
Method Article
レプトスピラの病原性種におけるCRISPR干渉(CRISPRi)の遺伝子サイレンシングへの応用
要約
ここで、 レプトスピラ 種における特異的遺伝子サイレンシングに対するCRISPR干渉(CRISPRi)の適用について説明する。 レプトスピラ 細胞は、dCas9(触媒的に「死んだ」Cas9)とシングルガイドRNA(sgRNA)を発現するプラスミドとの共役によって形質転換され、所望のゲノム標的への塩基対合を担う。遺伝子サイレンシングを検証する方法を紹介します。
要約
レプトスピラ症は世界的に放置された動物性感染症であり、年間少なくとも100万例、約6万人の死亡を引き起こしています。この疾患は、レプトスピラ属の病原性および有害な細菌によって引き起こされるもので、細菌と直接接触するか、汚染された水または土壌への暴露によって間接的に起こされる。家畜および野生動物は感染の貯蔵所として機能し、腎臓の植民地化された腎尿細管から尿を介して環境にレプトスピレスを流す。レプトスピラの変異株の生成は、感染の病原性メカニズムを評価し、理解するために重要です。CRISPR干渉(CRISPRi)は、病原性レプトスピラにおける遺伝子サイレンシングのための簡単で手頃な価格で特定のツールであることが証明されています。したがって、dCas9とガイドRNAの両方を含むプラスミド構築物を得る方法論的詳細、大腸菌株β2163との共役によるレプトスピラへのプラスミドの送達、およびトランスコンジュガント回収および評価、について説明する。さらに、最近説明されたHornsby-Alt-Nally(HAN)培地は、寒天プレート上の突然変異コロニーの比較的迅速な単離および選択を可能にする。
概要
レプトスピラ症は、レプトスピラ属の病原性および毒性の種によって引き起こされる無視された世界的な動物性である。ヒトでは、この病気は世界中で年間100万人以上の症例と60,000人の死亡を占めています。これまでのところ、この病気に対する長期的かつ効果的なワクチンはありません。病原性因子および病原性メカニズムの同定は、より良い治療および予防戦略の開発にとって極めて重要である。したがって、遺伝子変異を生成し、得られた表現型を評価する能力は、機能的ゲノム解析3にとって極めて重要である。
病原性 レプトスピラ における変異体の構築は、これまで、本質的に非効率的で、骨が折れ、高価で、実装が困難であると考えられていました。このシナリオは、最近のCRISPR干渉(CRISPRi)を窒息4 および病原性5 レプトスピレスに適用して大きく変化しました。
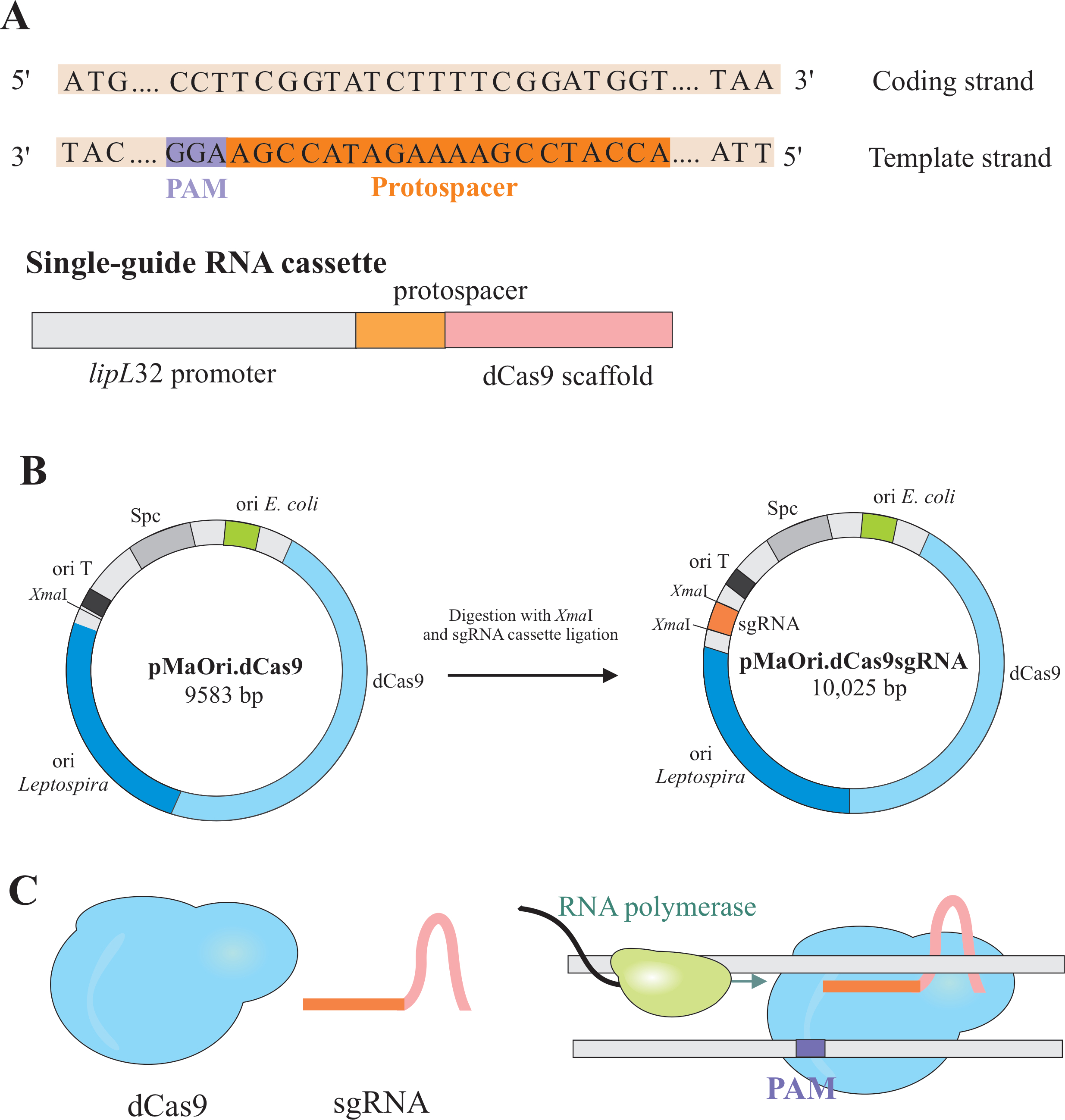
遺伝子サイレンシングは、CRISPR/Casの変種(c光沢のあるregularly interspaced s hortpalindromic repeat/CRISPRの2つの成分の発現によって達成される )酵素Cas9は、連鎖死Cas9(dCas9)およびシングルガイドRNA(sgRNA)と呼ばれる、所望の標的6、7、8に従って編集することができる。dCas9タンパク質は、sgRNAに結合すると、ワトソンとクリック塩基対合により特異的なDNA標的に向けられ、RNAポリメラーゼ伸長に対して立体的閉塞を引き起こし、遺伝子転写7の閉塞による遺伝子サイレンシングを生じる(図1)。
本稿は、dCas9およびsgRNAの両方を発現するためのプラスミドの構築、ドナー 大腸菌 β2163およびレシピエント レプトスピラ 細胞との間の結合、経協同化、および最後に、選択された突然変異コロニーの検証を記述することを目的とする。
プロトコル
1. プロトススペースの定義とプラスミド構造
注: このセクションでは、sgRNAを構築するための適切なプロトスペースを選択する最初のステップとpMaOri.dCas9へのさらにライゲーションを説明します(図1)。このプロトプスサー配列は、所望の標的に対する20ヌクレオチド配列を含む。
- GenBank(https://www.ncbi.nlm.nih.gov/genbank)でサイレンシングを目的とする遺伝子の塩基配列を取得する。「ファスタターゲット」を選択した後、ストレプトコッカス・ピオゲネスCas9とプロトスペーサー隣接モチーフNGGに定義されたパラメータを持つCHOPCHOPウェブサーバー(http://chopchop.cbu.uib.no/)に提出してください。パラメータを「CRISPR/Cas9」およびPAM(プロトスパサー隣接モチーフ)NGGに定義します。
- 得られた結果に基づいて、可能な限り最良のスコア(緑色の矢印)を有するプロトスペンサーを選択し、コーディング領域の5'末端にできるだけ近く位置し、そして最も重要なことに、sgRNAは完全な遺伝子サイレンシングのために遺伝子のコード鎖に対してペアでなければならないので、鋳型(マイナス)鎖に含まれる。
注: NGG モチーフは、最終的な sgRNA 配列には含まれていません。 - lipL32プロモーターを使用して、5'末端および保存されたdCas9足場配列で可変20ヌクレオチド配列を含む単一ガイドRNAを発現させます。プロトスパーサと呼ぶ20-nt配列を、lipL32プロモーター(その5'末端)およびsgRNA足場(3'末端)にマージする(図1B)。
注:明確に定義された lipL32プロモーターについては、TSSに-334を含むプロモーター領域を利用する(転写開始部位、ジュコバら9に基づく)。最終的な sgRNA カセットの 補足ファイル を確認します。 - シーケンシャルPCR5 によりsgRNAカセットを生成するか、市販業者によって合成した。
- カセットを入手した後、両端のXmaI制限部位でpMaOri.dCas9プラスミドにリゲートします(cccggg)4.
- SgRNAカセットとpMaOri.dCas9プラスミドをXmaI制限酵素で消化し、ライゲーションに進みます(図1B)。
- dT栄養性大腸菌株π11111でクローニングステップを実行し、pMaOri11(および拡張によりpMaOri.dCas9)複製の起源、R6K-γに起因する。
注: ライゲーションとクローン選択の詳細なプロトコルについては、フェルナンデスとナシメント12の以前の出版物を参照してください。sgRNA誘導dCas9は、選択した対象遺伝子のコード鎖に結合し、したがって、RNAポリメラーゼ伸長(図1C)を妨げ、遺伝子サイレンシングを生じる。
2. 共役による レプトスピラ 変換
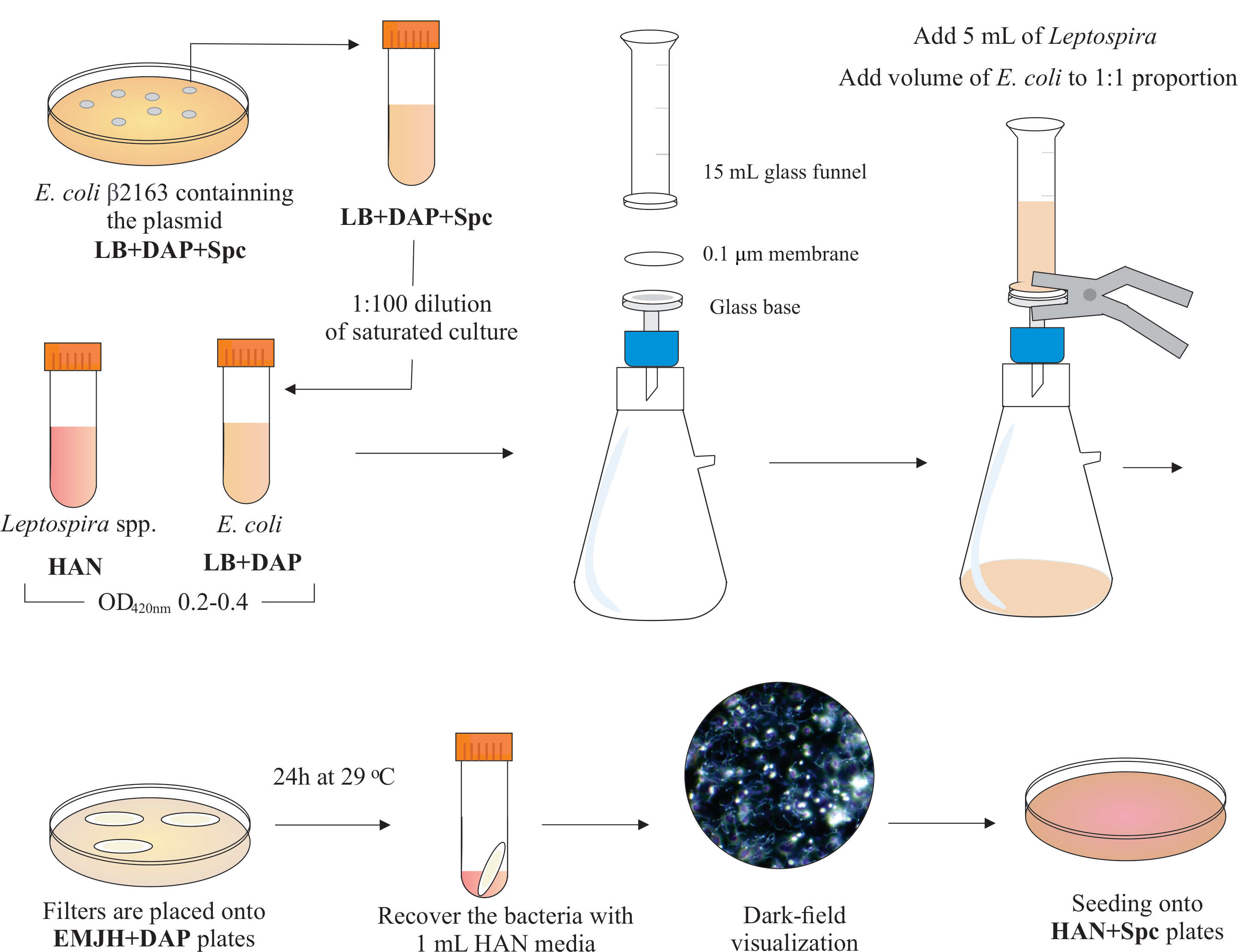
注: この手順のグラフィカルなスキームを図 2に示します。HANメディアとHANプレートを作る方法については、Hornsbyら13およびフェルナンデスら 5 を参照してください。
- 1:100で新鮮なHANで飽和培養物を希釈することによって攪拌下のHAN培地13で29または37 °Cで病原性レプトスピラ細胞を成長させる;典型的には、L. インターロガンスセロバル コペンハーゲン株フィオクルス L1-130 適切な細胞密度に到達するまでに 4-6 日かかります。
- 培養物は、結合に使用する前に、420 nm(2~5 x 108 細胞/mL)で0.2-0.4のO.D.に到達するようにしてください。
注意:HAN培地は、セル密度が増加するにつれて色が変化するため(DMEM培地に含まれるフェノールレッドによる)、遠心分離機(4,000 x g、15分、室温)1 mLの培養培地をレプトスピールを除去し、O.D.を測定するためのブランクとして上清を適用する。
- 培養物は、結合に使用する前に、420 nm(2~5 x 108 細胞/mL)で0.2-0.4のO.D.に到達するようにしてください。
- 結合性 大腸菌 株β2163を変換し、ジアミノピメル酸(DAP)の栄養要求性、プラスミドpMaOri.dCas9にsgRNAカセットを含む。 Eの場合. 大腸菌 変換は、熱衝撃プロトコルまたはエレクトロポレーションのいずれかを使用する。コントロールとしてsgRNAカセットを持たないプラスミドpMaOri.dCas9を用いた変換を含む。
- 熱ショック変換のために、プラスミドDNA(100 ng)を化学的に有能な 大腸菌 細胞と混合し、氷の上で30分間インキュベートします。42°Cで90sの熱ショックを行い、再び氷の上に5分間置きます。LB培地を1mL添加して細胞を回収し、37°Cで1時間インキュベートし、めっきに進みます。
- エレクトロポレーションの場合、100 ngのプラスミドDNAと混合した電気コンピテント細胞を使用してください。パルスに使用するパラメータは1.8kV、100 Ω、25μFです。
- 変換されたドナー 大腸菌 を、ジアミノピメル酸(DAP)(0.3mM)およびスペキノマイシン(40μg/mL)を添加したLB寒天培地にプレートを入れ、プラスミドを選択します。
- 結合のために、結合の日の前日の1つのプレートから1つのコロニーを選択する(これはレプトスピレスの培養のO.D.を監視することによって決定される)。
- 空のpMaOri.dCas9から 大腸菌 β2163のコロニーを1つ、pMaOri.dCas9sgRNAプレートから1コロニーを選択します。LB+DAPとスペクチノマイシンの10 mLで一晩37°Cで成長させます。
- 翌日、0.2-0.4のOD420nm まで、新鮮なLBプラスDAP(ここに抗生物質を含まない)の10mLで飽和培養1:100を希釈する。通常、 大腸菌 がこれらの密度に達するまでに2〜3時間かかります。
- BSL2バイオセーフティフードの内部に、ガラスベースの上部に直径25mm、0.1μmの細孔サイズ、混合セルロースエステル膜フィルターを配置して濾過装置を組み立てます。15 mLガラス漏斗を上部に置き、両方のピースをスプリングクランプで保持します。ガラスを真空ポンプに接続し、培養液を漏斗に加え、濾過します。
- 漏斗に レプトスピラ 培養液の5 mLを加えます。大 腸菌 の体積を加え、両培養のOD420nm 値に基づいて1:1の割合を構成する。真空ポンプをオンにし、濾過によって細胞を濃縮します。膜フィルター内の細胞濃度の後、慎重にそれを取り出す。培地が膜を通して濾過されていることを確認します。
メモ:ろ過は5〜10分かかります。 - DAP(0.3 mM)を補充した市販のEMJHプレート( 材料表を参照)にフィルターを置きます。バクテリア側がアップしていることを確認します。プレートを29°Cで24時間インキュベートします。
注意:HANまたは補足された社内EMJH14プレートを使用すると、大腸菌が増殖し、意図された1:1の割合を克服することができ、その結果、共役効率を低下させることができます5。 - 24時間後、プレートからフィルターを回収し、各フィルタを50 mLの円錐形チューブに入れ、
- 1 mLの液体 HAN 培地を使用して、広範なピペッティングとボルテックスによりフィルター表面から細胞を放出します。
- 回収された混合細菌溶液を暗視野顕微鏡で可視化し、細胞の生存率と運動性、 およびLeptospira:E.大腸菌 の割合を確認します。
注:この段階では、 大腸菌 と レプトスピラ の同等の数が見られます。 - この培養液の100~200μLを、0.4%不活性化ウサギ血清と40μg/mLスペチノマイシンを含むHANプレートに広げます。3%CO2雰囲気で37°Cのプレートをインキュベートします。
注:通常 、L.インターロガンス セロバルコペンハーゲン菌株フィオクルスL1-130細胞は、コントロールプレート上で5〜7日、スペチノマイシンプレート上で8〜10日でコロニーを形成します。この段階では、大 腸菌 はDAPの栄養性があるため成長しません。 - コントロールとして、104 レプトスピール/mLで培養を希釈し、抗生物質を使わずにプレートに100 μLを加え、レプトスパイラルの成長をモニタリングします。
3. コロニー選択とトランスコンジュガントの成長と検証
注:コロニーは10日目までに明らかにする必要があります。しかし、視覚化が容易に行えるわけではありません。通常、この時点で、HANプレートは、広がった乾燥細胞のために少し不透明であり、 レプトスピラ コロニーは白っぽい背景に対して透明なハローとして現れることができます。異なる光発生率を達成するために異なる角度でプレートを表示することをお勧めします, したがって、コロニーをより明白にします.より長いインキュベーション時間では、コロニーはより密な外観を得ることができ、この場合、それらは暗い背景に対して乳白色のハローとして提示する。
- 1.5 mLマイクロチューブに100μLの液体HAN培地を加え、変異体を回収します。各プレートから少なくとも3つのコロニーを取る。
- マイクロピペットの先端の助けを借りて、レプト渦巻きコロニーが地下にすることができるので、プレートからコロニーを取り出すために寒天を「掘る」。
注:寒天は、この段階で一緒に撮影される予定です。コロニーは、空のpMaOri.dCas9プラスミドを含むコントロールプレートと、標的遺伝子用に設計されたdCas9および単一ガイドRNAの両方を発現するプラスミドを含むレプトスピレスを含むプレートから採取する必要があります。 - 収集したコロニーを1.5 mLマイクロチューブで100μLのHAN培地に分配し、精力的に均質化します。この段階では、細胞を放出するために寒天の完全性の最大の破断を確保する。10 sのための懸濁液をボルテックス。
- マイクロピペットの先端の助けを借りて、レプト渦巻きコロニーが地下にすることができるので、プレートからコロニーを取り出すために寒天を「掘る」。
- ガラススライドに5μLの落下を加えて、200-400x倍率で暗視野顕微鏡で回収した細胞を可視化し、カバースリップですぐにサンプルを覆います。
- コロニーから回収された生き生きと生存可能なレプトスピレスの存在を確認する。
- 生存可能なレプトスピレスの可視化と確認の後、40 μg/mL スペチノマイシンを含む液体HAN培地に100 μLの細胞を移します。
- 液体HAN培地で増殖した後、プライマーpMaOri2 F(ACGCAATGTATCGATATGAC)およびR(ATAGGTGAAGTAGGCCCACCC)を用いてプラスミドの存在について培養を評価し、sgRNAカセットに隣接する領域を認識する。
- 培養物200μLを収集し、遠心分離機(4,000 x g、15分)、上清を捨て、得られたペレットを20μLの水に再懸濁します。
- この懸濁液を追加のPCRのテンプレートとして使用し、DNA抽出12を必要としない。
注:pMaOri.dCas9を有するセルは、723 bpのアンプリコンをレンダリングするsgRNAカセットを持つプラスミドを含むものと比較して、281 bpのアンプリコンをレンダリングします。
- 遺伝子サイレンシングの確認のために、pMaOri.dCas9(陰性対照)およびpMaOri.dCas9sgRNAのみを含むトランスコンジュガントからの細胞抽出物を利用して免疫ブロットを実行する。
- ドデシル硫酸ナトリウム(SDS)ポリアクリルアミドゲルのレーン当たり5 x 107 細胞に相当する量を塗布する。
- 適切な抗体を用いてインキュベーションするための膜への電気伝達タンパク質。サイレンシング用の標的遺伝子に対する抗体以外にも、負荷制御のために別のものを使用する。
- プラスミドを維持するためのHAN培地とスペクチノマイシンの変異培養を保持する。培地に抗生物質が適用されない場合、少なくとも3つの通路5について完全な遺伝子サイレンシングが観察される。
結果
レプトスピラspp. ゲノムの CG 含有量は通常約 35% です。事実上すべての遺伝子はPAM 5'NGG 3'を含む可能性が高い。このモチーフはテンプレートストランドで考慮する必要があります。CHOPCHOPの結果に基づいて遺伝子のコード配列(開始から停止コドンまで)を入力した後、プロトススペースはマイナス(-、テンプレート)鎖で選択されなければならない。NGGモチーフを20-nt sgRNAプロトススペースサーに含めないようにすることが重要である。
結合が1:1ドナー:レシピエント細胞比率で行われ、EMJH寒天プレートプラスDAPの表面に24時間、回収された細菌懸濁液の200μLがHANプラススペクチノマイシン寒天プレートに広がった場合、トランスコンジュガントコロニーは約8〜10日で見えるべきである。この体積の広がりは、通常、プレートあたり20〜40コロニーをもたらす(図3A)。結合後の細胞生存率をチェックするために、細胞は抗生物質を選択しないHANプレートに広がることができる。この場合、コロニーは7日ですぐに観察することができる。HANプレートは3%のCO2 雰囲気で淡黄色に変わります。
液体培地にプラススペクトルマイシンを加えてコロニーピッキングおよび成長した後、全細胞およびpMaOri2プライマーを用いたPCRは、トランスコンジュタントの初期品質チェックに使用することができる(図3B)。コントロールpMaOri.dCas9プラスミドを含むレプトスパイラル細胞は、281 bpのアンプリコンをもたらすはずですが、サイレンシング用のプラスミドを含む細胞、すなわちdCas9とsgRNAの両方を含む細胞は、723 bpアンプリコンをもたらすはずです。pMaOri2 FおよびRプライマーは、SgRNAカセット結紮中に使用される部位である XmaI制限部位に隣接するように設計された。
プラスミド存在の確認により、細胞を培地から回収し、PBSで2回洗浄し、次いで免疫ブロッティング用の全細胞抽出物を調製するために使用することができる。サイレンシングが発生した場合、標的タンパク質(この場合はLipL32またはLigAおよびLigBの両方)は、野生型細胞およびpMaOri.dCas9を含む細胞でのみ観察されるべきである。また、pMaOri.dCas9sgRNAを含む細胞では、対応するタンパク質を見るべきではありません(図3C)。
遺伝子サイレンシング後にレプト渦巻き毒性を評価する実験が計画されている場合、共役に使用される培養物は、低通路毒性 レプトスピラであるべきである。遺伝子サイレンシングが確認された後、いくつかのアリコートをバックアップとして凍結することができる。沈黙遺伝子が測定可能な表現型を有する場合、例えば、組換えタンパク質を用いた以前の研究に基づいて、培養物を検証に使用することができ、この場合、pMaOri.dCas9のみを含む細胞は、陰性対照として含めることができる。

図1:プラスミドを発現するdCas9とsgRNAの開発(A)A(A)長いプロトスペーサ、続いてS.pyogenes dCas9 PAM 5'-NGG-3'が標的遺伝子のテンプレート鎖内で選択され、その後のsgRNAがワトソンとクリックベースの組み合わせを対応するコード鎖に対して行うことができるように、完全な遺伝子サイレンシングをもたらす。(B)sgRNAカセットは、lipL32プロモーター、20-ntプロトスパーサー、およびdCas9足場から構成される。pMaOri.dCas9プラスミドは、XmaI制限部位におけるsgRNAカセット結紮のバックボーンとして使用される。得られたプラスミドは、pMaOri.dCas9sgRNAと呼ば、レプトスピレスに送達され、そしてdCas9およびsgRNAの両方の発現が遺伝子サイレンシングを担う。(C) sgRNA指向dCas9は、RNAポリメラーゼ伸長に対する物理的な障壁として作用し、したがって転写を妨げる。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:共役プロトコルの概略図望ましいレプトスピラ種は、420 nmで0.2-0.4(中間対数相)のO.D.まで、攪拌下のHAN培地で増殖する。結合の1日前に、目的のプラスミドを含む組換えドナー大腸菌β2163のコロニーは、細胞が同じ補充で液体LBで一晩成長するので、LB + DAP + Spc寒天プレートから摘み取られます。翌日、飽和E.大腸菌培養物はLB+DAPで希釈され、420nmで0.2-0.4のO.D.まで成長する。ドナー大腸菌とレプトスピラの両方が、陰圧下の濾過装置によって0.1μmフィルターの表面に1:1細胞プロポーションで混合される。次いで、フィルターをDAPを補ったEMJH寒天プレートの上に置き、29°Cで24時間インキュベーションを進行させる。 EMJHの使用は大腸菌の増殖を制限し、意図した1:1の割合は維持される。細菌は1 mLのHAN培地でピペット処理することによってフィルターから回収され、懸濁液は暗視野顕微鏡下で可視化される。最後に、各懸濁液の100〜200μLを0.4%のウサギ血清を含むHAN寒天プレートに播種し、3%CO2で37°Cでインキュベートした。この段階では、DAPは省略され、その結果、栄養性大腸菌は成長しない。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3:突然変異体の評価の代表的な結果:(A)空のpMaOri.dCas9(さらなる実験のための陰性対照)とプラスミドpMaOri.dCas9sgRNA(標的遺伝子が沈黙した状態)を含むプレートからのコロニーが摘み取られ、液体HAN中で精力的に均質化され、液体中で増殖する組換え細胞は、pMaOri.dCas9内のXmaI部位に隣接するプライマーを用いたPCRによって検証することができる。(B)この場合、pMaOri.dCas9を含む細胞は281bpのアンプリコンをもたらし、一方で、dCas9およびsgRNAの両方を含むサイレンシング用のプラスミドを含む細胞は、723 bpアンプリコンを示した。プラスミドの存在を確認した後、免疫ブロット分析により遺伝子サイレンシングが検証された。(C)標的タンパク質と負荷制御タンパク質の両方に対する抗体を用いてインキュベーションすることが推奨される。代表的な免疫ブロットでは、pMaOri.dCas9単独またはリプル32を標的とするsgRNAカセット(pMaOri.dCas9sgRNAlipL32)およびLigAおよびLigB(pMaOri.dCas9sgRNAligAB)遺伝子を含むトランスコンジュガントからの全細胞抽出物が表示される。抗LipL32、抗リガブおよび抗LipL41(非標的、ローディングコントロール)との共培養は、pMaOri.dCas9sgRNAlipL32およびリガとLigBの両方を含む細胞においてLipL32タンパク質の発現が廃止されることを確認する。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
補足ファイル: シングルガイドRNA(sgRNA)カセット配列。 sgRNA転写は、構成的な lipL32 プロモーター(太字ヌクレオチド)によって指示される。sgRNAは、プロトスペンサを指す20ヌクレオチドから構成され、標的遺伝子のコード鎖に塩基対合を担当し、dCas9足場配列(下線付きヌクレオチド)とを担う。 エクスマI制限部位(cccggg)は、pMaOri.dCas9プラスミドでのライゲーションの両端に含まれる。 このファイルをダウンロードするには、ここをクリックしてください。
ディスカッション
病原性15、16、17、18種の早期シーケンシングおよび天下19種の細胞化後、レプトスパイラル病因のいくつかの側面にゲノムのデータマイニングが光を当てた。ほとんどの場合、タンパク質機能は、遺伝子組換えのレプトスパイラル表面露出タンパク質と、その後のネイティブタンパク質機能20、21、22、23、24、25、26の組換え対応を用いて探索した。
変異体の生成、およびそれぞれの表現型の評価は、機能的ゲノム解析の主要な構成要素である。レプトスピラsppで変異体を生成する最初の試みは、ランダムトランスポゾン突然変異誘発27、28、29、30によって達成された。しかし、破壊された遺伝子の同一性を推測するための広範かつ骨の折れる分析の後、L. インターロガン・セロバー・マニラの全遺伝子の15%だけが27を破壊した。標的遺伝子ノックアウトは、自殺プラスミドを利用した相同組換えによってさらに達成され、目的の標的31,32内に相同の腕で横たわる抗生物質耐性カセットを送達する。
これらの技術を応用することにより、レプトスパイラル基礎生物学と病原性のいくつかの側面を探索した31、33、34、35 、36、37.大腸菌-レプトスピラ共役シャトルベクターの開発により、pMaOri 11は、エピソーム遺伝子サイレンシング用の成分の送達を可能にした。
Cas9誘導二本鎖破断はレプトスピラspp.に対して致死的であり、代わりに触媒的に不活性な酵素の変異体dCas9は、生理学的および病原性種4、5の両方で遺伝子サイレンシングを達成するために使用できることが示された。プラスミドpMaOri.dCas9をsgRNAカセットライゲーションのバックボーンとして使用することにより、dCas9とsgRNAの両方の発現により特異的かつ安定的な遺伝子サイレンシングが得られる。dCas9結合sgRNAは、ワトソン-クリック塩基対合により、所望の標的にタンパク質を導く。
完全な遺伝子サイレンシングのために、プロトスペースサーは、sgRNAの塩基対がコード鎖と共に起こるように、所望の遺伝子のテンプレート鎖に基づいて設計されるべきである。 レプトスピラ spp.の平均C+G含有量35%に基づいて、PAM 5'-NGG-3'は100 bpごとに少なくとも3回発生します。したがって、 レプトスピラ のゲノム内の事実上すべての遺伝子には、少なくとも1つのPAMが含まれます。しかし、モチーフNGGが見つからない場合、代替NAGモチーフを評価することができます。
以前の遺伝子サイレンシング技術は、ジンクフィンガーおよびTALE(転写活性化剤様エフェクター)のような、各標的に対して1つの異なるタンパク質の構築に依存し、これらの技術を骨の折れる、高価な38にする。CRISPRiの場合、可変成分はsgRNAであり、5'の終わりで20 bpだけを変更する必要があります。完全、安定、および標的遺伝子サイレンシングは、レプトスピラspp. 4,5だけでなく、他の細菌でも観察されている8,39,40,41.
HAN培地13の開発は、コロニー形成のためのインキュベーション時間を大幅に短縮し、レプトスピラが37o Cで成長することを可能にすることによって突然変異体の回復を支持した。しかし、共役工程中は、大腸菌がこの培地で激しく増殖し、ドナー細胞とレシピエント細胞の間で意図された1:1の割合を克服できるため、その使用はお勧めできません。この段階では、大腸菌はこのメディアで不十分に複製されるため、EMJHプラスDAPが良い選択です。一部の研究所は、大腸菌細胞の増殖をサポートする可能性のある追加のコンポーネントを含むことができる社内補足EMJHを作ることは言及する価値があります。
ここで提示される共役プロトコルは 、L.インターロガンス セロバルコペンハーゲン株フィオクルスL1-130に最適化され、また、土壌サンプル5から最近単離された病原性株の形質転換に有効であることが証明された。 L.ボルグペテルセニ 種の異なる血清バールを持つ最初の試みは、記載されたプロトコルとのより低い共役効率を示す。したがって、 レプトスピラの異なる種/血清体を扱う場合、結合のための最適な条件は、ドナー:レシピエント細胞の割合、初期細胞密度、共役培地および時間(24および48時間)を考慮して、経験的に決定されるべきである。レ プトスピラ 種とセロバルは、異なる共役プロトコルで異なる動作をすると仮定するのが妥当です。
窒息 性レプトスピラ コロニーはプレート上で比較的容易に視覚化できますが、病原性コロニーの観察はより困難です。通常、0.4%のウサギ血清およびスペチノマイシンを補ったHAN培地を用いることによって、トランスコンジュガントコロニーは10日目に観察され得る。私たちの経験では、コロニーは最初にメディア表面に透明なハローとして存在します。ビデオプロトコルでは、より密度の高いコロニーが、14日間の成長後、透明なものがフィルム化しにくいために示されている。この段階では、プレートを回転して異なる光発生率を達成し、白と暗い背景の間をシフトすると、コロニーを特定するのに役立ちます。
変異型の検証のために、イムノブロッティングは簡単なアプローチを提供します。しかし、抗体は標的タンパク質に対して常に利用できるとは限らないので、遺伝子サイレンシングを検証する代替戦略を追求することができます。sgRNA誘導dCas9は遺伝子転写の閉塞を担うため、標的遺伝子および構成制御にプライマーを用いた定量的な逆転写酵素PCR(qRT-PCR)は、遺伝子サイレンシングの検証に有効である。標的遺伝子がタンパク質ゲル中に明確に定義されたタンパク質バンドをコードする場合、SDS-PAGEはサイレンシングを実証することができ、またlipL32遺伝子に従ってサイレンシング5。LPS生合成遺伝子が沈黙している場合、LPS染色を採用することができます。明確に定義された基質を持つ酵素をコードする遺伝子をサイレンシングする場合、発火基質を用いた運動アッセイは有効な戦略である。β-ビフレクサにおけるβ-ガラクトシダーゼサイレンシングは、X-galおよびONPG(オルト・ニトロフェニル-βガラクトシド)基質4の使用により検証された。
遺伝子サイレンシングの確認後、さらに表現型を評価する実験を設計することができる。結合アッセイは、細菌の接着をサイレンシングする場合に行うことができる。血清チャレンジアッセイは、病原性 レプトスピラ5によって表示される血清生存におけるLigAおよびLigBの役割を確認した。変異体はまた、毒性の減衰を評価するために動物を接種するために使用することができます。この場合、変異体を接種した動物は、pMaOri.dCas9のみを含む細胞に感染したものと比較されるべきである。
結論として、現在のプロトコルは、10日以内に変異回復を促進するためにHAN培地を使用して病原性 レプトスピラ 種における遺伝子サイレンシングのためのCRISPRiの適用を記述する。遺伝子サイレンシングと機能ゲノム解析を組み合わせることで、 レプトスピラの病原性メカニズムの理解が深まり、最終的には疾病管理のためのより良い予防戦略の開発につながります。
開示事項
著者らは開示するものは何もない。
謝辞
USDAは機会均等プロバイダーと雇用主です。本書に記載されている商号や商用製品の言及は、特定の情報を提供する目的のみを目的としており、米国農務省の推薦または承認を意味するものではありません。ブラジルの機関FAPESP(助成金2014/50981-0)は、この作業を財政的に支援しました。LGVFはFAPESP(2017/06731-8および2019/20302-8)からのフェローシップで資金提供されています。資金提供者は、研究デザイン、データ収集と分析、出版の決定、または原稿の準備に何の役割も持っていませんでした。著者らはまた、ビデオプロトコルを撮影し、編集するためのUSDAビジュアルサービスのハンナ・ヒルとアレクサンダー・グライムズに感謝します。
資料
| Name | Company | Catalog Number | Comments |
| 0.1 µm pore size mixed cellulose esters membrane | Millipore | VCWP02500 | Filtration for bacterial conjugation |
| 2,6-Diaminopimelic acid (DAP) | Sigma | D1377 | Growth of auxotrophic E. coli β2163 |
| Agar Noble | BD & Company | 214230 | Used for preparation of solid EMJH and HAN plates |
| Bacto Agar | BD & Company | 214010 | Used for preparation of solid LB plates |
| Clarity Western ECL substrate | Biorad | 170-5060 | Chemiluminescent substrate |
| dNTP set | Thermo Fisher | 10297-018 | dNTPs for PCR reaction |
| Glass Microanalysis Filter Holder | Millipore | XX1012530 | Filtration for bacterial conjugation |
| Imaging System | Biorad | ChemiDoc MP | Chemiluminescence detection |
| LB broth, Miller | BD & Company | 244620 | Lysogenic liquid medium for E. coli culturing |
| Leptospira Enrichment EMJH | BD & Company | 279510 | Supplementation of EMJH media |
| Leptospira Medium Base EMJH | BD & Company | 279410 | EMJH medium for Leptospira |
| Mini-PROTEAN TGX Gels 12% | Biorad | 4568043 | Used for polyacrylamide gel eletrophoresis |
| Optical density reader | Molecular Devices | SpectraMax M2 | For optical density measurements of bacterial cultures |
| Phosphate Buffered Saline 7.4 | Sigma | 806552 | Saline solution for washing bacterial pellets |
| Spectinomycin | Sigma | S0692 | Selection of pMaOri backbone plasmids |
| Taq DNA Polymerase | Thermo Fisher | EP0402 | Enyme, buffer and MgCl2 for PCR reaction |
| Thermocycler | Applied Biosystem | GeneAmp PCR System 9700 | Used for PCR reaction cycling |
| Thymidine (dT) | Sigma | T9250 | Growth of auxotrophic E. coli π1 |
| XmaI restriction enzyme | New Englan BioLabs | R0180L | Digestion of plasmids and inserts |
参考文献
- Bharti, A. R., et al. Leptospirosis: A zoonotic disease of global importance. Lancet Infectious Disease. 3 (12), 757-771 (2003).
- Costa, F., et al. Global morbidity and mortality of Leptospirosis: A systematic review. PLoS Neglected Tropical Disease. 9 (9), 0003898 (2015).
- Shapiro, R. S., et al. A CRISPR-Cas9-based gene drive platform for genetic interaction analysis in Candida albicans. Nature Microbiology. 3 (1), 73-82 (2018).
- Fernandes, L. G. V., et al. Gene silencing based on RNA-guided catalytically inactive Cas9 (dCas9): a new tool for genetic engineering in Leptospira. Science Reports. 9 (1), 1839 (2019).
- Fernandes, L. G. V., Hornsby, R. L., Nascimento, A. L. T. O., Nally, J. E. Genetic manipulation of pathogenic Leptospira: CRISPR interference (CRISPRi)-mediated gene silencing and rapid mutant recovery at 37 C. Science Reports. 11 (1), 1768 (2021).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Qi, L. S., et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 152 (5), 1173-1183 (2013).
- Choudhary, E., Thakur, P., Pareek, M., Agarwal, N. Gene silencing by CRISPR interference in mycobacteria. Nature Communication. 6, 6267 (2015).
- Zhukova, A., et al. Genome-wide transcriptional start site mapping and sRNA identification in the pathogen. Frontiers in Cell and Infectious Microbiology. 7, 10 (2017).
- Demarre, G., et al. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPalpha) conjugative machineries, and their cognate Escherichia coli host strains. Research in Microbiology. 156 (2), 245-255 (2005).
- Pappas, C. J., Benaroudj, N., Picardeau, M. A replicative plasmid vector allows efficient complementation of pathogenic Leptospira strains. Applied Environmental Microbiology. 81 (9), 3176-3181 (2015).
- Fernandes, L. G. V., Nascimento, A. L. T. O. Specific gene silencing in Leptospira biflexa by RNA-guided catalytically inactive Cas9 (dCas9). Methods in Molecular Biology. 2134, 109-122 (2020).
- Hornsby, R. L., Alt, D. P., Nally, J. E. Isolation and propagation of leptospires at 37 °C directly from the mammalian host. Science Reports. 10 (1), 9620 (2020).
- Haake, D. A. Hamster model of leptospirosis. Current Protocols in Microbiology. , (2006).
- Nascimento, A. L., et al. Comparative genomics of two Leptospira interrogans serovars reveals novel insights into physiology and pathogenesis. Journal of Bacteriology. 186 (7), 2164-2172 (2004).
- Nascimento, A. L., et al. Genome features of Leptospira interrogans serovar Copenhageni. Brazillian Journal of Medical Biology Research. 37 (4), 459-477 (2004).
- Ren, S. X., et al. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature. 422 (6934), 888-893 (2003).
- Bulach, D. M., et al. Genome reduction in Leptospira borgpetersenii reflects limited transmission potential. Proceedings of the National Academy of Science U. S. A. 103 (39), 14560-14565 (2006).
- Picardeau, M., et al. Genome sequence of the saprophyte Leptospira biflexa provides insights into the evolution of Leptospira and the pathogenesis of leptospirosis. PLoS One. 3 (2), 1607 (2008).
- Fernandes, L. G., et al. OmpL1 is an extracellular matrix- and plasminogen-interacting protein of Leptospira spp. Infections and Immunity. 80 (10), 3679-3692 (2012).
- Fernandes, L. G., et al. Leptospira spp.: Novel insights into host-pathogen interactions. Veterinary Immunology and Immunopathology. 176, 50-57 (2016).
- Castiblanco-Valencia, M. M., et al. Leptospiral immunoglobulin-like proteins interact with human complement regulators factor H, FHL-1, FHR-1, and C4BP. Journal of Infectious Diseases. 205 (6), 995-1004 (2012).
- Choy, H. A., et al. The multifunctional LigB adhesin binds homeostatic proteins with potential roles in cutaneous infection by pathogenic Leptospira interrogans. PLoS One. 6 (2), 16879 (2011).
- Siqueira, G. H., et al. The recombinant LIC10508 is a plasma fibronectin, plasminogen, fibrinogen and C4BP-binding protein of Leptospira interrogans. Pathogen and Diseases. 74 (2), (2016).
- Teixeira, A. F., et al. Features of two new proteins with OmpA-like domains identified in the genome sequences of Leptospira interrogans. PLoS One. 10 (4), 0122762 (2015).
- Kochi, L. T., et al. The interaction of two novel putative proteins of Leptospira interrogans with E-cadherin, plasminogen and complement components with potential role in bacterial infection. Virulence. 10 (1), 734-753 (2019).
- Murray, G. L., et al. Genome-wide transposon mutagenesis in pathogenic Leptospira species. Infections and Immunity. 77 (2), 810-816 (2009).
- Bourhy, P., Louvel, H., Saint Girons, I., Picardeau, M. Random insertional mutagenesis of Leptospira interrogans, the agent of leptospirosis, using a mariner transposon. Journal of Bacteriology. 187 (9), 3255-3258 (2005).
- Pětrošová, H., Picardeau, M. Screening of a Leptospira biflexa mutant library to identify genes involved in ethidium bromide tolerance. Applied Environmental Microbiology. 80 (19), 6091-6103 (2014).
- Slamti, L., Picardeau, M. Construction of a library of random mutants in the spirochete Leptospira biflexa using a mariner transposon. Methods in Molecular Biology. 859, 169-176 (2012).
- Croda, J., et al. Targeted mutagenesis in pathogenic Leptospira species: Disruption of the LigB gene does not affect virulence in animal models of leptospirosis. Infections and Immunity. 76 (12), 5826-5833 (2008).
- Picardeau, M., Brenot, A., Saint Girons, I. First evidence for gene replacement in Leptospira spp. Inactivation of L. biflexa flaB results in non-motile mutants deficient in endoflagella. Molecular Microbiology. 40 (1), 189-199 (2001).
- King, A. M., et al. High-temperature protein G is an essential virulence factor of Leptospira interrogans. Infections and Immunity. 82 (3), 1123-1131 (2014).
- Lambert, A., et al. FlaA proteins in Leptospira interrogans are essential for motility and virulence but are not required for formation of the flagellum sheath. Infections and Immunity. 80 (6), 2019-2025 (2012).
- Murray, G. L., et al. Leptospira interrogans requires heme oxygenase for disease pathogenesis. Microbes and Infections. 11 (2), 311-314 (2009).
- Ristow, P., et al. The OmpA-like protein Loa22 is essential for leptospiral virulence. PLoS Pathogens. 3 (7), 97 (2007).
- Sasaki, Y., et al. Leptospiral flagellar sheath protein FcpA interacts with FlaA2 and FlaB1 in Leptospira biflexa. PLoS One. 13 (4), 0194923 (2018).
- Gaj, T., Gersbach, C. A., Barbas, C. F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends in Biotechnology. 31 (7), 397-405 (2013).
- Cress, B. F., et al. Rapid generation of CRISPR/dCas9-regulated, orthogonally repressible hybrid T7-lac promoters for modular, tuneable control of metabolic pathway fluxes in Escherichia coli. Nucleic Acids Research. 44 (9), 4472-4485 (2016).
- Zhao, C., Shu, X., Sun, B. Construction of a gene knockdown system based on catalytically inactive ("dead") Cas9 (dCas9) in Staphylococcus aureus. Applied Environmental Microbiology. 83 (12), (2017).
- Zhao, Y., et al. CRISPR/dCas9-mediated multiplex gene repression in Streptomyces. Biotechnology Journal. , 1800121 (2018).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved