
Method Article
Высококачественная подготовка ядер головного мозга и костного мозга для мультиомных одноядерных анализов
В этой статье
Резюме
Успех транскриптомики одноклеточных/одноядерных и мультиомиксов во многом зависит от качества клеток/ядер. Поэтому выделение клеток/ядер из ткани и их очистка должны быть строго стандартизированы. Этот протокол описывает подготовку ядер головного мозга и костного мозга для последующего одноядерного мультиомного анализа.
Аннотация
Одноклеточный анализ стал предпочтительным подходом для разгадки сложности биологических процессов, требующих оценки вариабельности индивидуальных клеточных реакций на лечение или инфекцию с одноклеточным разрешением.
За последние 10 лет было разработано множество методов молекулярного профилирования отдельных клеток, и несколько специализированных технологий были коммерциализированы. Каплеточное профилирование одиночных клеток 10X Genomics — это широко распространенная технология, которая предлагает готовые к использованию реагенты для транскриптомного и мультиомного профилирования одиночных клеток. Технология включает в себя рабочие процессы для секвенирования одноклеточной и одноядерной РНК (scRNA-Seq и snRNA-Seq соответственно), scATAC-Seq, одноклеточного иммунного профилирования (секвенирование BCR/TCR) и мультиома. Последний сочетает в себе транскрипционную (scRNA-Seq) и эпигенетическую (scATAC-Seq) информацию, поступающую из одной и той же клетки.
Качество (жизнеспособность, целостность, чистота) одноклеточных или одноядерных суспензий, выделенных из тканей и проанализированных любым из этих подходов, имеет решающее значение для получения высококачественных данных. Поэтому протоколы пробоподготовки должны быть адаптированы к особенностям каждой биологической ткани и обеспечивать получение высококачественных клеточных и ядерных суспензий.
В этой статье описаны два протокола подготовки образцов головного мозга и костного мозга для последующего конвейера multiome 10X Genomics. Протоколы выполняются поэтапно и охватывают диссоциацию тканей, сортировку клеток, выделение ядер и контроль качества готовой суспензии ядер, которая используется в качестве исходного материала для разделения клеток и штрихкодирования, подготовки библиотек и секвенирования. Эти стандартизированные протоколы позволяют создавать высококачественные библиотеки ядер, а также надежные и надежные данные.
Введение
В течение многих лет одноклеточные методы были золотым стандартом анализа биологических процессов. Первоначально они были ограничены фенотипированием отдельных клеток с помощью микроскопии, проточной цитометрии и аналогичных анализов. Прорыв в анализе одиночных клеток произошел с разработкой подходов к молекулярному профилированию отдельных клеток, в частности секвенирования одноклеточной РНК (scRNA-Seq), позволившего охарактеризовать весь транскриптом отдельных клеток. Очень мощная scRNA-Seq генерирует информацию о транскрипционном статусе клетки в определенном состоянии и в определенный момент времени. Тем не менее, он не дает представления о регуляции генов, которые управляют транскрипцией, или о молекулярных модификациях, которые происходят с течением времени. Чтобы преодолеть это ограничение, было вложено много усилий в разработку одноклеточных мультиомиксных анализов, которые позволяют анализировать множество факторов и процессов, происходящих из одной и той же клетки 1,2,3,4. Первое успешное измерение двух модальностей в одиночных клетках было получено путем связывания мультиплексных паттернов экспрессии поверхностных белков с полным транскриптомом отдельных клеток в подходе CITE-Seq5. Более поздние эволюции сочетают экспрессию генов с доступностью хроматина (анализ на транспозазный хроматин с использованием секвенирования, ATAC-Seq), тем самым одновременно захватывая транскриптомные и эпигеномные модальности в одних и тех же клетках (например, sci-CAR)6. Первые коммерческие решения, позволившие ассоциировать транскриптомику с клеточным фенотипом или с эпигенетическими изменениями одной и той же клетки, были разработаны компанией 10X Genomics.
Эксперименты по молекулярному профилированию отдельных клеток включают следующие этапы: (1) диссоциация тканей или приготовление одноклеточных суспензий; (2) очистка клеток и/или выделение ядер; (3) секционирование и штрихкодирование; (4) библиотечное строительство и контроль качества; (5) секвенирование нового поколения; (6) Анализ данных. Хотя шаги (3)-(6) могут значительно различаться в зависимости от используемой технологии, начальные шаги, как правило, являются общими для всех них. Качество приготовленной суспензии клетка/ядро будет определять общий результат эксперимента. В зависимости от типа ткани получение высококачественных одноклеточных/ядерных суспензий может быть сложной задачей. Особенности некоторых тканей, таких как сердце, мышцы, мозг, легкие, кишечник и другие, требуют методов разрушения тканей и выделения ядер, адаптированных к каждому типу образца, чтобы гарантировать получение высококачественных ядер для молекулярного анализа 7,8,9,10 . Методы разрушения тканей и протоколы диссоциации могут быть механическими, ферментативными (например, смесь коллагеназ и ДНКазы) или их комбинацией, и могут выполняться вручную или с помощью инструментов (например, Qiagen DSC-400, gentleMACS).
Одноклеточные методы стали предпочтительным инструментом для биомедицинских исследований. В нейробиологии разнообразие клеток в головном мозге и сложность их функций требуют анализа с высоким разрешением и высокой пропускной способностью для визуализации редких клеточных популяций и оценки их гетерогенности 11,12,13,14. Связывание клеточной идентичности и механизмов регуляции генов отдельных клеток дает представление о развитии и физиологии мозга. Другим примером являются исследования иммунного ответа в контексте инфекционных, аутоиммунных или раковых заболеваний, которые в значительной степени основаны на анализе отдельных клеток. Гетерогенность субпопуляций иммунных клеток и сложность их активности и взаимодействия с другими типами клеток требуют разрешения отдельных клеток для расшифровки механизмов, лежащих в основе иммунного ответа. Иммунные клетки происходят из костного мозга, где гемопоэтические предшественники состоят из постепенно дифференцируемых клеток, которые приобретают и теряют маркеры клеточной поверхности в течение поэтапного процесса, прежде чем выйти из костного мозга в дом на периферии. Анализ отдельных клеток позволяет детально охарактеризовать стадии клеточного развития. Это может быть достигнуто с помощью фенотипирования одной клетки, традиционно выполняемого с помощью многопараметрической проточной цитометрии. Тем не менее, было показано, что транскриптомные сигнатуры одиночных клеток позволяют более точно идентифицировать подтипы клеток-предшественников, поскольку эти клетки распределены в кластерах, которые попадают друг в друга и, следовательно, могут быть неправильно идентифицированы при использовании метода маркеров грубой клеточной поверхности15. Все большее число исследований раскрывает эпигенетические модификации, которые гемопоэтические стволовые и прогениторные клетки (HSPC) могут приобретать при воздействии различных агентов, что приводит к значительному влиянию на долгосрочную чувствительность иммунной системы 16,17,18,19. Новые мультиомиксные технологии позволяют изучать эти процессы с одноклеточным разрешением.
Для образцов головного мозга 11,20,21,22 и образцов костного мозга23,24 описано множество протоколов выделения клеток и ядер. Для минимизации систематической ошибки, обусловленной экспериментальной изменчивостью, необходимо валидировать оптимизированные протоколы подготовки одноядерных ядер для совместного одноклеточного транскриптомного и эпигеномного секвенирования, тем самым обеспечивая воспроизводимость одноклеточных мультиомных анализов.
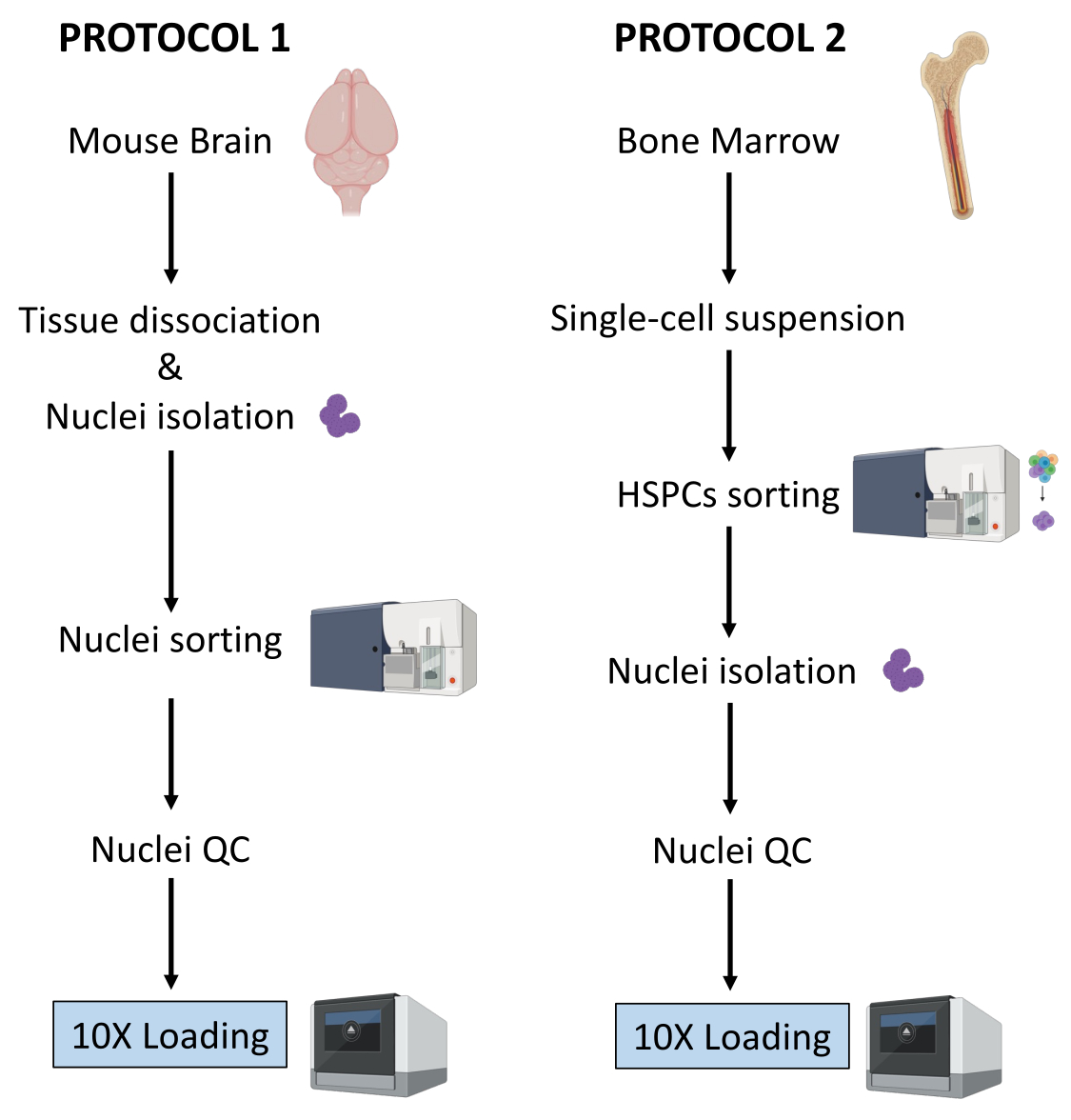
В данной работе описаны два надежных протокола подготовки ядер из (1) свежезамороженной ткани мозга и (2) свежих ГСПК костного мозга для последующего анализа одноклеточного мультиома (рис. 1).

Рисунок 1: Схематическое изображение протоколов выделения ядер из свежезамороженных тканей головного мозга и костного мозга. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
{kind=link}
протокол
Экспериментальные процедуры проводились в строгом соответствии с протоколами, утвержденными Комитетом по этике экспериментов на животных (CETEA). Для выделения ядер головного мозга использовали 3-месячных мышей C57BL/6. Для выделения костного мозга использовали 8-недельных самок мышей C57BL/6J массой 18 г.
1. Очищение ядер мозга мыши
ПРИМЕЧАНИЕ: Во время процедуры всегда надевайте латексные или нитриловые перчатки. Настоятельно рекомендуется, чтобы эксперимент выполняли два человека, шаги с 1 по 3 (т.е. приготовление одноядерной суспензии) выполнял один человек, а шаг 4 (т.е. подготовка сортировщика) выполнял параллельно другой человек. Поскольку протокол очень чувствителен ко времени, крайне важно свести к минимуму время обработки пробы, подготовив сортировщик сразу после приготовления одноядерной суспензии.
- Подготовка реагентов и материалов
- Тщательно простерилизовать инструменты для вскрытия в автоклаве (при 121 °C в течение 20 мин) и промыть их этиловым спиртом 70% непосредственно перед использованием. Приготовьте одну чашку Петри для каждого образца, наполненную 2-3 мл ледяного 1x фосфатно-солевого буфера (DPBS) Dulbecco.
- Охладите микроцентрифугу до 4 °C, наполните ведро льдом и поставьте стеклянный гомогенизатор на лед.
- Приготовьте буфер для лизиса ядер, добавив дигитонин для конечной концентрации 0,01%, 10 мл на образец.
- Приготовьте буфер для окрашивания, добавив ингибиторы РНКазы в буфер для окрашивания клеток для получения конечной концентрации 0,2 Ед/мкл, 20 мл на образец.
- Приготовьте DPBS 0,04% BSA, добавив ингибиторы РНКазы для получения конечной концентрации 0,2 U/мкл, 2 мл на образец.
- Приготовьте 1 мл буфера для разбавленных ядер в соответствии с протоколом Multiome25.
- Храните все реактивы и образцы на льду.
- Рассечение тканей
- Приносите в жертву мышей, используя протоколы, утвержденные учреждением. В этом протоколе мыши были обезглавлены после передозировки кетамина/ксилазина.
- Отрежьте голову мыши ножницами и извлеките мозг из черепа, как описано в Meyerhoff et al.26. Немедленно перенесите мозг в чашку Петри, приготовленную с помощью ледяного 1x DPBS, под стереомикроскопом со светодиодной подсветкой.
- Разрежьте ткань мозга скальпелем, чтобы отделить интересующие области мозга (например, энторинальную кору, гиппокамп, префронтальную кору) и перенесите каждую область в отдельную чашку Петри, содержащую ледяной 1x DPBS. Держите на льду.
- Скальпелем измельчите ткань на кусочки <0,5 см, чтобы облегчить гомогенизацию на следующем этапе.
- С помощью микропипетки P1000 перенесите измельченную ткань и 1x DPBS из чашки Петри в пробирку объемом 1,5 мл. Обязательно используйте пробирки из пластика с низким связыванием белка. Дайте кусочкам ткани отделиться под действием силы тяжести. Осторожно удалите излишки 1x DPBS с помощью микропипетки P1000.
ПРИМЕЧАНИЕ: После этого шага измельченную ткань можно мгновенно заморозить, перенеся пробирки с низким связыванием белка в сухой лед, а затем храня при -80 °C до выделения ядер.
- Выделение ядер
- Наполните стеклянную пробку 2 мл ледяного буфера для лизиса ядер с 0,01% дигитонина. Добавьте кусочки ткани в отскочку.
ПРИМЕЧАНИЕ: при работе со свежезамороженными тканями добавляйте измельченную замороженную ткань непосредственно в буфер лизиса ядер 0,01% дигитонина; Не допускайте оттаивания ткани до этого. - Гомогенизируйте с помощью гомогенизатора для салфеток со стеклянным отскоком 25 раз пестиком А, а затем 25 раз пестиком Б. Перелейте гомогенат в пробирку объемом 15 мл.
- Добавьте дополнительно 2 мл ледяного буфера для лизиса ядер с 0,01% дигитонином и инкубируйте на льду в течение 5 минут. Ядра центрифуги при 500 x g в течение 5 мин при 4 °C.
- Удалить надосадочную жидкость с помощью микропипетки и добавить 4 мл ледяного буфера для лизиса ядер с 0,01% дигитонина. Инкубируют на льду в течение 5 минут и процеживают через ситечко с ячейками 40 мкм.
- Центрифужируют ядра при 500 х г в течение 5 мин при 4 °C и удаляют надосадочную жидкость с помощью микропипетки.
- Добавьте 4 мл окрашивающего буфера для промывания ядер и центрифуги при 500 x g в течение 5 мин при 4 °C. Удалите надосадочную жидкость с помощью микропипетки и повторно суспендируйте гранулу в 4 мл окрашивающего буфера.
- Процеживают через ситечко с ячейками 40 мкм и центрифугу при 500 x g в течение 5 мин при 4 °C. Ресуспендировать в 1 мл PBS с 0,04% BSA.
- Подсчитайте ядра, чтобы обеспечить согласованность подготовки ткани/ядер в разных образцах. Ожидается, что будет получено аналогичное количество ядер из одних и тех же областей мозга:
- Добавьте 10 мкл 0,4% трипанового синего в пустую пробирку объемом 0,5 мл. Добавьте 10 мкл ядер и перемешайте 5 раз пипетированием.
- Подсчитайте ядра с помощью автоматического счетчика клеток, следуя рекомендациям поставщика. Держите ядра на льду.
- Подготовьте ядра к сортировке.
ПРИМЕЧАНИЕ: Экстрагированные ядра содержат 7-AAD, и это окрашивание используется для их очистки с помощью флуоресцентно-активированного клеточного сортировщика (FACS).- Перенесите 100 мкл ядер в пробирку FACS для контроля неокрашенности. Добавьте 10 мкл 7-AAD к оставшимся ядрам и держите 5 мин при 4 °C.
- Отсортируйте минимум 0,5 x 10,6 ядер по FACS, чтобы исключить дублеты и мусор.
- Наполните стеклянную пробку 2 мл ледяного буфера для лизиса ядер с 0,01% дигитонина. Добавьте кусочки ткани в отскочку.
- Сортировка ядер с помощью FACS
ПРИМЕЧАНИЕ: Несмотря на то, что сортировка ядер может выполняться на самых разных клеточных сортировщиках, процедура использования приборов BD FACSAria Fusion или BD FACSAria III описана здесь. Настоятельно рекомендуется, чтобы калибровка и настройка сортировщика ячеек выполнялись под наблюдением или опытным пользователем прибора. Чтобы сократить время обработки пробы, очень важно, чтобы сортировщик был готов сразу после приготовления одноядерной суспензии.- Калибровка прибора FACS
- Включите сортировщик ячеек и компьютер. После подключения программного обеспечения к прибору запустите процедуру запуска жидкости. Выберите Cytometer > Fluidic start-up в главном меню и выполните четыре шага. Нажмите « Готово » после завершения каждого из них.
- Вставьте сопло 70 мкм, включите струю и оставьте струю стабилизироваться на 15 минут. Отрегулируйте амплитуду, чтобы получить образование капель, и нажмите на Sweet Spot.
- Установите фильтр нейтральной плотности (N.D) 1.0 и откройте интерфейс настройки и отслеживания цитометра (CST).
- Ежедневный контроль качества: Разведите гранулы CST в среде FACS (см. рекомендации поставщика) и выполните контроль CST. После завершения замените N.D 1.0 на N.D 2.0.
- Разбавьте Accudrops в среде FACS (см. рекомендации поставщика) и выполните задержку капли, как описано в шагах 6–10.
- В шаблоне эксперимента выберите эксперимент «Задержка падения капли » и откройте макет сортировки для пробирки.
- В нижнем окне камеры нажмите « Напряжение », а затем « Оптический фильтр », чтобы включить нанесение заряда на отклоняющие пластины и использование специального оптического фильтра перед камерой. Убедитесь, что квадрант справа показывает 100. При необходимости отрегулируйте красный лазерный винт для оптимизации лазерного воздействия.
- Отрегулируйте скорость потока, чтобы достичь скорости от 1 000 до 3 000 событий в секунду.
- Нажмите « Сортировать и отменить». Убедитесь, что левый квадрант равен 100, а правый — 0. Если левый квадрант ниже 95, выполните автоматическую задержку.
- Нажмите « Напряжение», затем «Тестовая сортировка». Контролируйте качество боковых потоков, осаждаемых в сборных трубах. При необходимости отрегулируйте положение боковых потоков, перемещая ползунки.
- Настройка прибора FACS для сортировки ядер.
- Приступайте к приобретению неокрашенных ядер. Они используются для определения прямого и бокового рассеяния, а также напряжения детектора для параметра 7-ADD. Задайте параметры таким образом, чтобы сигнал 7-AAD неокрашенного образца попадал в первую декаду логарифмической шкалы на точечной диаграмме.
- Начните собирать трубку с 7-AAD-окрашенными ядрами и определите популяции ядер с помощью стратегии стробирования, основанной на (1) FSC-A/SCC-A, а затем FSC-H/SSC-H для размера и гранулярности, (2) FSC-H/FSC-A для различения дублетов и (3) SSC-A/7-AAD для 7-AAD положительных ядер (см. рис. 2A).
- Убедитесь, что струя и отклонение стабильны.
- В боковой потоковой камере включите тестовую сортировку, включите напряжение и подтвердите точную сортировку капель в пробирке объемом 1,5 мл, установленной с левой стороны.
- В окне Макет сортировки (Sorting Layout ) выберите интересующую совокупность, как определено на шаге 2 (выше). В разделе Целевые события выберите пороговое значение в поле Непрерывный , чтобы получить минимум 0,5 x 106 ядер на образец. В разделе « Точность» выберите «4-сторонняя чистота».
- Когда все будет готово, нажмите «Сортировать » и «ОК », чтобы начать сортировку ядер.
- Калибровка прибора FACS
- Контроль качества и подсчет очищенных ядер
ПРИМЕЧАНИЕ: Этот шаг должен быть выполнен только во время пилотного эксперимента по оптимизации этапов пробоподготовки с целью проверки чистоты отсортированных ядер, которые будут загружены на хромовый чип 10X. После того, как протокол полностью оптимизирован, не рекомендуется выполнять этот этап контроля качества в последующих экспериментах, чтобы избежать ненужных потерь собранных ядер, которые могут быть доступны в небольших количествах.- Контроль чистоты методом проточной цитометрии
- Перенесите 10 мкл отсортированных ядер в новую пробирку FACS, содержащую 90 мкл DPBS с 2% термически инактивированной фетальной бычьей сывороткой (HI-FBS).
- Собирайте и записывайте данные после сортировки, чтобы убедиться в чистоте и жизнеспособности сортировки. Убедитесь, что не менее 98% ядер находятся в интересующем вас затворе, как определено в 4.2 (см. рис. 2B).
- Подсчет очищенных ядер
- Центрифугой сортируют ядра в течение 5 мин при 500 х г и при 4 °С и осторожно полностью удаляют надосадочную жидкость с помощью микропипетки. Ресуспендировать в 100 мкл разбавленного буфера ядер.
- Добавьте 10 мкл 0,4% трипанового синего в пустую пробирку объемом 0,5 мл. Добавьте 10 мкл отсортированных ядер и перемешайте 5 раз пипетированием.
- Подсчитайте ядра с помощью автоматического счетчика клеток, следуя рекомендациям поставщика. Отрегулируйте концентрацию ядер до 3,5 x 106/мл, т.е. 16 000 ядер на 5 мкл.
- Контроль качества очищенных ядер методом микроскопии
ПРИМЕЧАНИЕ: Этот шаг должен быть выполнен только во время пилотного эксперимента для оптимизации этапов пробоподготовки для проверки качества ядер, которые будут загружены на хромистый чип 10X. После того, как протокол полностью оптимизирован, не рекомендуется выполнять этот этап контроля качества в последующих экспериментах, чтобы избежать ненужных потерь собранных ядер, которые могут быть доступны в небольших количествах.- Убедитесь, что предметные стекла микроскопа и крышки чистые и не содержат пыли. При необходимости вымойте и ополосните покровные стекла абсолютным этанолом и высушите их безворсовыми салфетками.
- Распределите 25 мкл поли-l-лизина в скользящих лунках, которые будут использоваться, и инкубируйте в течение 10 минут при комнатной температуре (RT), защищенной от пыли.
- Удалите излишки поли-l-лизина и добавьте 10 мкл очищенной суспензии ядер. Инкубируют в течение 5 мин при РТ, предохраняя от пыли.
- Добавьте по капле монтажной среды в каждую лунку, избегая пузырьков.
- Сверху на засеянные лунки положите покровный листок. Накройте бумажными салфетками и плотно прижмите защитный листок, чтобы удалить излишки монтажного материала. Будьте осторожны, чтобы не сдвинуть покровное стекло, и не очищайте излишки монтажного материала.
- Сделайте несколько снимков с помощью инвертированного микроскопа с яркопольным светом и минимальным увеличением 40х.
- Контроль чистоты методом проточной цитометрии
- Проведите мультиомный анализ.
- Немедленно перейдите к руководству пользователя Chromium Next GEM Single Cell Multiome ATAC + Gene Expression (CG000338 - Rev F)25.
2. Очистка ядер от гемопоэтических стволовых и прогениторных клеток костного мозга мышей (HSPC)
ПРИМЕЧАНИЕ Этот протокол описывает очистку ядер из трех подгрупп ГСПК костного мозга: lineage-c-Kit+Sca-1+ гемопоэтические стволовые клетки (ГСК), lineage-c-Kit+Sca-1-CD34+FcγR-распространенные миелоидные предшественники (CMP) и lineage-c-Kit+Sca-1-CD34+FcγR+ гранулоцитарно-моноцитарные предшественники (GMP). Во время процедуры всегда надевайте латексные или нитриловые перчатки. Этот протокол является адаптацией протокола 10X Genomics Demonstrated Protocol - Nuclei Isolation for Single Cell Multiome ATAC + GEX sequencing (CG000365 - Rev C)27. В первоначальный протокол были внесены изменения для максимального извлечения ядер. Настоятельно рекомендуется, чтобы эксперимент проводили два человека, чтобы был шаг 1. до 3. (т.е. приготовление одноячеистого раствора) выполняется одним человеком, а шаг 4 (т.е. приготовление сортировщика) выполняется параллельно другим человеком. Поскольку протокол очень чувствителен ко времени, очень важно свести к минимуму время обработки пробы, подготовив сортировщик сразу после приготовления одноячеистой суспензии.
- Подготовка реагентов и материалов
- Наполните ведро льдом.
- Приготовьте буфер FACS: DPBS с 2% раствором HI-FBS (примерно 500 мл на 6 образцов) и отфильтруйте через фильтр 0,2 мкм.
- Подготовьте среду для сбора: DPBS с 10% раствором HI-FBS (500 мкл на образец) и отфильтруйте через фильтр 0,2 мкм.
- Выделение клеток костного мозга
- Приносите в жертву мышей, используя протоколы, утвержденные учреждением. В этом эксперименте мыши были принесены в жертву вывихом шейки матки после передозировки кетамина/ксилазина.
- Опрыскайте живот и задние лапы мышей 70% этиловым спиртом.
- С помощью стерильных щипцов и ножниц сделайте небольшой разрез посередине нижней части живота и вскройте брюшину от основания задних лап до диафрагмы (дополнительный рисунок 1).
- Сделайте дополнительный надрез для каждой задней ноги перпендикулярно открытой брюшине, затем возьмитесь за любую сторону одного из этих дополнительных надрезов и потяните его, чтобы снять кожу с обеих задних лап мимо голеностопного сустава, чтобы обнажить мышцы обеих задних ног (дополнительный рисунок 1A).
- Проведите ножницами вдоль позвоночника в тазобедренном суставе одной задней ноги, чтобы вырезать ногу, не разрезая бедренную кость (дополнительный рисунок 1B, C). Повторите то же самое для другой ноги.
- Чтобы изолировать бедренную кость, вырежьте большую часть мышечной ткани, затем держите бедренную и большеберцовую кости в каждой руке кончиками пальцев в суставе (дополнительный рисунок 1D, E). Осторожно согните ногу относительно естественного сгиба, чтобы вывихнуть большеберцовую кость из бедренной кости (дополнительный рисунок 1E), а затем осторожно разрежьте соединительную ткань ножницами, чтобы отделить бедренную и большеберцовую кости.
- Используйте ножницы легкими скручивающими движениями, чтобы вывихнуть кусочек позвоночника из верхнего конца бедренной кости (дополнительный рисунок 1E).
- Очистите изолированную бедренную кость папиросной бумагой, чтобы удалить оставшуюся мышечную и соединительную ткань.
- Хранить в холодном состоянии на льду в 12-луночной пробирке, наполненной 2 мл DMEM (1x) + GlutaMAX-I.
- После того, как все бедренные кости собраны, убедитесь, что мышечная и волокнистая ткани полностью удалены из кости. Не разрезайте кость, чтобы: а) сохранить костный мозг стерильным и (б) избежать потери клеток в лунке. Используйте следующие шаги для промывания клеток из двух бедренных костей одной мыши, адаптированные из Haag и Murthy28.
- Приготовьте одну пробирку объемом 1,5 мл и одну пробирку объемом 0,5 мл. Добавьте 150 мкл буфера FACS в пробирку объемом 1,5 мл, затем проделайте отверстие в нижней части пробирки объемом 0,5 мл с помощью иглы 18 г и вставьте пробирку объемом 0,5 мл в пробирку объемом 1,5 мл.
- Вскрыть дистальную часть каждой бедренной кости с помощью хирургических ножниц мыши (Дополнительный рисунок 1F): Зафиксируйте дистальный эпифиз между лопатками и слегка надавите, переворачивая ножницы, чтобы плавно отделить дистальный эпифиз, не разрезая кость резко. В случае успеха на обнаженном конце физиса должны быть видны 4 выступа (дополнительный рисунок 1G).
- Вставьте две бедренные кости открытым концом вниз в подготовленную пробирку объемом 0,5 мл, помещенную в пробирку объемом 1,5 мл, содержащую буфер FACS (дополнительный рисунок 1H).
- Поместите ситечко с ячейками 70 мкм на пробирку объемом 50 мл и предварительно смочите ситечко 2 мл буфера FACS.
- Чтобы промыть костный мозг, центрифугируйте пробирки (крышки открыты) при 12 000 x g до тех пор, пока центрифуга не достигнет значения 12 000 x g , затем немедленно остановите центрифугу.
- Убедитесь, что клетки костного мозга гранулированы в пробирку объемом 1,5 мл, а бедренные кости белые (до промывки клеток они красные) (дополнительный рисунок 1I). Откажитесь от пробирок объемом 0,5 мл с 2 бедренными костью.
- Выбросьте 150 мкл надосадочной жидкости с помощью пипетки.
- Ресуспендировать гранулу с помощью микропипетки в 1 мл буфера для лизировки аммония-хлорид-калия (ACK) в течение 1-2 мин при РТ для лизировки эритроцитов. Избегайте более длительных инкубационных периодов, так как они могут привести к снижению жизнеспособности ядросодержащих клеток.
- Перелейте в пробирку объемом 50 мл через предварительно смоченный ситечко размером 70 мкм.
- Добавьте 10 мл буфера FACS, чтобы разбавить буфер для лизинга ACK и тем самым остановить лизис.
- Центрифуга при 400 x g в течение 5 мин при 4 °C. Ресуспендируйте в буфере FACS объемом 10 мл, сначала суспендировав в 1 мл, а затем долейте 9 мл.
- Подготовьте клетки к подсчету, как описано в 1.3.8.
- Подсчитайте ячейки с помощью автоматического счетчика ячеек в соответствии с рекомендациями поставщика. Ожидается, что он соберет около 40 миллионов клеток из 2 бедренных костей.
- Окрашивание костного мозга HSPC
- Центрифугируют клетки при 400 x g в течение 5 мин при 4 °C и ресуспендируют гранулы с помощью микропипетки в буфере FACS до конечной концентрации 1 x 107 клеток/мл.
- С помощью микропипетки P1000 перенесите суспензию в пробирку FACS, отфильтровав через колпачок сетчатого фильтра 35 мкм.
- Для каждого антитела, перечисленного в таблице 1 , подготовьте одиночные образцы пробирок для настройки компенсаций флуорохромов на клеточном сортировщике:
- Подготовьте одну пробирку FACS на каждое антитело и заполните пробирки 200 мкл PBS.
- Добавьте 15 мкл компенсационных гранул флуорохрома в каждую пробирку FACS с флуорохром-конъюгированным антителом. В пробирках FACS для неокрашенных и для живых/мертвых одиночных окрашенных клеток добавьте 500 000 ячеек вместо шариков.
- Добавьте 1 мкл каждого флуорохром-конъюгированного антитела (см. таблицу 1) в соответствующую пробирку FACS. Добавьте 0,5 мкл живого/мертвого пятна в пробирку Live/Dead с одним пятном FACS.
- Держать на льду 15 минут в защищенном от света месте.
- Приготовьте смеси 1 и 2, как указано в таблице 2.
ПРИМЕЧАНИЕ: Объемы антител, указанные в таблице 2 , действительны для антител, указанных в таблице материалов. Они должны быть оптимизированы для любого нового референса антител или другой партии того же эталона антител. - Добавьте 300 мкл смеси 1 в пробирку с образцом, ресуспендируйте и храните в течение 15 минут на льду в защищенном от света месте.
- Добавьте 300 мкл смеси 2 в пробирку с образцом, ресуспендируйте и храните в течение 20 минут на льду в защищенном от света месте.
- Добавьте 3 мл буфера FACS в пробирки с однократным окрашиванием и пробирки со смешанным окрашиванием. Отжим при 400 x g в течение 5 минут при 4°C.
- Осторожно выбросьте надосадочную жидкость с помощью микропипетки и повторно суспендируйте гранулу в 500 мкл буфера FACS.
- Приготовьте пробирку объемом 1,5 мл, предварительно заполненную 500 мкл среды для сбора.
ПРИМЕЧАНИЕ: Смесь 1 готовится в DPBS, так как она содержит краситель Live/Dead, на который значительно повлиял HI-FBS. После того, как клетки окрашены Live/Dead, добавляется смесь 2, которая содержит флуорохром-конъюгированные антитела, ресуспендированные в буфере FACS, содержащем HI-FBS. Единственным исключением является антитело к CD16/32, которое включено в смесь антител 1 в качестве блокатора Fc-рецептора, предотвращающего неспецифическое связывание других антител, добавленных на следующем этапе.
- Сортировка клеток с помощью FACS
ПРИМЕЧАНИЕ: Несмотря на то, что сортировка клеток может выполняться на самых разных сортировщиках, здесь описана процедура использования приборов BD FACSAria Fusion или BD FACSAria III. Настоятельно рекомендуется, чтобы калибровка и настройка сортировщика ячеек выполнялись под наблюдением или опытным пользователем прибора.- Калибровка прибора FACS: см. Протокол 1, шаг 4.1.
- Настройка прибора FACS для сортировки клеток:
- Приступайте к приобретению неокрашенных клеток. Они используются для определения прямого и бокового рассеяний, а также напряжения детектора для каждого флуорофора. Задайте параметры таким образом, чтобы флуоресцентный сигнал каждого флуорофора попадал в пределах первой декады логарифмической шкалы на точечной диаграмме.
- Используйте одноцветные элементы управления для настройки компенсаций вручную (медиана положительной и отрицательной совокупностей должна быть выровнена) или используйте программное обеспечение для автоматического расчета (измерения наклона). Убедитесь, что регуляторы компенсации соответствуют экспериментальным флуорохромам и настройкам детектора. Запишите 10 000 событий для ячеек и 5 000 событий для бусин.
- Используйте пробирку с образцом (т.е. многоцветные клетки) для определения интересующих клеточных популяций с помощью стратегии стробирования, показанной на рисунке 3A. Выполните шаги 4 - 6 (ниже).
- Для идентификации трех интересующих ГСПК костного мозга (ГСК, КМП и ГМП) начните стробирование с использования размера (FSC-A) и зернистости (SSC-A) для гейта лейкоцитов, а затем FSC-H/FSC-A для различения дублетов.
- На основе маркера SSC-A/мертвых клеток, затвор живых клеток. Используйте Lineage/c-Kit для выбора клеток, которые являются отрицательными по линии и экспрессируют промежуточные или высокие уровни c-Kit. Через c-Kit/Sca-1 установить ворота на ГСК lineage-c-Kit+ Sca-1+ (LKS+), одну из трех популяций, представляющих интерес.
- Среди миелоидных предшественников (lineage-c-Kit+Sca-1-) используют FcγR/CD34 для исключения CD34-FcγR-мегакариоцитов и эритроидных предшественников (MEP), включая CD34+ FcγR-CMP, а также CD34+FcγR+ GMP в клеточные популяции, подлежащие сортировке.
- Убедитесь, что поток и отклонение стабильны.
- В камере бокового потока включите тестовую сортировку, включите напряжение и подтвердите точную сортировку капель в пробирке объемом 1,5 мл, установленной с левой стороны.
- В окне Макет сортировки (Sorting Layout ) выберите интересующие популяции (например, "LKS+" и "CD34+ миелоидные предшественники", показанные в этом примере). В разделе «Устройство» выберите «2 трубки». В разделе Точность выберите Чистота. В разделе Целевые события выберите Непрерывный , чтобы отсортировать от 160 000 до 200 000 миелоидных предшественников LKS+ и CD34+ .
- Добавьте 500 мкл буфера FACS в клеточную суспензию и перенесите 1 мл образца путем фильтрации в новую пробирку FACS с 35 мкм клетками, чтобы убедиться, что все клетки находятся в одной суспензии непосредственно перед получением. Это устраняет скопления клеток, которые могут засорить прибор.
- Когда все будет готово, нажмите «Сортировка » и «ОК », чтобы начать сортировку. Отрегулируйте скорость потока , чтобы поддерживать скорость ниже 10 000 событий в секунду.
ПРИМЕЧАНИЕ: Ожидаемое соотношение миелоидных предшественников LKS+ и CD34+ составляет 1:3 для взрослой (8-12 недель) самки мыши C57BL/6J в равновесном состоянии. Целевые номера отсортированных ячеек обычно достигаются в течение 30 минут после сортировки.
- Контроль качества и подсчет отсортированных клеток
ПРИМЕЧАНИЕ: Этот этап должен выполняться только во время пилотного эксперимента по оптимизации этапов пробоподготовки с целью проверки чистоты отсортированных клеток, которые будут использоваться для выделения ядер. После того, как протокол полностью оптимизирован, не рекомендуется выполнять этот этап контроля качества в последующих экспериментах, чтобы избежать ненужных потерь исходного материала, который может быть доступен в небольших количествах для выделения ядер.- Контроль чистоты методом проточной цитометрии
- Перенесите 10 мкл отсортированных клеток в новую пробирку FACS, содержащую 90 мкл буфера FACS.
- Собирайте и записывайте данные после сортировки, чтобы убедиться в чистоте и жизнеспособности сортировки. Убедитесь, что по крайней мере 95% ячеек находятся в интересующем вас гейте, как определено в пунктах 3–6 и проиллюстрировано на рисунке 3B.
- Контроль чистоты методом проточной цитометрии
- Выделение ядер из сортированных ГППК костного мозга
- Используйте протокол "Low Cell Input Nuclei Isolation" Приложения из 10X Genomics Demonstrated Protocol - Nuclei Isolation for Single Cell Multiome ATAC + GEX sequencing (CG000365 - Rev C)27 со следующими изменениями, внесенными для оптимизации извлечения ядер:
- Время лизиса: Проведите пилотный эксперимент для этого протокола, чтобы определить наилучшее время лизиса для выделения ядер. Обеспечьте полный лизис клеток при сохранении неповрежденных ядер.
ПРИМЕЧАНИЕ: Шаг f вышеупомянутого протокола 10X Genomics27 предписывает «инкубировать [в буфере лизиса] в течение 3-5 минут на льду». Во время пилотного эксперимента тестируйте не менее 3 мин, 4 мин и 5 мин и оценивайте количество восстановленных ядер путем подсчета и качество с помощью проточной цитометрии и микроскопии, чтобы выбрать оптимальную продолжительность лизиса (см. описание этих проверок контроля качества ниже). Чтобы сэкономить реагенты, замените разбавленный буфер ядер на PBS 0,04% BSA в пилотном эксперименте. Для ГСПК костного мозга оптимальная продолжительность лизиса была определена как 3 мин. - Центрифугирование клеток: Для всех центрифугирования клеточной суспензии центрифугирование при 300 x g в течение 7 мин (вместо 5 мин в CG000365 - Rev C)27 при 4 °C.
- Центрифугирование ядер: Выполняйте все центрифугирования суспензии ядер при 500 x g в течение 5 мин в соответствии с CG000365 - Rev C27.
- Сбор ядер: На этапе b, после ресуспендирования в 50 мкл PBS 0,04% BSA и переноса в пробирку объемом 0,2 мл, добавьте 50 мкл PBS 0,04% BSA в исходную пробирку и пипетку, чтобы собрать оставшиеся клетки. Перелейте в пробирку объемом 0,2 мл, чтобы получить общий объем 100 мкл.
- Отныне общий объем будет составлять 100 мкл вместо 50 мкл протокола. Соответствующим образом отрегулируйте последующие этапы (например, для стадии d удалите 90 мкл вместо 45 мкл; для стадии e добавьте 90 мкл лизисного буфера вместо 45 мкл).
- Для стадии m ресуспендируйте гранулы ядер в 12 мкл буфера разбавленных ядер вместо 7 мкл.
- Подсчитайте изолированные ядра. В пустую пробирку объемом 0,5 мл добавьте 10 мкл 0,4% трипанового синего и 8 мкл PBS 0,04% BSA.
- Добавьте 2 мкл ядер в пробирку и подсчитайте ядра, как описано в 1.3.8. Используйте автоматический счетчик ячеек, следуя рекомендациям поставщика.
- Время лизиса: Проведите пилотный эксперимент для этого протокола, чтобы определить наилучшее время лизиса для выделения ядер. Обеспечьте полный лизис клеток при сохранении неповрежденных ядер.
- Используйте протокол "Low Cell Input Nuclei Isolation" Приложения из 10X Genomics Demonstrated Protocol - Nuclei Isolation for Single Cell Multiome ATAC + GEX sequencing (CG000365 - Rev C)27 со следующими изменениями, внесенными для оптимизации извлечения ядер:
- Контроль чистоты методом проточной цитометрии
ПРИМЕЧАНИЕ: Этот шаг должен быть выполнен только во время пилотного эксперимента для оптимизации этапов подготовки образцов для проверки чистоты ядер, которые будут загружены на чип 10X Chromium. После того, как протокол полностью оптимизирован, не рекомендуется выполнять этот этап контроля качества в последующих экспериментах, чтобы избежать ненужных потерь собранных ядер, которые могут быть доступны в небольших количествах.- После завершения выделения ядер перенесите 6 мкл ресуспензии ядер в новую пробирку FACS, предварительно заполненную 150 мкл буфера FACS. Добавьте 3 мкл 7-AAD и инкубируйте в течение 5 минут на льду.
- Собирайте и записывайте данные после сортировки, чтобы убедиться в чистоте и жизнеспособности сортировки. Убедитесь, что не менее 95% ядер находятся в интересующих вас воротах, как определено в шаге 4.2 Протокола 1 (см. рисунок 4).
- Контроль качества очищенных ядер методом микроскопии:
ПРИМЕЧАНИЕ: Этот шаг должен быть выполнен только во время пилотного эксперимента для оптимизации этапов пробоподготовки для проверки качества ядер, которые будут загружены на чип 10X Chromium. После того, как протокол полностью оптимизирован, не рекомендуется выполнять этот этап контроля качества в последующих экспериментах, чтобы избежать ненужных потерь собранных ядер, которые могут быть доступны в небольших количествах.- Действуйте, как описано в шаге 1.5.3.
- Проведение мультиомного анализа
- Немедленно перейдите к руководству пользователя Chromium Next GEM Single Cell Multiome ATAC + Gene Expression (CG000338 - Rev F)25.
Результаты
Два протокола, описанные выше, подробно описывают выделение ядер, начиная с двух разных типов тканей. Различия и сходства между двумя протоколами схематично представлены на рисунке 1.
Очищение ядер мозга мыши
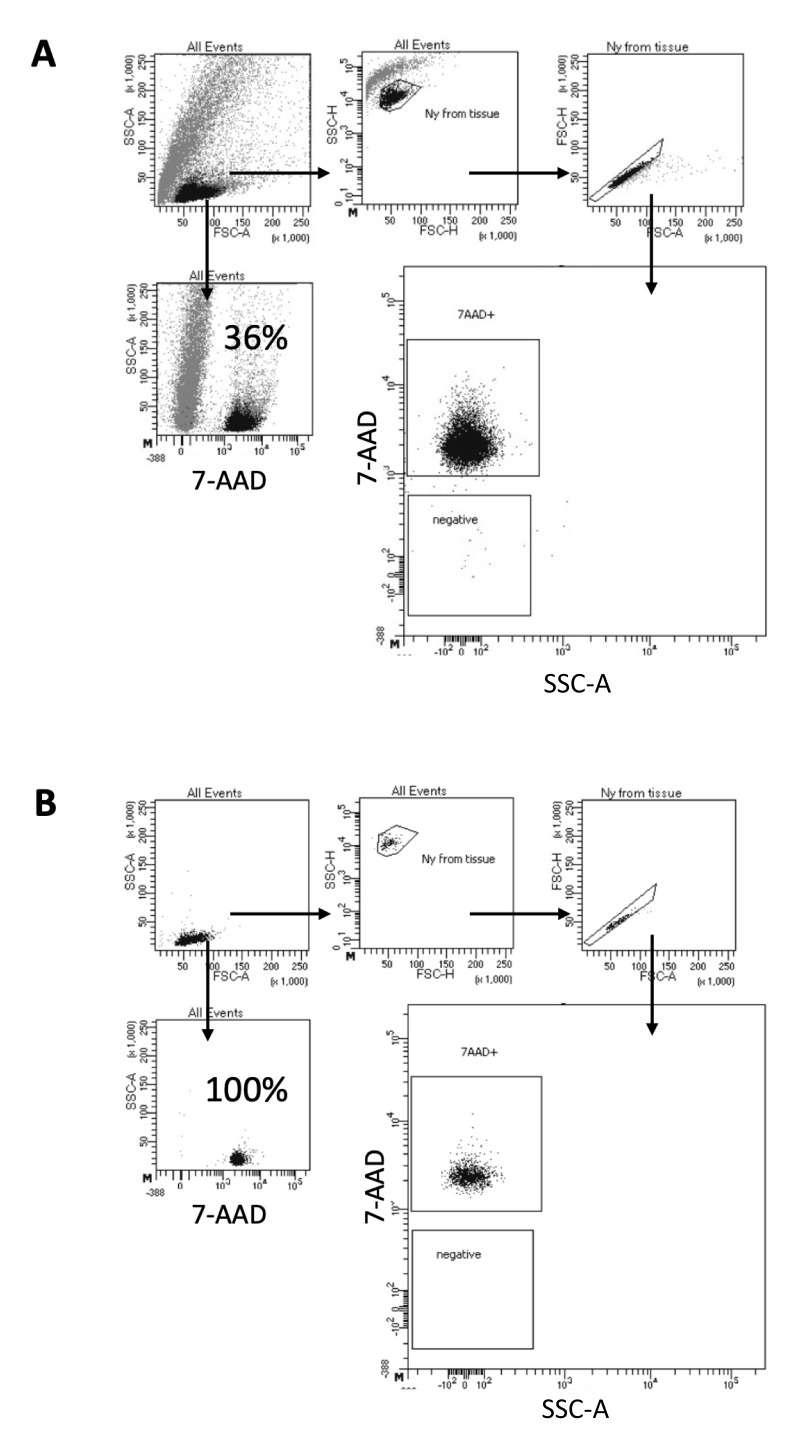
В протоколе, описанном здесь, мы предлагаем щадящий метод получения ядер из образцов мозга. Он начинается с механической диссоциации ткани мозга в лизисном буфере, за которой следуют этапы промывки и фильтрации ситечком, которые удаляют оставшуюся ткань из суспензии. Последующее удаление мусора, нелизированных клеток и мелких частиц достигается с помощью FACS, чтобы гарантировать, что только очищенные ядра загружаются для последующего протокола Multiome. На рисунке 2 показан профиль ядер до и после сортировки. После фильтрации и перед сортировкой ядер образец содержит большое количество мусора, причем более 99% «синглетов» положительны на ядерное окрашивание (7-AAD), что указывает на оптимальный лизис клеток (рис. 2A). Ядра сортируются по 7-AAD положительному затвору. Фракция отсортированного материала отбирается для проверки чистоты подготовленных ядер. На рисунке 2Б показан профиль ядер мозга после сортировки. Сортировка ядер позволила увеличить чистоту ядер с начальных 36% (рис. 2А) до почти 100% (рис. 2Б).

Рисунок 2: Стратегия литникования для сортировки ядер и испытания на чистоту после сортировки. Ядра окрашивали 7-AAD и собирали с помощью клеточного сортировщика. (A) Ядра сначала стробируются в зависимости от их размера и зернистости (FSC-A и SSC-A, соответственно). Затем отдельные частицы отбираются на основе их свойств FSC-A/FSC-H и окрашивания 7-AAD. (Б) После сортировки клеток часть ядер из пробирки для сбора проверяется на чистоту с использованием той же стратегии стробирования, что и в А. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
{kind=link}
Очистка прогениторов гемопоэтических стволовых клеток костного мозга мышей (HSPC)
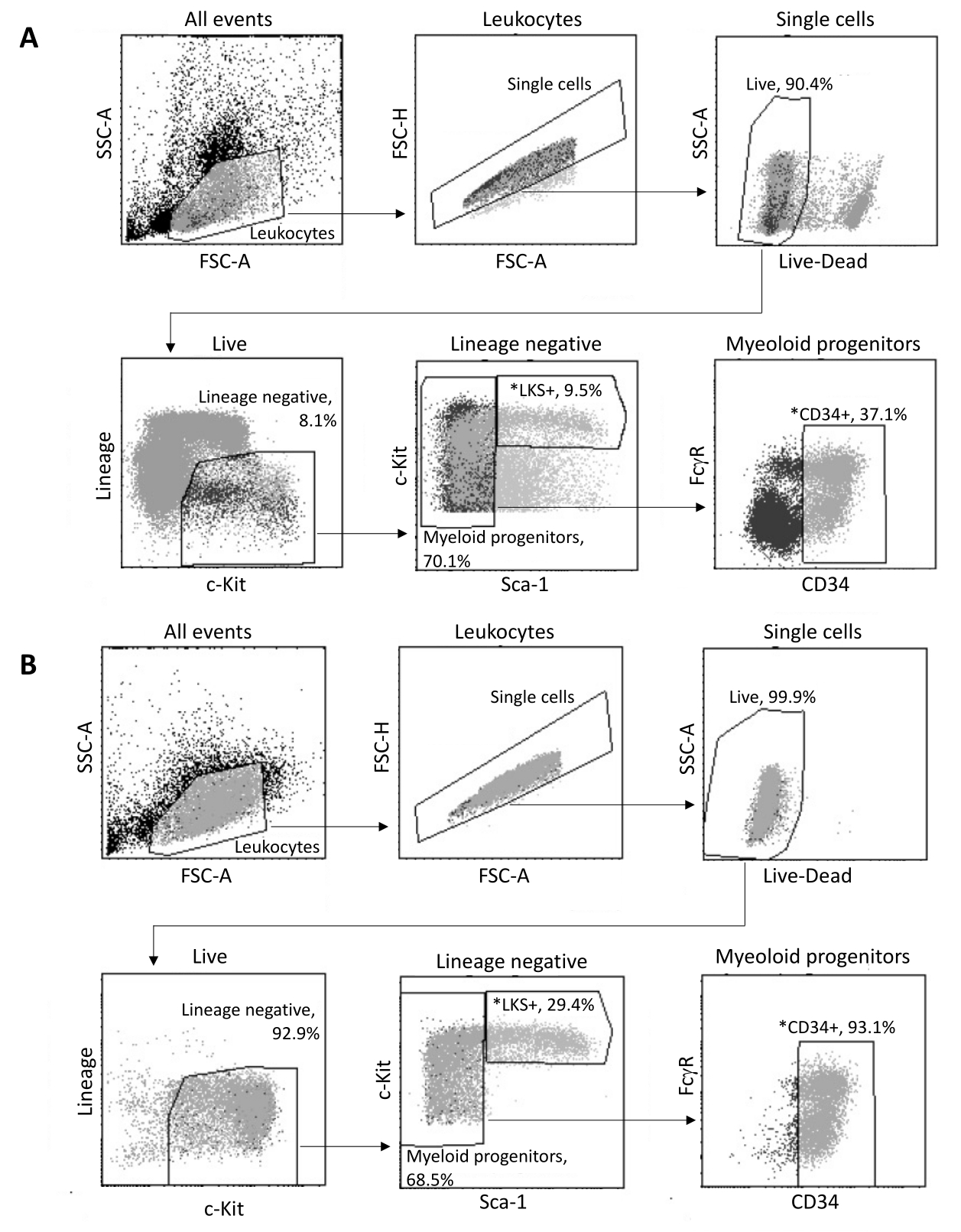
После выделения из костного мозга до 2 x 105 HSPC сортируются с помощью FACS в соответствии со стратегией стробирования, показанной на рисунке 3A. Оценивается эффективность сортировки и чистота пробы (рис. 3Б).

Рисунок 3: Стратегия стробирования для сортировки ТГСК костного мозга. (A) Репрезентативная стратегия стробирования FACS для сортировки жизнеспособных LKS+ гемопоэтических стволовых клеток и CD34+ миелоидных предшественников для выделения ядер. (B) Репрезентативные участки FACS, используемые для проверки чистоты отсортированной клеточной популяции. Показаны пропорции различных подмножеств клеток по отношению к родительской популяции. * Две отсортированные популяции. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
{kind=link}
Протокол "Low Cell Input Nuclei Isolation" позволяет выделять ядра из образцов с максимальным количеством клеток 105 . Он включает в себя малое количество этапов центрифугирования, тем самым сводя к минимуму потери клеток/ядер. Мы отрегулировали объем лизисных и промывочных буферов пропорционально входу в клетку и увеличили время центрифугирования для максимального извлечения ядер. Проведен пилотный эксперимент по оценке количества восстановленных ядер методом подсчета и их качества методом проточной цитометрии и микроскопии. На рисунке 4 показан образец HSPCs после лизиса клеток. Этот протокол генерировал высококачественные ядра, как показано на рисунке 5A, без какого-либо мусора, который мог бы повлиять на нижележащий протокол мультиома.

Рисунок 4: Сортировка по тесту чистоты изолированных ядер HSPC костного мозга. Ядра окрашивали 7-AAD и собирали с помощью клеточного сортировщика. Сначала ядра были подвергнуты стробированию на основе их размера и зернистости (FSC-A и SSC-A, соответственно) для оценки чистоты образца. Доля ядер указана по отношению к родительской популяции. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
{kind=link}
| Номер трубки | Наименование трубки | Запятнанная сущность | Количество окрашенных объектов | Антитело/краситель (мкл) | Буфер сбора (мкл) |
| 1 | Безупречный | Клетки | 5,00,000 | Н/Д | 200 |
| 2 | ЖИВОЙ/МЕРТВЫЙ Поправимое пятно от мертвых клеток Aqua | Клетки | 5,00,000 | 0.5 | |
| 3 | APC/Cyanine 7 против мышей CD16/32 (FcγR) | Бусины OneComp eBeads | 15 мкл | 1 | |
| 4 | Тихоокеанский синий антимышиный коктейль Lineage | Бусины OneComp eBeads | 15 мкл | 1 | |
| 5 | ПЭ антимышиный Ly-6A/E (Sca-1) | Бусины OneComp eBeads | 15 мкл | 1 | |
| 6 | APC для защиты от мыши CD117 (c-Kit) | Бусины OneComp eBeads | 15 мкл | 1 | |
| 7 | FITC против мышей CD34 | Бусины OneComp eBeads | 15 мкл | 1 |
Таблица 1: Контроль одиночных пятен для настроек компенсации на проточном цитометре. Указываются необходимые средства контроля одиночного окрашивания, количество клеток или шариков, подлежащих окрашиванию, и количество антител.
| Мастер-микс | Реагент | Окончательное разбавление | Антитело/краситель (мкл) | Тип буфера | Буфер (мкл) |
| Микс 1 | APC/Cyanine 7 против мышей CD16/32 (FcγR) | 1/500 | 1.2 | ДПБС | 300 |
| ЖИВОЙ/МЕРТВЫЙ Поправимое пятно от мертвых клеток Aqua | 1/250 | 2.4 | |||
| Микс 2 | Тихоокеанский синий антимышиный коктейль Lineage | 1/20 | 30 | Буфер FACS | 300 |
| ПЭ антимышиный Ly-6A/E (Sca-1) | 1/200 | 3 | |||
| APC для защиты от мыши CD117 (c-Kit) | 1/200 | 3 | |||
| FITC против мышей CD34 | 1/50 | 12 | |||
| Общий объем окрашивания | 600 |
Таблица 2: Состав красящей смеси для костного мозга ГПСК. Показаны объемы реагентов, необходимых для окрашивания одного образца, содержащего 40 миллионов клеток. Для окрашивания большего количества образцов умножьте указанный объем на необходимое количество образцов и добавьте половину дополнительного объема образца, чтобы обеспечить достаточный объем мастер-смеси.
Дополнительный рисунок 1: Протокол выделения клеток костного мозга. (А) Откройте брюшину. Белыми пунктирными линиями обозначена линия, по которой нужно пройти. (B) После снятия кожи с задней ноги проведите ножницами вдоль позвоночника в тазобедренном суставе, чтобы вырезать ногу, не разрезая бедренную кость. (В) Внешний вид ноги, отделенной от тела до удаления мышц. (D) Внешний вид ноги после удаления мышцы. (E) Процедура отделения бедренной кости в коленном суставе, затем в тазобедренном суставе, с соблюдением осторожности, чтобы не разрезать бедренную кость. Белые изогнутые стрелки показывают необходимое движение. Белыми пунктирными стрелками обозначена область, которую нужно аккуратно отделить ножницами. (F) Процедура вскрытия дистальной части бедренной кости (т.е. части, ранее прикрепленной к большеберцовой кости в коленном суставе) путем надежного захвата хряща и дистального эпифиза ножницами и переворачивания его назад, чтобы обнажить костный мозг. (G) Четыре выступа, обозначенные черными стрелками, должны быть видны на открытом конце физиса. (H) Появление бедренной кости открытым концом, обращенным вниз в подготовленную пробирку объемом 0,5 мл, помещенную в пробирку объемом 1,5 мл, содержащую 150 мкл буфера FACS. (I) Внешний вид гранулированных клеток костного мозга и теперь белой бедренной кости после быстрого центрифугирования при 12 000 x g. Пожалуйста, нажмите здесь, чтобы скачать этот файл.
Обсуждение
Приготовление высококачественной клеточной или ядерной суспензии имеет решающее значение для успеха одноклеточного или одноядерного РНК-Seq и одноклеточного мультиомного анализа 29,30,31. Здесь мы описали протоколы пробоподготовки и выделения ядер для мультиомных анализов из двух типов тканей: головного мозга и костного мозга.
Протокол работы мозга, описанный в этой статье, позволяет извлекать высококачественные ядра из свежезамороженных тканей мозга. Он включает в себя следующие этапы: разрушение замороженных тканей, выделение ядер, очистка ядер и контроль качества подготовленного материала. Ткань мозга состоит из множества различных типов клеток, и процедура диссоциации тканей и выделения ядер должна сохранять пропорции клеточных популяций, присутствующие в исходной ткани. Здесь был оптимизирован состав лизисного буфера и время инкубации, чтобы обеспечить полный и щадящий лизис всех клеточных популяций, составляющих ткань.
Протокол HSPCs костного мозга несколько отличается, поскольку он требует одного дополнительного шага в начале эксперимента, чтобы выделить интересующую клеточную популяцию из гетерогенной клеточной суспензии. После забора свежей ткани эритроциты лизируются, и образец обогащается для интересующей подгруппы клеток. Клетки-мишени лизируются, ядра изолируют, а качество приготовленного материала контролирует.
10X Genomics предоставляет несколько протоколов, валидированных для выделения ядер в различных тканях32,33. Компания также выпускает на рынок набор для выделения ядер с простым конвейером для выделения ядер из валидированных тканей34. Однако эти протоколы нуждаются в дополнительной оптимизации, чтобы адаптировать особенности определенных образцов. В качестве примера можно привести примеры, требующие работы с малым количеством входных данных. Для этих образцов наиболее сложными этапами являются центрифугирование, которое должно быть достаточно строгим для очистки образца и достаточно щадящим, чтобы избежать потери клеток/ядер. В соответствии с протоколом, описанным здесь, мы адаптировали протокол 10X Genomics Demonstrated Protocol - Nuclei Isolation for Single Cell Multiome ATAC + GEX Sequencing (CG000365 - Rev C)27, чтобы найти тонкий баланс между этими двумя требованиями. Как показано на примере получения ядер из отсортированных ГСПЦ, мы улучшили извлечение ядер без ущерба для качества образца.
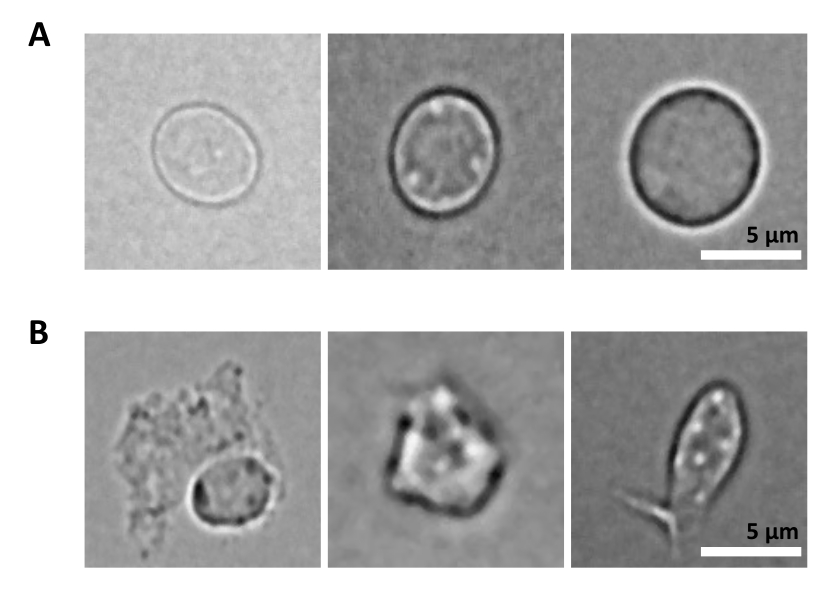
Дополнительной проблемой является стадия лизиса очищенных клеток для выделения ядер. Более жесткие условия лизиса и более длительный инкубационный период могут повредить ядра и тем самым повлиять на качество данных секвенирования. На рисунке 5 показана репрезентативная визуализация ядер из образцов костного мозга при различном времени инкубации с лизисным буфером и показано, насколько различным может быть состояние ядер в зависимости от лизиса клетки. На примере ГСПК мы определили 3-минутный лизис как условие, которое приводит к наибольшей доле здоровых, интактных ядер и наименьшей доле поврежденных ядер. Время инкубации лизиса должно быть оптимизировано для каждого нового типа образца.

Рисунок 5: Контроль качества ядер с помощью микроскопии. Показаны репрезентативные светлопольные изображения изолированных ядер из костного мозга мыши с (А) интактными и (Б) поврежденными ядрами. Масштабная линейка 5 мкм. Изображения были получены с помощью инвертированного микроскопа с использованием 40-кратного объектива ELWD NA 0.60 и 1,5-кратного цифрового зума. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
{kind=link}
Оба протокола, подробно описанные в этой работе, основаны на очистке целевых клеток или ядер с помощью высокопроизводительных приборов FACS. Этот этап имеет решающее значение для протоколов подготовки одноклеточных/ядерных клеток, где редкие подмножества клеток должны быть выделены из гетерогенных суспензий. В этих случаях, как и в приведенном здесь примере для сортировки HSPC, может потребоваться многомерная панель проточной цитометрии для включения «стробирования» на интересующей клеточной популяции. Сортировка происходит очень быстро и точно, что приводит к чистоте отсортированных подмножеств клеток более 95%. При таком подходе клеточная суспензия подвергается давлению до 70 фунтов на квадратный дюйм и, следовательно, может быть ограничением для сортировки хрупких клеток (например, дендритных клеток, нейтрофилов), поскольку это может привести к разрыву их клеточной мембраны. В этих случаях следует выбирать альтернативные решения для клеточной очистки, в том числе магнитную сортировку, применение приборов нового поколения (например, CellenOne, Cellenion; MACSQuant Tyto, Miltenyi)35,36 или системы на основе капель (например, ODIN, Sensific)37. Тем не менее, медленная скорость сортировки этих технологий, когда сортировка клеток длится несколько часов, а не минут, является сильным ограничивающим фактором для применения этих подходов при подготовке жизнеспособных клеток для Multiome и других одноклеточных приложений, основанных на анализе большого количества клеток.
Для очистки ядер, выделенных из ткани, предпочтительным методом является FACS из-за его пропускной способности и чистоты выделенного материала. Ядра не чувствительны к давлению, а отфильтрованные тканевые изоляты можно легко очистить с помощью клеточного сортировщика. Если лаборатория не оснащена прибором FACS, существуют другие альтернативы, несколько менее эффективные, но достаточно хорошие. В качестве примера можно привести ультрацентрифугирование или использование небольшого оборудования, такого как MARS (Applied Cell), которое разделяет частицы на основе их разницы в размерах, используя акустические волны; ламинарная промывочная машина CURIOX, использующая гидрофобные свойства клеточно-ядерных суспензий; или LEVITAS bio, который опирается на физические свойства клеток (левитация), чтобы отделить их от мусора.
Здесь мы опишем протоколы для получения большого количества ядер и наилучшей чистоты для последующего протокола Multiome. Сортировка FACS и повторные этапы центрифугирования приводят к значительным потерям исходного материала. По этой причине в протоколе подготовки ядер из мозга, который мы здесь описываем, требуется достаточно обильный исходный материал, чтобы в результате после сортировки FACS было собрано не менее 500 000 ядер. Альтернативные протоколы должны применяться, если этот критерий не может быть сопоставлен. При работе с редкими клеточными популяциями или небольшими участками тканей лимитирующим фактором может быть доступное количество исходного материала. Для решения этой проблемы можно улучшить извлечение ядер путем (а) уменьшения объема лизиса, (б) уменьшения объема промывки, (в) использования однократной промывки с увеличенным временем центрифугирования, чтобы попытаться улучшить извлечение, как указано в протоколах 10X Genomics для выделения ядер с низким входом клеток. Для мультиомного анализа материалов с низким содержанием стоит рассмотреть приложения на основе планшетов, такие как scNMT, SNARE-seq и Paired-seq38 , которые требуют гораздо меньшего количества входных образцов.
Таким образом, мы описали два надежных протокола подготовки ядер головного мозга и костного мозга для последующего анализа Multiome. Эти протоколы применимы в любом научном проекте, требующем высококачественных одноядерных суспензий из этих двух типов тканей, независимо от поставленного научного вопроса. Наша группа применяет протокол выделения ядер мозга в исследованиях развития мозга при инактивации различных генов-мишеней, а также в исследованиях иммунного ответа на фоне неврологических заболеваний. Мы используем протокол выделения ядер костного мозга для расшифровки участия различных кроветворных субпопуляций в формировании иммунной системы.
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Ана Джимин Чой получила стипендию от Международной программы докторантуры Парижского университета Пастера (PPU).
Материалы
| Name | Company | Catalog Number | Comments |
| 18 G x 1 ½ (1.2 mm x 38 mm) Agani needles | Terumo | AN*1838S1 | |
| 15 mL tubes | Falcon | 352097 | |
| 5 mL round bottom FACS tube with cell strainer cap 35 µm | falcon | 352235 | |
| 50 mL tubes | Falcon | 352070 | |
| 7-AAD | BD pharmagen | 559925 | |
| ACK Lysing Buffer | Gibco | A10492-01 | |
| APC anti-mouse CD117 (c-Kit) | BioLegend | 105812 | Clone: 2B8 |
| APC/Cyanine 7 anti-mouse CD16/32 (FcγR) | BioLegend | 101328 | Clone: 93 |
| BD FACSAria III | BD Biosciences | non-applicable | |
| BD FACSDiva Software v8.0.1 | BD Biosciences | non-applicable | |
| Bovine Serum Albumin stock solution 10% | Miltenyi Biotec | 130-091-376 | |
| Cell staining buffer | Biolegend | 420201 | |
| CFI Suprplan Fluor ELWD 40XC ON 0.6 | Nikon | non-applicable | |
| CMOS camera Prime 95B 25 mm | Photometrix | non-applicable | |
| Countess II FL Automated Cell Counter | Invitrogen | AMQAF1000 | |
| Countess cell counting chamber slide | Invitrogen | C10283 | |
| Coverglass 24 mm x 24 mm 0.13-0.17 mm | Brand | BR470819 | |
| Digitonine 5% | Invitrogen | BN2006 | |
| Disposable Scalpels | Swann-Morton | 0508 | |
| DMEM (1x) + GlutaMAX-I | Gibco | 31966-021 | |
| DPBS (10x) | Gibco | 14200-067 | |
| DTT | Sigma aldrich | 646563 | |
| Epifluorescence inverted microscope Nikon Ti2 -E | Nikon | non-applicable | |
| Eppendorf Safe-Lock Tubes 0.5 mL | Eppendorf | 30123603 | |
| Ethanol 70% | VWR | 83801.290 | |
| FITC anti-mouse CD34 | Invitrogen | 11-0341-85 | Clone: RAM34 |
| Forceps for dissection | FST | 11152-10 | |
| Heat-inactivated Fetal Bovine Serum (FBS) | Gibco | 11533387 | |
| Dounce Homogeniser 2 mL | Bellco glass | 1984-10002 | Pestle “A” Large Clearance: .0030-.0050″ and Pestle “B” Small Clearance: .0005-.0025″ |
| LIVE/DEAD fixable aqua dead cell stain kit | Invitrogen | L34957 | |
| Magnesium chloride solution 1 M | Sigma aldrich | M1028 | |
| Microcentrifuge | Eppendorf | 5424R | |
| Mounting medium Fluoromount-G | invitrogen | 00-4958-02 | |
| Nonidet P40 substitute | Sigma aldrich | 74385 | |
| Nuclease free water | ThermoFischer | AM9932 | |
| Nuclei buffer 20x | 10X Genomics | 2000153/2000207 | |
| Nuclei isolation kit EZ prep | Sigma Aldrich | NUC-101 | |
| OneComp eBeads compensation beads | Invitrogen | 01-1111-41 | |
| Pacific Blue anti-mouse lineage cocktail (including anti-mouse CD3, Ly-6G/Ly-6C, CD11b, CD45R/B220, TER-119) | BioLegend | 133310 | Clones (in the same order as the antibodies listed): 17A2, RB6-8C5, M1/70, RA3-6B2, Ter-119 |
| PCR Tube Strips 0.2 mL | Eppendorf | 951010022 | |
| PE anti-mouse Ly-6A/E (Sca-1) | BioLegend | 122507 | Clone: E13-161.7 |
| Petri dish 100 mm x 20 mm OPTILUX | Falcon | 353003 | |
| Ply-L-lysine 0.01% sterile-filtered suitable for cell culture | Sigma | P4707 | |
| Printed microscope slides 8 well 6 mm numbered | Epredia | ER-301B-CE24 | |
| Protein LoBind Tubes 1.5 mL | Eppendorf | 30108116 | |
| Recombinant Rnase inhibitor 5000 U | Takara | 2313A | |
| Scissors for dissection | FST | 14090-09 | |
| Sodium chloride solution 5 M | Sigma aldrich | 59222C | |
| Syringe filters, PES, 0.2 µm | Fisher Scientific | 15206869 | |
| Transparent nail polish | any | non-applicable | |
| Trizma Hydrochloride solution pH 7.4 | Sigma aldrich | T2194 | |
| Trypan Blue 0.4% | gibco | 15250061 | |
| Tween 20 | Biorad | 1662404 | |
| UltraPure Distilated Water Dnase/Rnase Free | Invitrogen | 10977-035 |
Ссылки
- Clark, S. J., et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nature Communications. 9 (1), 781(2018).
- Lee, J., Hyeon, D. Y., Hwang, D. Single-cell multiomics: technologies and data analysis methods. Experimental & Molecular Medicine. 52 (9), 1428-1442 (2020).
- Cerrizuela, S., et al. High-throughput scNMT protocol for multiomics profiling of single cells from mouse brain and pancreatic organoids. STAR Protocols. 3 (3), 101555(2022).
- Dimitriu, M. A., Lazar-Contes, I., Roszkowski, M., Mansuy, I. M. Single-cell multiomics techniques: From conception to applications. Frontiers in Cell and Developmental Biology. 10, 854317(2022).
- Stoeckius, M., et al. Simultaneous epitope and transcriptome measurement in single cells. Nature Methods. 14 (9), 865-868 (2017).
- Cao, J., et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science (New York, N.Y.). 361 (6409), 1380-1385 (2018).
- Narayanan, A., et al. Nuclei Isolation from Fresh Frozen Brain Tumors for Single-Nucleus RNA-seq and ATAC-seq. Journal of Visualized Experiments: JoVE. (162), e61542(2020).
- Kim, M., et al. Single-nucleus transcriptomics reveals functional compartmentalization in syncytial skeletal muscle cells. Nature Communications. 11 (1), 6375(2020).
- Santos, M. D., et al. Extraction and sequencing of single nuclei from murine skeletal muscles. STAR Protocols. 2 (3), 100694(2021).
- Safabakhsh, S., et al. Isolating nuclei from frozen human heart tissue for single-nucleus RNA sequencing. Current Protocols. 2 (7), e480(2022).
- Lau, S. -F., Cao, H., Fu, A. K. Y., Ip, N. Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 117 (41), 25800-25809 (2020).
- Armand, E. J., Li, J., Xie, F., Luo, C., Mukamel, E. A. Single-cell sequencing of brain cell transcriptomes and epigenomes. Neuron. 109 (1), 11-26 (2021).
- Morabito, S., et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer's disease. Nature Genetics. 53 (8), 1143-1155 (2021).
- Chen, S., et al. Spatially resolved transcriptomics reveals genes associated with the vulnerability of middle temporal gyrus in Alzheimer's disease. Acta Neuropathologica Communications. 10 (1), 188(2022).
- Paul, F., et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. 163 (7), 1663-1677 (2015).
- Kaufmann, E., et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. 172 (1-2), 176-190.e19 (2018).
- Christ, A., et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. 172 (1-2), 162-175.e14 (2018).
- Moorlag, S. J. C. F. M., et al. β-Glucan Induces protective trained immunity against mycobacterium tuberculosis infection: A key role for IL-1. Cell Reports. 31 (7), 107634(2020).
- de Laval, B., et al. C/EBPβ-dependent epigenetic memory induces trained immunity in hematopoietic stem cells. Cell Stem Cell. 26 (5), 657-674.e8 (2020).
- Renthal, W., et al. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nature Neuroscience. 21 (12), 1670-1679 (2018).
- Yang, A. C., et al. A human brain vascular atlas reveals diverse mediators of Alzheimer's risk. Nature. 603 (7903), 885-892 (2022).
- Lee, D. R., Zhang, Y., Rhodes, C. T., Petros, T. J. Generation of single-cell and single-nuclei suspensions from embryonic and adult mouse brains. STAR Protocols. 4 (1), 101944(2022).
- Corces, M. R., et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nature Genetics. 48 (10), 1193-1203 (2016).
- Ranzoni, A. M., et al. Integrative single-cell RNA-seq and ATAC-seq analysis of human developmental hematopoiesis. Cell Stem Cell. 28 (3), 472-487.e7 (2021).
- 10X Genomics Chromium Next GEM Single Cell Multiome ATAC + Gene Expression User Guide, Document Number CG000338 Rev F. , At https://www.10xgenomics.com/support/single-cell-multiome-atac-plus-gene-expression/documentation/steps/library-prep/chromium-next-gem-single-cell-multiome-atac-plus-gene-expression-reagent-kits-user-guide (2022).
- Meyerhoff,, et al. Microdissection of mouse brain into functionally and anatomically different regions. Journal of Visualized Experiments: JoVE. (168), e61941(2021).
- 10X Genomics 10X Genomics Demonstrated Protocol - Nuclei Isolation for Single Cell Multiome ATAC + GEX sequencing (CG000365 - Rev C). , At https://www.10xgenomics.com/support/single-cell-multiome-atac-plus-gene-expression/documentation/steps/sample-prep/nuclei-isolation-for-single-cell-multiome-atac-plus-gene-expression-sequencing (2022).
- Haag, S., Murthy, A. Murine monocyte and macrophage culture. Bio-Protocol. 11 (6), e3928(2021).
- Haque, A., Engel, J., Teichmann, S. A., Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Medicine. 9 (1), 75(2017).
- Jiang, P. Quality control of single-cell RNA-seq. Methods in Molecular Biology (Clifton, N.J). 1935, 1-9 (2019).
- Regan, C., Preall, J. Practical considerations for single-cell genomics. Current Protocols. 2 (8), e498(2022).
- 10X Genomics 10X Genomics Demonstrated Protocol - Nuclei Isolation for Single Cell ATAC Sequencing (CG000169 - Rev E). , At https://www.10xgenomics.com/support/single-cell-atac/documentation/steps/sample-prep/nuclei-isolation-for-single-cell-atac-sequencing (2022).
- 10X Genomics 10X Genomics Demonstrated Protocol - Nuclei Isolation from Complex Tissues for Single Cell Multiome ATAC + Gene Expression Sequencing. (CG000375 - Rev C). , At https://www.10xgenomics.com/support/single-cell-multiome-atac-plus-gene-expression/documentation/steps/sample-prep/nuclei-isolation-from-complex-tissues-for-single-cell-multiome-atac-plus-gene-expression-sequencing (2022).
- 10X Genomics 10X Genomics - Chromium Nuclei Isolation Kit (CG000505 - Rev A). , At https://www.10xgenomics.com/support/single-cell-gene-expression/documentation/steps/sample-prep/chromium-nuclei-isolation-kit-sample-prep-user-guide (2022).
- Shomroni, O., et al. A novel single-cell RNA-sequencing approach and its applicability connecting genotype to phenotype in ageing disease. Scientific Reports. 12 (1), 4091(2022).
- Ocañas, S. R., Pham, K. D., Blankenship, H. E., Machalinski, A. H., Chucair-Elliott, A. J., Freeman, W. M. Minimizing the ex vivo confounds of cell-isolation techniques on transcriptomic and translatomic profiles of purified microglia. eNeuro. 9 (2), (2022).
- Gérard, A., et al. High-throughput single-cell activity-based screening and sequencing of antibodies using droplet microfluidics. Nature Biotechnology. 38 (6), 715-721 (2020).
- Vandereyken, K., Sifrim, A., Thienpont, B., Voet, T. Methods and applications for single-cell and spatial multi-omics. Nature Reviews. Genetics. 24, 494-515 (2023).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены