.
주 : 결합 TF들 (32)의 신원을 확인하기 위해 질량 분석을 통해 용출 샘플을 분석한다. 이후, 웨스턴 블롯 (33)이어서 소듐 도데 실 황산나트륨 폴리 아크릴 아미드 겔 전기 영동 (SDS-PAGE)을 통해 결과를 확인 단백체. 질량 분석기를 사용할 수없는 경우, 유전자형 의존적 결합을 나타내는 단백질 (들)의 크기를 결정하는 대신 웨스턴 블롯의 표준 기술을 사용하여 실버 염색을 실행. 소개에 자세히 계산 방법에서 예측과 TF의 목록을 좁힐이 정보를 사용합니다.
결과
해물의 품질을 특징으로 관련이있는 EMSA 또는 방위 사업 청, 그리고 다양성을 수행 할 때이 절에서 무엇을 기대해야하는지의 대표적인 결과가 제공된다. 예를 들어, 여러 번 동결 융해 단백질 시료 것은 변성 될 수 있다는 것을 제안하고있다. 이러한 "동결 - 해동"사이클의 맥락에서 EMSA 분석 재현성을 탐색하기 위해, 하나 이상의 유전 적 변형에 다른 두 개의 35 염?...
토론
시퀀싱 및 유전형 기술의 발전이 크게 질병과 관련된 유전자 변형을 식별하는 우리의 능력을 강화하고 있지만, 이러한 변형의 영향을 기능적 메커니즘을 이해하는 능력이 떨어지고있다. 문제의 주된 원인 가능성 유전자 발현을 제어하기 어려워-간 예측 메커니즘에 영향을하는 부호화 게놈 영역 많은 질병 관련 변종 N에 위치된다는 것이다. 여기서는 가능성이 많은 비 - 코딩 변이체의 기능에...
공개
저자는 공개 아무것도 없어.
감사의 말
We thank Erin Zoller, Jessica Bene, and Lindsey Hays for input and direction in protocol development. MTW was supported in part by NIH R21 HG008186 and a Trustee Award grant from the Cincinnati Children's Hospital Research Foundation. ZHP was supported in part by T32 GM063483-13.
자료
| Name | Company | Catalog Number | Comments |
| Custom DNA Oligonucleotides | Integrated DNA Technologies | http://www.idtdna.com/site/order/oligoentry | |
| Potassium Chloride | Fisher Scientific | BP366-500 | KCl, for CE buffer |
| HEPES (1 M) | Fisher Scientific | 15630-080 | For CE and NE buffer |
| EDTA (0.5M), pH 8.0 | Life Technologies | R1021 | For CE, NE, and annealing buffer |
| Sodium Chloride | Fisher Scientific | BP358-1 | NaCl, for NE buffer |
| Tris-HCl (1M), pH 8.0 | Invitrogen | BP1756-100 | For annealing buffer |
| Phosphate Buffered Saline (1x) | Fisher Scientific | MT21040CM | PBS, for cell wash |
| DL-Dithiothreitol solution (1 M) | Sigma | 646563 | Reducing agent |
| Protease Inhibitor Cocktail | Thermo Scientific | 87786 | Prevents catabolism of TFs |
| Phosphatase Inhibitor Cocktail | Thermo Scientific | 78420 | Prevents dephosphorylation of TFs |
| Nonidet P-40 Substitute | IBI Scientific | IB01140 | NP-40, for nuclear extraction |
| BCA Protein Assay Kit | Thermo Scientific | 23225 | For measuring protein concentration |
| Odyssey EMSA Buffer Kit | Licor | 829-07910 | Contains all necessary EMSA buffers |
| TBE Gels, 6%, 12 Wells | Invitrogen | EC6265BOX | For EMSA |
| TBE Buffer (10x) | Thermo Scientific | B52 | For EMSA |
| FactorFinder Starting Kit | Miltenyi Biotec | 130-092-318 | Contains all necessary DAPA buffers |
| Licor Odyssey CLx | Licor | Recommended scanner for DAPA/EMSA | |
| Antibiotic-Antimycotic | Gibco | 15240-062 | Contains 10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of Fungizone® Antimycotic |
| Fetal Bovine Serum | Gibco | 26140-079 | FBS, for culture media |
| RPMI 1640 Medium | Gibco | 22400-071 | Contains L-glutamine and 25 mM HEPES |
참고문헌
- Hindorff, L. A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 106 (23), 9362-9367 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Ward, L. D., Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 30 (11), 1095-1106 (2012).
- Paul, D. S., Soranzo, N., Beck, S. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays. 36 (2), 191-199 (2014).
- Zhang, F., Lupski, J. R. Non-coding genetic variants in human disease. Hum Mol Genet. , (2015).
- Lee, T. I., Young, R. A. Transcriptional regulation and its misregulation in disease. Cell. 152 (6), 1237-1251 (2013).
- Slatkin, M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 9 (6), 477-485 (2008).
- Bush, W. S., Moore, J. H. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 8 (12), e1002822 (2012).
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 491 (7422), 56-65 (2012).
- Chang, C. C., et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
- Purcell, S., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81 (3), 559-575 (2007).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16 (1), 123-131 (2006).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 10 (12), 1213-1218 (2013).
- Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R., Lieb, J. D. FAIRE Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17 (6), 877-885 (2007).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Martens, J. H., Stunnenberg, H. G. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 98 (10), 1487-1489 (2013).
- Liu, T., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12 (8), R83 (2011).
- Griffon, A., et al. Integrative analysis of public ChIP-seq experiments reveals a complex multi-cell regulatory landscape. Nucleic Acids Res. 43 (4), e27 (2015).
- Staudt, L. M., et al. A lymphoid-specific protein binding to the octamer motif of immunoglobulin genes. Nature. 323 (6089), 640-643 (1986).
- Singh, H., Sen, R., Baltimore, D., Sharp, P. A. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature. 319 (6049), 154-158 (1986).
- Weirauch, M. T., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 158 (6), 1431-1443 (2014).
- Ward, L. D., Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40 (Database issue), D930-D934 (2012).
- Boyle, A. P., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22 (9), 1790-1797 (2012).
- Hume, M. A., Barrera, L. A., Gisselbrecht, S. S., Bulyk, M. L. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 43 (Database issue), D117-D122 (2015).
- Mathelier, A., et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42 (Database issue), 142-147 (2014).
- Smith, M. F., Delbary-Gossart, S. Electrophoretic Mobility Shift Assay (EMSA). Methods Mol Med. 50, 249-257 (2001).
- Franza, B. R., Josephs, S. F., Gilman, M. Z., Ryan, W., Clarkson, B. Characterization of cellular proteins recognizing the HIV enhancer using a microscale DNA-affinity precipitation assay. Nature. 330 (6146), 391-395 (1987).
- . BCA Protein Assay Kit: User Guide Available from: https://tools.thermofisher.com/content/sfs/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf (2014)
- Wijeratne, A. B., et al. Phosphopeptide separation using radially aligned titania nanotubes on titanium wire. ACS Appl Mater Interfaces. 7 (21), 11155-11164 (2015).
- Silva, J. M., McMahon, M. The Fastest Western in Town: A Contemporary Twist on the Classic Western Blot Analysis. J. Vis. Exp. (84), (2014).
- Lu, X., et al. Lupus Risk Variant Increases pSTAT1 Binding and Decreases ETS1 Expression. Am J Hum Genet. 96 (5), 731-739 (2015).
- Ramana, C. V., Chatterjee-Kishore, M., Nguyen, H., Stark, G. R. Complex roles of Stat1 in regulating gene expression. Oncogene. 19 (21), 2619-2627 (2000).
- Fillebeen, C., Wilkinson, N., Pantopoulos, K. Electrophoretic Mobility Shift Assay (EMSA) for the Study of RNA-Protein Interactions: The IRE/IRP Example. J. Vis. Exp. (94), e52230 (2014).
- Heng, T. S., Painter, M. W. Immunological Genome Project, C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 9 (10), 1091-1094 (2008).
- Wu, C., et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10 (11), R130 (2009).
- Wu, C., Macleod, I., Su, A. I. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 41 (Database issue), D561-D565 (2013).
- Wang, J., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22 (9), 1798-1812 (2012).
- Holden, N. S., Tacon, C. E. Principles and problems of the electrophoretic mobility shift assay. J Pharmacol Toxicol Methods. 63 (1), 7-14 (2011).
- Xu, J., Liu, H., Park, J. S., Lan, Y., Jiang, R. Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development. 141 (7), 1442-1452 (2014).
- Yang, T. -. P., et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 26 (19), 2474-2476 (2010).
- Fort, A., et al. A liver enhancer in the fibrinogen gene cluster. Blood. 117 (1), 276-282 (2011).
- Solberg, N., Krauss, S. Luciferase assay to study the activity of a cloned promoter DNA fragment. Methods Mol Biol. 977, 65-78 (2013).
- Rahman, M., et al. A repressor element in the 5'-untranslated region of human Pax5 exon 1A. Gene. 263 (1-2), 59-66 (2001).
- Mali, P., et al. RNA-Guided Human Genome Engineering via Cas9. Science. 339 (6121), 823-826 (2013).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유