
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Screening für Funktionelle nichtkodierenden genetischen Varianten Mit elektrophoretische Mobilitäts-Shift-Assay (EMSA) und DNA-Affinitätspräzipitation Assay (DAPA)
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
We present a strategic plan and protocol for identifying non-coding genetic variants affecting transcription factor (TF) DNA binding. A detailed experimental protocol is provided for electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genotype-dependent TF DNA binding.
Zusammenfassung
Population and family-based genetic studies typically result in the identification of genetic variants that are statistically associated with a clinical disease or phenotype. For many diseases and traits, most variants are non-coding, and are thus likely to act by impacting subtle, comparatively hard to predict mechanisms controlling gene expression. Here, we describe a general strategic approach to prioritize non-coding variants, and screen them for their function. This approach involves computational prioritization using functional genomic databases followed by experimental analysis of differential binding of transcription factors (TFs) to risk and non-risk alleles. For both electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genetic variants, a synthetic DNA oligonucleotide (oligo) is used to identify factors in the nuclear lysate of disease or phenotype-relevant cells. For EMSA, the oligonucleotides with or without bound nuclear factors (often TFs) are analyzed by non-denaturing electrophoresis on a tris-borate-EDTA (TBE) polyacrylamide gel. For DAPA, the oligonucleotides are bound to a magnetic column and the nuclear factors that specifically bind the DNA sequence are eluted and analyzed through mass spectrometry or with a reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blot analysis. This general approach can be widely used to study the function of non-coding genetic variants associated with any disease, trait, or phenotype.
Einleitung
Sequenzierung und Genotypisierung basierte Studien, einschließlich der genomweiten Assoziationsstudien (GWAS), Kandidaten-Locus Studien und tiefSequenzierungsUntersuchungen haben viele genetische Varianten identifiziert, die statistisch im Zusammenhang mit einer Krankheit, Zug oder Phänotyp. Im Gegensatz zu dem frühen Vorhersagen, die meisten dieser Varianten (85-93%) werden in nicht-codierenden Regionen und die Aminosäuresequenz von Proteinen nicht 1,2 ändern. Interpretieren der Funktion dieser nicht-kodierenden Varianten und Bestimmung der biologischen Mechanismen , um sie zu dem zugeordneten Krankheit verbindet, trait oder Phänotyp hat Herausforderung erwiesen 3-6. Wir haben eine allgemeine Strategie entwickelt, um die molekularen Mechanismen zu identifizieren, die Varianten zu einem wichtigen Zwischen Phänotyp verknüpfen - Genexpression. Diese Pipeline wird speziell zu identifizieren, die Modulation der TF-Bindung durch genetische Varianten entwickelt. Diese Strategie kombiniert Rechenansätze und Techniken der Molekularbiologie Ziel vorherzusagenbiologische Wirkungen von Kandidatenvarianten in silico, und überprüfen diese Prognosen empirisch (Abbildung 1).

Abbildung 1:.. Strategischer Ansatz für die Analyse von nicht-kodierenden genetischen Varianten Schritte, die in der detaillierten Protokoll sind nicht mit diesem Manuskript assoziiert enthalten sind grau schattiert Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
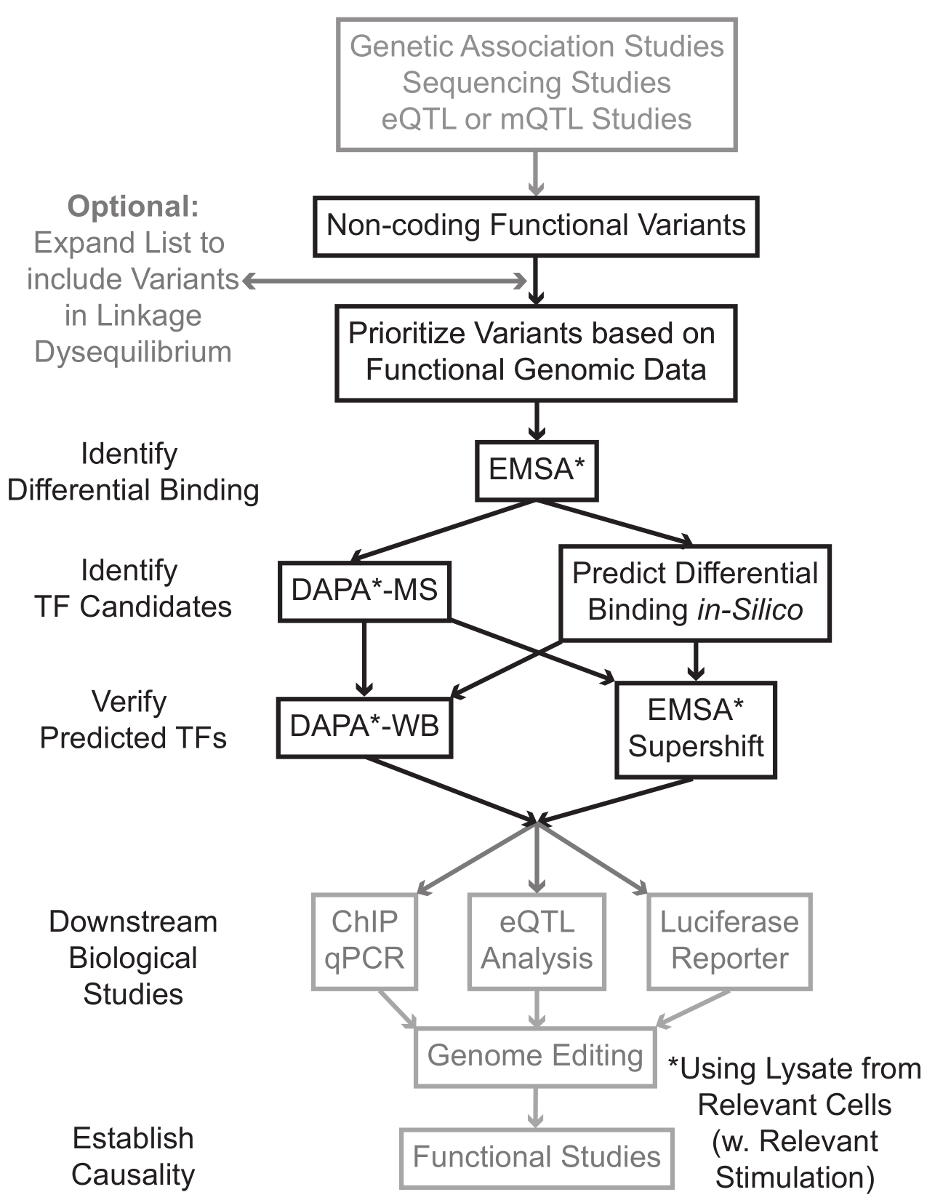{kind=link}
In vielen Fällen ist es wichtig, durch eine Erweiterung der Liste der Varianten zu beginnen alle, die in hohen Gestänge-Ungleichgewichts (LD) mit jeder statistisch assoziiert Variante enthalten. LD ist ein Maß der nicht-zufällige Assoziation von Allelen an zwei verschiedenen chromosomalen Stellen, die durch die r 2 -Statistik 7 gemessen werden kann. r 2 ist ein Maß für die linKage Ungleichgewichts zwischen zwei Varianten, mit einem r 2 = 1 bezeichnet perfekte Verbindung zwischen zwei Varianten. Allelen in hohen LD gefunden auf dem Chromosom über Vorfahren Populationen zusammen entmischen. Aktuelle Genotypisierung Arrays enthalten nicht alle bekannten Varianten im menschlichen Genom. Stattdessen verwerten sie die LD innerhalb des menschlichen Genoms und sind eine Teilmenge der bekannten Varianten , die 8 für andere Varianten innerhalb einer bestimmten Region des LD als Stellvertreter fungieren. Somit kann mit einer bestimmten Krankheit assoziiert sein eine Variante ohne biologische Folge kann, da es in LD mit dem kausalen varianten der Variante mit einer sinnvollen biologischen Wirkung. Prozedural, wird empfohlen , die neueste Version der 1000 Genome Projekt 9 Variante Call - Dateien (VCF) in binäre Dateien zu konvertieren kompatibel mit PLINK 10,11, einem Open-Source - Tool für Genom - Assoziations - Analyse. Anschließend werden alle anderen genetischen Varianten mit LD r 2> 0,8 mit jedem Eingang genetischen variant als Kandidaten identifiziert werden. Es ist wichtig , die entsprechende Referenzpopulation für diese Schritt- zB zu verwenden, wenn eine Variante , bei Themen von europäischer Abstammung identifiziert wurde, Daten von Patienten mit ähnlicher Herkunft sollte für LD Expansion verwendet werden.
LD Expansion resultiert oft in Dutzenden von Kandidatenvarianten, und es ist wahrscheinlich, dass nur ein kleiner Bruchteil von diesen zu Krankheitsmechanismus beizutragen. Oft ist es nicht durchführbar, experimentell jede dieser Varianten einzeln untersuchen. Es ist daher sinnvoll, die Tausende von öffentlich verfügbaren funktionellen genomischen Datensätze als Filter zu nutzen, um die Varianten zu priorisieren. Zum Beispiel 12 das ENCODE - Konsortium hat Tausende von ChIP-seq Experimente beschreiben die Bindung von Transkriptionsfaktoren und Co-Faktoren, und Histonmarkierungen in einem breiten Spektrum von Kontexten, zusammen mit Chromatin Zugänglichkeit von Daten von Technologien wie DNase-seq 13, ATAC ausgeführt -SEQ 14 und FAIRE-seq 15. DatabAses und Web - Servern wie dem UCSC Genome Browser 16, Roadmap Epigenomics 17, Epigenome Blueprint 18, Cistrome 19 und ReMap 20 bieten freien Zugang zu Daten , die durch diese und andere experimentelle Techniken in einer Vielzahl von Zelltypen und Bedingungen hergestellt. Wenn es zu viele Varianten sind experimentell zu untersuchen, können diese Daten verwendet werden, die innerhalb wahrscheinlich regulatorischen Regionen in entsprechenden Zell- und Gewebetypen angeordnet zu priorisieren. Ferner wird in Fällen, in denen eine Variante für ein spezifisches Protein in einer ChIP-seq peak ist, können diese Daten Potential führt als der spezifischen TF (s) oder Cofaktoren schaffen, deren Bindung könnte beeinflussen.
Als nächstes werden die resultierenden priorisiert Varianten gescreent experimentell vorhergesagt Genotyp-abhängige Protein zu validieren Bindungs 21,22 mittels EMSA. EMSA misst die Veränderung in der Migration des oligo auf einem nicht-reduzierenden TBE-Gel. Fluoreszierend markiertes Oligo mit der inkubiertenKern Lysat, und die Bindung von Kernfaktoren wird verzögern die Bewegung des Oligo auf dem Gel. Auf diese Weise Oligo, die mehr Kernfaktoren gebunden hat, als ein stärkeres Fluoreszenzsignal beim Abtasten präsentieren wird. Bemerkenswert ist, erfordert keine EMSA Prognosen über die spezifischen Proteine, deren Bindung betroffen.
Sobald Varianten identifiziert werden, die innerhalb der erwarteten regulatorischen Regionen befinden und in der Lage sind unterschiedlich Bindung Kernfaktoren sind Berechnungsverfahren die spezifische TF (s), dessen vorherzusagen verwendet Bindung sie beeinflussen könnten. Wir bevorzugen CIS-BP 23,24, RegulomeDB 25, UNIProbe 26 zu verwenden, und JASPAR 27. Sobald Kandidaten TFs identifiziert werden, können diese Prognosen ausdrücklich Antikörper gegen diese TFs (EMSA-supershifts und DAPA-Westerns) getestet werden. Eine EMSA-Supershift beinhaltet die Zugabe eines TF-spezifischen Antikörpers an das Kern Lysat und Oligo. Ein positives Ergebnis in einem EMSA-Supershift ist represented als eine weitere Verschiebung der EMSA Band oder einem Verlust des Bandes (in Bezug 28 überprüft). In der komplementären DAPA, eine 5'-biotinylierten Oligokomplexes die Variante und das 20 Basenpaar-flankierenden Nukleotide enthalten, werden mit nuklearen Lysat von relevanten Zelltyp (en) inkubiert alle Kernfaktoren speziell zur Erfassung der Oligos binden. Die Oligokomplexes-nuclear factor-Komplex wird durch Streptavidin-Mikrokügelchen in einer magnetischen Säule immobilisiert. Die gebundenen Kernfaktoren werden direkt durch Elution 29,48 gesammelt. Bindungsvorhersagen können dann durch Western-Blot untersucht werden, um Antikörper, spezifisch für das Protein verwendet wird. In Fällen, in denen es keine offensichtlichen Prognosen oder zu viele Vorhersagen, die Elutionen von Variante Pull-downs der DAPA Experimente können zu einem Proteomik Kern gesendet werden, um Kandidaten TFs mit Massenspektrometrie identifizieren, die anschließend validiert werden können mit diesen zuvor beschriebenen Methoden.
In dem Rest des article wird das detaillierte Protokoll für EMSA und DAPA Analyse genetischer Varianten vorgesehen.
Protokoll
1. Herstellung von Lösungen und Reagenzien
- Bestellen Sie kundenspezifische DNA-Oligonukleotid-Sonden zur Verwendung in EMSA und DAPA.
- Zur Reduzierung der unspezifischen Proteinbindung, entwerfen kurze Oligos (zwischen 35-45 Basenpaare (bp) in der Länge) 30, und legen Sie die Variante von Interesse direkt in der Mitte , flankiert von seinen 17 bp endogene genomische Sequenz. Für EMSA Oligos, eine 5'-Fluorophor hinzuzufügen. Für DAPA Oligos, eine 5'-Biotin-Tag hinzufügen.
- Um sowohl die Sense-Strang und seine Umkehrkomplement Strang. Alternativ bestellen Duplex (vorgeglüht) Oligos. Wenn die Oligos zu benennen, stellen Sie die Nomenklatur, die auf einem festgelegten Referenzgenom.
Hinweis: "Risiko" und "Nicht-Risiko" -Bezeichnung können Krankheit und projektspezifische, während "Referenz" und "non-reference" sind universell relevant sein. - Bei der Ankunft der Oligos, drehen kurz den Inhalt nach unten und resuspendieren in Nuklease-freies Wasser bis zu einer endgültigen Konzentration von 100 μ; M. Shop suspendiert Lager bei -20 ° C. Schützen Oligos mit einem Fluorophor aus Licht markiert, indem sie mit Alufolie wickeln.
| Name | Sequenz |
| rs76562819_REF_FOR | GTAATGCCTTAATGAGAGAG A GTTAGTCATCTTCTCACTTC |
| rs76562819_REF_REV | GAAGTGAGAAGATGACTAAC T CTCTCTCATTAAGGCATTAC |
| rs76562819_NONREF_FOR | GTAATGCCTTAATGAGAGAG G GTTAGTCATCTTCTCACTTC |
| rs76562819_NONREF_REV | GAAGTGAGAAGATGACTAAC C CTCTCTCATTAAGGCATTAC |
Tabelle 1: Beispiel EMSA / DAPA Oligonukleotid - Entwurf , der eine SNP für Differential bin zu testending. "REF" steht für die Referenz - Allels, während "NONREF" für die Nicht-Referenz - Allels steht. "FOR" steht für den vorderen Strang, während "REV" sein Komplement an. Die SNP wird in rot gesehen.
- Vorbereitung zytoplasmatischen Extraktion (CE) Puffer mit einer Endkonzentration von 10 mM HEPES (pH 7,9), 10 mM KCl und 0,1 mM EDTA in deionisiertem Wasser.
- Vorbereitung Kernextraktion (NE) Puffer mit einer Endkonzentration von 20 mM HEPES (pH 7,9), 0,4 M NaCl und 1 mM EDTA in deionisiertem Wasser.
- Vorbereitung Anelierungspuffer mit einer Endkonzentration von 10 mM Tris (pH 7,5-8,0), 50 mM NaCl und 1 mM EDTA in deionisiertem Wasser.
2. Herstellung von Kern Lysate aus kultivierten Zellen
Hinweis: Dieses experimentelle Protokoll wurde optimiert B-lymphoblastoiden Zelllinien, hat aber in mehreren anderen unabhängigen adhärenten / Suspensions-Zelllinien und arbeitet universell getestet.
- Culture B-lymphoblastoiden Zellen in Roswell Park Memorial Institute (RPMI) 1640 mit 2 mM L-Glutamin, 10% fötalem Rinderserum und 1x Antibiotikum-Antimykotikum, enthaltend 100 Einheiten / ml Penicillin, 100 ug / ml Streptomycin und 250 ng / ml Amphotericin B.
- Seed in einem Bereich von 200.000-500.000 lebenden Zellen / ml und Inkubation Kolben bei 37 ° C mit 5% Kohlendioxid in einer aufrechten Position mit belüfteten oder lose Kappen.
Hinweis: Das Wachstum der B-lymphoblastoiden Zellen verlangsamt, wenn sie über 1.000.000 Zellen / ml erreichen. Break up-Zell-Blasten durch Auf- und Abpipettieren mehrmals und das Rück Zellen zu 200.000-500.000 Zellen / ml eine schnelle Wachstumsrate aufrecht zu erhalten.
- Seed in einem Bereich von 200.000-500.000 lebenden Zellen / ml und Inkubation Kolben bei 37 ° C mit 5% Kohlendioxid in einer aufrechten Position mit belüfteten oder lose Kappen.
- Zweimal waschen kultivierten Zellen mit 10 ml eiskaltem Phosphat-gepufferte Salzlösung (PBS), Spin-down bei 4 ° C, 300 g für 5 min und entfernen PBS durch Absaugen.
- Zählen von Zellen mit einem Hämozytometer und Pellet als 1 ml eiskaltem PBS pro 10 7 Zellen.
Hinweis: Wenn beispielsweise Lysieren von 2 x 10 7 </ Sup> Zellen, Resuspendieren in 2 ml PBS. - Aliquot 1 ml bis 1,5 ml Mikrozentrifugenröhrchen so daß jedes Rohr 10 7 Zellen in PBS enthält. Zentrifuge bei 3.300 × g für 2 min 4 ° C und absaugen PBS.
- Vor der Verwendung hinzufügen 1 mM Dithiothreitol (DTT), 1x Phosphatase-Inhibitor und 1x Protease-Inhibitor zu einem Arbeits Lager von CE-Puffer. Resuspendieren Zellpellet mit 400 ul CE-Puffer und Inkubation für 15 Minuten auf Eis.
- In 25 ul 10% Nonidet P-40 und mischen durch Pipettieren. Zentrifuge bei 4 ° C, max Geschwindigkeit für 3 min. Dekantieren und den Überstand verwerfen.
- Vor der Verwendung hinzufügen 1 mM DTT, 1x Phosphatase-Inhibitor und 1x Protease-Inhibitor zu einem Arbeits Lager von NE-Puffer. Resuspendieren Zellpellet mit 30 ul Puffer NE und durch Vortexen mischen.
- Inkubieren bei 4 ° C in einem Rohr Rotator oder auf Eis für 10 min. Zentrifuge 3.300 xg für 2 min 4 ° C.
- Sammeln Sie die klare Überstand (nuclear Lysat) und aliquoten vor der Lagerung bei -806; C mehrere Gefrier-Auftau-Zyklen zu vermeiden, die das Protein abbauen kann. Lassen Sie eine 10 - ul - Aliquot Proteinkonzentration unter Verwendung des bichoninic Säure - Assay (BCA) 31 zu messen.
3. elektrophoretische Mobilität Shift Assay (EMSA)
- Bereiten Sie Oligo-Funktion Stock und EMSA Gel.
- Wenn Oligos im Duplex bestellt wurden, tauen die 100 uM Lager und verdünnt 1: 2000 in Anelierungspuffer 50 nM Arbeits Lager zu erreichen.
- Wenn Oligos wurden einsträngige bestellt, tauen die 100 uM Aktien und verdünnen 1:10 in Anelierungspuffer 100 nM Arbeitsvorräte zu erreichen. Kombinieren von 100 ul der 100 nM Komplement Strang Lösung miteinander in einem Mikrozentrifugenröhrchen.
- In einem Heizblock bei 95 ° C für 5 min. Schalten Sie die Hitze Block aus und lassen Sie die Oligos langsam für mindestens eine Stunde auf Raumtemperatur abkühlen vor der Verwendung.
- die EMSA Gel Pre-run.
- Entfernen Sie die Folie aus einem vorgegossenen 6% TBE Gel und spülen Sie unter entsalztes Wasser mehrmals jeden Puffer aus den Vertiefungen zu entfernen. Vorbereitung 1 l TBE-Puffer 0,5x durch Zugabe von 50 ml 10x TBE zu 950 ml deionisiertem Wasser hergestellt.
- Montieren Sie das Gel Elektrophorese-Vorrichtung und auf Dichtigkeit prüfen durch die innere Kammer füllt mit 0,5x TBE-Puffer. Wenn kein Puffer in die äußere Kammer austritt, füllen die äußere Kammer etwa zwei Drittel des Weges.
- Pre-run für 60 Minuten das Gel bei 100 V.
- Spülen Sie jede Vertiefung mit 200 ul 0,5x TBE-Puffer.
- Bereiten Sie Buffer Master Mix Bindung.
- Bereiten 10x Puffer mit einer Endkonzentration von 100 mM Tris-Bindungs, 500 mM KCl, 10 mM DTT; pH-Wert 7,5 in entmineralisiertem Wasser.
- In einem Mikrozentrifugenröhrchen, schaffen eine Mastermix der Reagenzien bestehend gemeinsam für alle Reaktionen (10 ul 10fach Bindungspuffer, 10 ul DTT / Polysorbat, 5 & mgr; l Poly d (IC), und 2,5 ul Lachssperma - DNA; Tabelle 2). Bereiten Sie eine zusätzliche 10%zur Volumenverlust durch Pipettieren zu berücksichtigen.
| Reagens | Endkonz. | Rxn 1 # | Rxn 2 # | Rxn 3 # | Rxn 4 # |
| Reinstwassersysteme | bis 20 & mgr; l Vol. | 13,5 ul | 11.98 ul | 13.5μl | 11.98 ul |
| 10x Binding Buffer | 1x | 2 ul | 2 ul | 2 ul | 2 ul |
| DVB - T / TW-20 | 1x | 2 ul | 2 ul | 2 ul | 2 ul |
| Lachssperma - DNA | 500 ng / ul | 0,5 ul | 0,5 ul | 0,5 ul | 0,5 ul |
| 1 ug / ul Poly d (IC) | 1 & mgr; g | 1 ul | 1 ul | 1 ul | 1 ul |
| Kernextrakt (5,26 ug / ul) | 8 & mgr; g | - | 1,52 ul | - | 1,52 ul |
| NE - Puffer | 1,52 ul | - | 1,52 ul | - | |
| Referenz - Allels Oligo | 50 fmol | 1 ul | 1 ul | - | - |
| Nicht-Referenz - Allels Oligo | 50 fmol | - | - | 1 ul | 1 ul |
Tabelle 2: Beispiel EMSA Reaktion setup. Die Tabelle zeigt ein Beispiel EMSA die Hypothese zu testen , dass Genotyp-abhängig ist , die Bindung von TFs zu einem spezifischen SNP.
- In Nuklease-freies Wasser in jedes Mikrozentrifugenröhrchen, so dass das endgültige Volumen nach Zugabe aller Reagenzien 20 ul sein wird.
- Fügen Sie den entsprechenden Betrag (5,5 ul) Master-Mix zu jedem Reaktionsgefäß.
- In 8 ug Kern Lysat zu den entsprechenden Mikrozentrifugenröhrchen. Fügen Sie Röhren die Oligo ohne Kernextrakt als Negativkontrollen (zB Tabelle 2, Rxn # 1 und Rxn # 3) enthält.
Anmerkung: Die optimale Menge an Lysat pro Reaktion experimentell durch Titration bestimmt werden. Im Allgemeinen wird ein Bereich von 2-10 & mgr; g Lysat Titrieren ausreichend. - In 50 fmol Oligo an die entsprechenden Mikrozentrifugenröhrchen. Flick zu mischen und kurz den Inhalt auf den Boden der Röhre drehen. Inkubieren für 20 min bei Raumtemperatur.
Hinweis: Wenn attemptieinen Supershift ng, inkubiere für 20 min mit Antikörper bei Raumtemperatur vor der Zugabe von Oligos des Lysats Mischung. Es wird empfohlen, für die besten Ergebnisse 1 ug eines ChIP-Grade-Antikörper zu verwenden. - In 2 ul 10x orange Loading Dye zu jedem Reaktionsgefäß. Pipette nach oben und unten zu mischen.
- Legen Sie die Proben in den Vorlauf 6% TBE-Gel durch Pipettieren nach oben und unten zu mischen und dann jede Probe in einem separaten gut vertreiben. Führen Sie das Gel bei 80 V bis die orange Farbstoff 2/3 bis 3/4 des Weges durch das Gel gewandert ist. Dies sollte etwa 60 bis 75 Minuten dauern.
- Entfernen Sie das Gel aus dem Plastikkassette durch sie offen mit einem Gel Messer neugierigen und legen Sie das Gel in einen Behälter mit 0,5% TBE-Puffer, damit er nicht austrocknet.
- Legen Sie das Gel auf der Oberfläche eines Infrarot und Chemilumineszenz-Bildgebungssystem, wobei Sie alle Blasen oder Verunreinigungen zu beseitigen, die das Bild stören wird.
- Mit dem Scan-System-Software, klicken Sie auf den "Acquire" Registerkarteund dann wählen Sie "Neu Zeichnen", um ein Feld ziehen rund um den Bereich entsprechend, wo das Gel befindet sich auf der Oberfläche des Scanners.
- In der "Kanäle" des "Acquire", wählen Sie den Kanal auf der Wellenlänge des Fluorophor-Markierung auf dem Oligo entspricht. In der "Scanner" Abschnitt, klicken Sie auf "Vorschau", um eine minderwertige Scan-Vorschau erhalten. Stellen Sie den Scanbereich durch das blaue Feld ziehen, das erworbene Vorschaubild bis auf den Teil des Gels umgebenden abgebildet werden.
Hinweis: Wenn zum Beispiel Oligos, markiert mit einem 700 nm Fluorophore verwenden, stellen Sie sicher, dass die "700 nm" Kanal vor dem Scannen ausgewählt wird. - In der "Scan-Steuerungen" Abschnitt, wählen Sie die "84 & mgr; M" Auflösungsoption und das "Medium" Qualität Option. Setzen Sie den Fokus auf die Hälfte der Dicke des Gels zu kompensieren. Hinweis: Zum Beispiel kann ein 1 mm Gel mit einer 0,5 mm Fokusversatz verwenden würde.
- In der "Scanner" Abschnitt, klicken Sie auf "Start" zu bEGIN den Scanvorgang.
Hinweis: Während des Scans die Helligkeit, Kontrast und Farbschema oft manuell angepasst werden je nach Hersteller des Abtastsystems werden kann. - Nachdem die Prüfung abgeschlossen ist, wählen Sie die Registerkarte "Bild" und klicken Sie auf "Drehen oder Kippen" im Abschnitt "Create", um die Ausrichtung zu korrigieren. Speichern Sie die Bilddatei von "Export" im Hauptmenü klicken und dann auf "Einzelbildansicht."
4. DNA-Affinitätsreinigung Assay (DAPA)
- Herstellung von 5 & mgr; M Oligo-Funktion Stock.
- Wenn Oligos im Duplex bestellt wurden, tauen die 100 uM Lager und verdünnen 01.20 in Anelierungspuffer 5 uM Arbeits Lager zu erreichen.
- Wenn Oligos wurden einsträngige bestellt, tauen die 100 uM Aktien und verdünnen 1:10 in Anelierungspuffer 10 uM Arbeitsvorräte zu erreichen. Mähdrescher 10 & mgr; l der 10 & mgr; M komplementäre Stränge miteinander. In einem Heizblock bei 95 ° C für5 Minuten. Schalten Sie die Hitze Block aus und lassen Sie die Oligos langsam auf Raumtemperatur abkühlen vor der Verwendung.
- Vor dem Start aufzuwärmen Bindungspuffer, niedriger Stringenz-Waschpuffer, hochstringenten Waschpuffer und Elutionspuffer auf Raumtemperatur.
Hinweis: eine Endkonzentration von 50 ng / ml Poly d (IC) mit dem Bindungspuffer, niedriger Stringenz-Waschpuffer hinzugefügt und hoher Stringenz-Waschpuffer potentielle nicht-spezifische Bindung von Proteinen an die Oligos zu reduzieren. - Bereiten Sie die Bindung Mischungen für jede Variante.
- 1 Volumenteil der Kern Lysat mit 2 Volumina Bindungspuffer.
Anmerkung: Die erforderliche Menge an Lysat muss experimentell bestimmt werden aufgrund unterschiedlicher Fülle von TFs. zwischen 100-250 ug Kern Lysat pro Spalte ist in den meisten Fällen ausreichend. - In 1x Phosphatase-Inhibitor, 1x Proteaseinhibitor und 1x Bindungsverstärker (optional) und mischen durch das Rohr mehrmals schnippen.
Hinweis: 100x BindungEnhancer besteht aus 750 mM MgCl 2 und 300 mM ZnCl 2. Hinzufügen Bindungsverstärker, wenn die Bindung des TF an DNA auf Cofaktoren oder Reduktionsmittel abhängig ist. Wenn diese Informationen nicht bekannt ist, fügen Sie den Bindungsverstärker. - In 10 ul 5 uM biotinylierten Fänger-DNA (50 pmol) zu jeder jeweiligen Bindungsmischung. Inkubieren für 20 min bei Raumtemperatur.
Hinweis: Die Inkubationszeit und Temperatur variieren in Abhängigkeit von der TF. Die optimalen Werte müssen experimentell bestimmt werden.
- 1 Volumenteil der Kern Lysat mit 2 Volumina Bindungspuffer.
- 100 l Streptavidin-Mikrokügelchen. Inkubieren für 10 min bei Raumtemperatur.
- Für jede Oligo-Sonde getestet, legen Sie eine verbindliche Spalte in der Magnetscheider. Legen Sie ein Mikrozentrifugenröhrchen direkt unter jeder Bindungssäule und anwenden 100 ul Bindungspuffer der Säule zu spülen.
- Pipettieren den Inhalt der einzelnen Bindungsmischung in separaten Spalten und die Flüssigkeit vollständig durch die Säule in die Mikro fließenRohr, bevor Sie fortfahren. Achten Sie darauf, die Spalten mit der Variante Oligo zu kennzeichnen, die in der Bindungsmischung verwendet wurde. Beschriften Sie die Flow-Through-Proben und ersetzen sie durch neue Mikrozentrifugenröhrchen, die mit geringer Stringenz Waschungen zu sammeln.
- Bewerben 100 ul niedrig stringenten Waschpuffer auf die Säule; warten, bis die Säule Behälter leer ist. Wiederholen Sie waschen 4x. Beschriften Sie die niedrigen Stringenz Waschproben und ersetzen sie durch neue Mikrozentrifugenröhrchen, die mit hoher Stringenz Waschungen zu sammeln.
- Anwenden 100 ul hoher Stringenz-Waschpuffer auf die Säule; warten, bis die Säule Behälter leer ist. Wiederholen Sie waschen 4x. Beschriften Sie die mit hoher Stringenz Waschproben und ersetzen sie durch neue Mikrozentrifugenröhrchen die Vorelution zu sammeln.
- In 30 ul nativen Elutionspuffer auf die Säule und lassen Sie sich für 5 Minuten stehen. Beschriften Sie die Vorelution Proben und ersetzen sie durch neue Mikrozentrifugenröhrchen die Elution zu sammeln.
Hinweis: Das ist nicht das gebundene Protein nicht eluieren; es wäscht die verbleibende hoher Stringenzaus dem Säulenpuffer und ersetzt sie durch Elutionspuffer die Effizienz der Elution zu maximieren. - Fügen Sie einen zusätzlichen 50 ul nativen Elutionspuffer die gebundenen TFs zu eluieren. Für eine höhere Ausbeute, aber weniger konzentrierte Eluat, fügen Sie eine zusätzliche 50 ul nativen Elutionspuffer und die Durchfluss sammeln.
Hinweis: Analysieren Elution Proben durch Massenspektrometrie 32 die Identität des gebunden TFs zu bestimmen. Anschließend überprüfen Sie die Proteom - Ergebnisse durch Natriumdodecyl- Sulfit - Polyacrylamid - Gelelektrophorese (SDS-PAGE) , gefolgt von einem Western - Blot - 33. Wenn Massenspektrometrie nicht verfügbar ist, führen Sie eine Silberfärbung unter Verwendung von Standard-Technik anstelle eines Western-Blot, der die Größe des Proteins (e) zeigt Genotyp-abhängige Bindung zu bestimmen. Verwenden Sie diese Informationen zu verengen die Liste der vorhergesagten TFs von den Rechenansätze in der Einleitung beschrieben.
Ergebnisse
In diesem Abschnitt repräsentative Ergebnisse von dem, was zu erwarten, zur Verfügung gestellt, wenn ein EMSA oder DAPA Durchführung und die Variabilität in Bezug auf die Qualität des Lysats gekennzeichnet ist. Beispielsweise wurde vorgeschlagen, dass mehrere Male Einfrieren und Auftauen Proteinproben in Denaturierung führen kann. Um die Reproduzierbarkeit der EMSA-Analyse im Rahmen dieser "Frost-Tau" Zyklen, zwei 35 bp Oligos unterschiedlichen an einer genetischen Varian...
Diskussion
Obwohl Fortschritte in der Sequenzierung und Genotypisierung Technologien stark unsere Fähigkeit, mit der Krankheit genetische Varianten assoziiert zu identifizieren verbessert haben, unsere Fähigkeit, die Funktionsmechanismen von diesen Varianten betroffen zu verstehen, hinkt. Eine wichtige Ursache des Problems ist, dass viele krankheitsassoziierten Varianten in n angeordnet sind on-kodierenden Regionen des Genoms, die wahrscheinlich Einfluss auf schwieriger zu sagen voraus Mechanismen Genexpression steuern. Hi...
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
We thank Erin Zoller, Jessica Bene, and Lindsey Hays for input and direction in protocol development. MTW was supported in part by NIH R21 HG008186 and a Trustee Award grant from the Cincinnati Children's Hospital Research Foundation. ZHP was supported in part by T32 GM063483-13.
Materialien
| Name | Company | Catalog Number | Comments |
| Custom DNA Oligonucleotides | Integrated DNA Technologies | http://www.idtdna.com/site/order/oligoentry | |
| Potassium Chloride | Fisher Scientific | BP366-500 | KCl, for CE buffer |
| HEPES (1 M) | Fisher Scientific | 15630-080 | For CE and NE buffer |
| EDTA (0.5M), pH 8.0 | Life Technologies | R1021 | For CE, NE, and annealing buffer |
| Sodium Chloride | Fisher Scientific | BP358-1 | NaCl, for NE buffer |
| Tris-HCl (1M), pH 8.0 | Invitrogen | BP1756-100 | For annealing buffer |
| Phosphate Buffered Saline (1x) | Fisher Scientific | MT21040CM | PBS, for cell wash |
| DL-Dithiothreitol solution (1 M) | Sigma | 646563 | Reducing agent |
| Protease Inhibitor Cocktail | Thermo Scientific | 87786 | Prevents catabolism of TFs |
| Phosphatase Inhibitor Cocktail | Thermo Scientific | 78420 | Prevents dephosphorylation of TFs |
| Nonidet P-40 Substitute | IBI Scientific | IB01140 | NP-40, for nuclear extraction |
| BCA Protein Assay Kit | Thermo Scientific | 23225 | For measuring protein concentration |
| Odyssey EMSA Buffer Kit | Licor | 829-07910 | Contains all necessary EMSA buffers |
| TBE Gels, 6%, 12 Wells | Invitrogen | EC6265BOX | For EMSA |
| TBE Buffer (10x) | Thermo Scientific | B52 | For EMSA |
| FactorFinder Starting Kit | Miltenyi Biotec | 130-092-318 | Contains all necessary DAPA buffers |
| Licor Odyssey CLx | Licor | Recommended scanner for DAPA/EMSA | |
| Antibiotic-Antimycotic | Gibco | 15240-062 | Contains 10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of Fungizone® Antimycotic |
| Fetal Bovine Serum | Gibco | 26140-079 | FBS, for culture media |
| RPMI 1640 Medium | Gibco | 22400-071 | Contains L-glutamine and 25 mM HEPES |
Referenzen
- Hindorff, L. A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 106 (23), 9362-9367 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Ward, L. D., Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 30 (11), 1095-1106 (2012).
- Paul, D. S., Soranzo, N., Beck, S. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays. 36 (2), 191-199 (2014).
- Zhang, F., Lupski, J. R. Non-coding genetic variants in human disease. Hum Mol Genet. , (2015).
- Lee, T. I., Young, R. A. Transcriptional regulation and its misregulation in disease. Cell. 152 (6), 1237-1251 (2013).
- Slatkin, M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 9 (6), 477-485 (2008).
- Bush, W. S., Moore, J. H. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 8 (12), e1002822 (2012).
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 491 (7422), 56-65 (2012).
- Chang, C. C., et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
- Purcell, S., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81 (3), 559-575 (2007).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16 (1), 123-131 (2006).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 10 (12), 1213-1218 (2013).
- Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R., Lieb, J. D. FAIRE Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17 (6), 877-885 (2007).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Martens, J. H., Stunnenberg, H. G. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 98 (10), 1487-1489 (2013).
- Liu, T., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12 (8), R83 (2011).
- Griffon, A., et al. Integrative analysis of public ChIP-seq experiments reveals a complex multi-cell regulatory landscape. Nucleic Acids Res. 43 (4), e27 (2015).
- Staudt, L. M., et al. A lymphoid-specific protein binding to the octamer motif of immunoglobulin genes. Nature. 323 (6089), 640-643 (1986).
- Singh, H., Sen, R., Baltimore, D., Sharp, P. A. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature. 319 (6049), 154-158 (1986).
- Weirauch, M. T., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 158 (6), 1431-1443 (2014).
- Ward, L. D., Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40 (Database issue), D930-D934 (2012).
- Boyle, A. P., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22 (9), 1790-1797 (2012).
- Hume, M. A., Barrera, L. A., Gisselbrecht, S. S., Bulyk, M. L. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 43 (Database issue), D117-D122 (2015).
- Mathelier, A., et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42 (Database issue), 142-147 (2014).
- Smith, M. F., Delbary-Gossart, S. Electrophoretic Mobility Shift Assay (EMSA). Methods Mol Med. 50, 249-257 (2001).
- Franza, B. R., Josephs, S. F., Gilman, M. Z., Ryan, W., Clarkson, B. Characterization of cellular proteins recognizing the HIV enhancer using a microscale DNA-affinity precipitation assay. Nature. 330 (6146), 391-395 (1987).
- . BCA Protein Assay Kit: User Guide Available from: https://tools.thermofisher.com/content/sfs/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf (2014)
- Wijeratne, A. B., et al. Phosphopeptide separation using radially aligned titania nanotubes on titanium wire. ACS Appl Mater Interfaces. 7 (21), 11155-11164 (2015).
- Silva, J. M., McMahon, M. The Fastest Western in Town: A Contemporary Twist on the Classic Western Blot Analysis. J. Vis. Exp. (84), (2014).
- Lu, X., et al. Lupus Risk Variant Increases pSTAT1 Binding and Decreases ETS1 Expression. Am J Hum Genet. 96 (5), 731-739 (2015).
- Ramana, C. V., Chatterjee-Kishore, M., Nguyen, H., Stark, G. R. Complex roles of Stat1 in regulating gene expression. Oncogene. 19 (21), 2619-2627 (2000).
- Fillebeen, C., Wilkinson, N., Pantopoulos, K. Electrophoretic Mobility Shift Assay (EMSA) for the Study of RNA-Protein Interactions: The IRE/IRP Example. J. Vis. Exp. (94), e52230 (2014).
- Heng, T. S., Painter, M. W. Immunological Genome Project, C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 9 (10), 1091-1094 (2008).
- Wu, C., et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10 (11), R130 (2009).
- Wu, C., Macleod, I., Su, A. I. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 41 (Database issue), D561-D565 (2013).
- Wang, J., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22 (9), 1798-1812 (2012).
- Holden, N. S., Tacon, C. E. Principles and problems of the electrophoretic mobility shift assay. J Pharmacol Toxicol Methods. 63 (1), 7-14 (2011).
- Xu, J., Liu, H., Park, J. S., Lan, Y., Jiang, R. Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development. 141 (7), 1442-1452 (2014).
- Yang, T. -. P., et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 26 (19), 2474-2476 (2010).
- Fort, A., et al. A liver enhancer in the fibrinogen gene cluster. Blood. 117 (1), 276-282 (2011).
- Solberg, N., Krauss, S. Luciferase assay to study the activity of a cloned promoter DNA fragment. Methods Mol Biol. 977, 65-78 (2013).
- Rahman, M., et al. A repressor element in the 5'-untranslated region of human Pax5 exon 1A. Gene. 263 (1-2), 59-66 (2001).
- Mali, P., et al. RNA-Guided Human Genome Engineering via Cas9. Science. 339 (6121), 823-826 (2013).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten