
Method Article
Purification of H3 and H4 Histone Proteins and the Quantification of Acetylated Histone Marks in Cells and Brain Tissue
In This Article
Summary
The purpose of this article is to provide a comprehensive, systematic guide to the efficient purification of histones H3 and H4 and the quantification of acetylated histone residues.
Abstract
In all eukaryotic organisms, chromatin, the physiological template of all genetic information, is essential for heredity. Chromatin is subject to an array of diverse posttranslational modifications (PTMs) that mostly occur in the amino termini of histone proteins (i.e., histone tail) and regulate the accessibility and functional state of the underlying DNA. Histone tails extend from the core of the nucleosome and are subject to the addition of acetyl groups by histone acetyltransferases (HATs) and the removal of acetyl groups by histone deacetylases (HDACs) during cellular growth and differentiation. Specific acetylation patterns on lysine (K) residues on histone tails determine a dynamic homeostasis between transcriptionally active or transcriptionally repressed chromatin by (1) influencing the core histone assembly and (2) recruiting synergistic or antagonistic chromatin-associated proteins to the transcription site. The fundamental regulatory mechanism of the complex nature of histone tail PTMs influences the majority of chromatin-templated processes and results in changes in cell maturation and differentiation in both normal and pathological development. The goal of the current report is to provide novices with an efficient method to purify core histone proteins from cells and brain tissue and to reliably quantify acetylation marks on histones H3 and H4.
Introduction
The term epigenetics refers to heritable changes in gene activity that occur independently from changes in the DNA sequence1,2. Gene transcription and repression are determined by (1) the accessibility of the chromosomal DNA wrapped around an octamer of core histone proteins (two copies each of H2A, H2B, H3, and H4) and by (2) the availability of transcription factors and scaffold proteins recruited to specific promoter sites3,4. Gene transcription is regulated by enzyme-mediated modifications of specific DNA promoter sites and the PTMs of histone tails5,6,7. The N-termini of histone H3 and H4 are among the most highly conserved sequences known in eukaryotic organisms3, and their posttranslational modifications have been extensively documented to play a central role in determining chromatin structure and function8,9. PTMs at the histone tails (i.e., acetylation, methylation, phosphorylation, and ubiquitination) change the interaction potential of the tails, influence the structural state and folding of chromatin fiber, and thereby, regulate DNA accessibility and processing4,10,11,12. Acetyl groups are added to and removed from K residues on histone tails by a set of specific histone-interacting epigenetic enzymes, namely HATs and HDACs, respectively13. For example, the acetylation of histone H4 at lysine 12 (H4K12ac) has been previously shown to activate the transcription of genes related to memory acquisition and consolidation14. Additionally, several lines of evidence suggest that the enzyme-mediated epigenetic control of gene transcription is a crucial aspect of healthy cellular growth and differentiation6,15. Alternation in the epigenetic regulation of gene expression, either by epigenetic modifications of DNA or by a mutation of the epigenetic enzymes themselves, has been shown to be dysregulated in human diseases where change in a particular gene activity is a hallmark of the pathology (e.g., cancer)6,16,17. Thus, the evaluation of changes in core histone PTMs is emerging as a high-value target for potential therapeutic interventions. However, determining the abundance, interacting partners, and specific roles of histones PTMs has been proven challenging18.
In the current report, an optimized, medium-throughput strategy to purify core histones from cells and brain tissues in a single fraction, and a complete protocol for the quantification of histones H3 and H4 PTMs is described. Of note, although currently published acid-based purification techniques and antibody-based histone detection strategies have been widely adopted for histone characterization, they lack descriptive details regarding critical steps of the procedure, thus hindering quick and replicable histone extraction and quantification. For example, the processing of cell extract and tissue biopsies requires different tools and technologies for successful extraction. Moreover, the optimized protocol presented in the current manuscript demonstrates a practical, medium-throughput approach. Core histones are extracted as a single, pure fraction, which enables reliable downstream antibody-mediated PTM detection without any interference from impurities. Furthermore, in the current manuscript, the challenges regarding histone detection due to their small molecular weight have been circumvented. Typically, the lack of compatibility between purification, quantification, and gel electrophoresis protocols hinder scientists from obtaining replicable and conclusive results. Here, an optimized workflow to purify core histones from cells and tissue and prepare them for downstream PTM analyses via western blot is presented.
The current protocol enables the purification of core histone proteins while preserving their native posttranslational modifications (i.e., acetylation, methylation, and phosphorylation). Figure 1 depicts the timeline of the histone purification protocol.
Protocol
All mice were housed in a humidity- and temperature-controlled, AAALAC-accredited animal facility at the University of Miami Miller School of Medicine. All experiments were approved by the University of Miami Miller School of Medicine Institutional Animal Care and Use Committee (IACUC) and conducted according to specifications of the NIH.
1. Preparation of the Sample Extract
- Adherent cells
- Plate cells in 10 cm dishes in the appropriate culture media (1 x 106 to 1 x 109 cells per dish for cell lines, such as BV2, HEK-293, and SH-SY5Y, but ~1 x 1015 cells per dish for primary cells, such as primary cortical neurons). Assure that the cells are equally distributed on the entire surface of the plate and allow the cells to grow for 48 h to reach ~100% confluence (37 °C, 5% CO2).
- Once the cells have reached the desired confluency, gently aspirate the culture media and wash the cells 2x with prewarmed serum-free media under a tissue culture hood.
- Aspirate the serum-free media from the dish and add 1 mL of ice-cold extraction buffer (0.4 M sulfuric acid, 1 mM KCl, 1 mM MgCl2, 50 mM Tris-HCl [pH 8.0] and 1x protease inhibitor cocktail) to each dish.
- Use a plastic cell scraper to collect all cells in the extraction buffer (by scraping) and transfer them to a 1.5 mL labeled tube with a 1,000 µL pipette. Pipet the cells up and down 3x to facilitate homogenization.
- Close all tubes and immediately put them on ice.
- Brain tissue
- If frozen tissue is being used, place the tissue in a prechilled 1.5 mL tube and briefly thaw it on ice. If fresh tissue is being used, proceed immediately to step 1.2.2.
NOTE: The current protocol describes procedures using frozen mouse brain and mouse prefrontal cortex samples. - Homogenize the tissue using a handheld Dounce homogenizer using the appropriate amount of extraction buffer and the recommended number of strokes (Table 1). To avoid excessive chromatin shredding, do not exceed the number of recommended strokes.
- Using a single channel 1,000 µL pipette, transfer the homogenate to a prechilled 1.5 mL tube. Close all tubes and immediately put them on ice.
- If frozen tissue is being used, place the tissue in a prechilled 1.5 mL tube and briefly thaw it on ice. If fresh tissue is being used, proceed immediately to step 1.2.2.
2. Preparation of the Crude Histone Extract
- Place the 1.5 mL tubes containing the cells or tissue suspended in extraction buffer on a rotating platform and rotate at 15 rpm at 4 °C to allow the extraction of crude histones.
NOTE: The extraction time may vary for different cell and tissue types and must be optimized for each procedure. The current protocol presents results obtained following 15 min, 2 h, and 24 h of extraction (Figure 2, Figure 3, Figure 4, and Figure 5). - Prechill the microcentrifuge to 4 °C. After the desired extraction time has passed, centrifuge the tubes at maximum speed for 10 min at 4 °C.
- Transfer the supernatant including the crude histones to a new, prechilled 1.5 mL tube. Discard the pellet.
- Store the supernatant at -80 °C (the extraction can be stopped at this step, see “Stop step” in Figure 1) or immediately proceed to the next step.
- Neutralize the crude histones with a 1/4 volume of 5x neutralization buffer (e.g., add 250 µL of 5x neutralization buffer to 1 mL of crude histones). Mix well by pipetting up and down 6x.
- Check the pH of the mixture with pH strips. Adjust accordingly by adding more neutralization buffer to reach a pH of 7.
- Evaluate the presence of histone and nonhistone protein in the crude histone extract as follows (Figure 2).
- Add 37.5 µL of the sample to 12.5 µL of 4x (Laemmli) sample buffer and denature for 10 min at 99 °C.
- Load the sample onto an SDS-PAGE gel and run the gel for 1 h at 100 V.
- Stain the gel overnight with Coomassie Brilliant Blue R-250 staining solution and destain during three consecutive washes (1 h/wash) with Coomassie Brilliant Blue R-250 destaining solution.
NOTE: Crude histones (Figure 2) can be compared with eluted and purified histones (i.e., column input [Figure 5A]).
3. Purification of Core Histones
- Spin column equilibration
- Add 500 µL of equilibration buffer to each spin column being used. Do not touch the column membrane.
- Centrifuge at 4 °C for 3 min at 800 x g. Discard the flow-through. Repeat 1x.
- Histone Purification
- Add 500 µL of the sample of interest from step 2.6 to the column. Centrifuge at 4 °C for 3 min at 800 x g. Collect the flow-through.
- Repeat the previous step as many times as necessary to load the entire sample onto the column. Do not overfill the spin column.
- Combine the flow-through from each centrifugation step to analyze the column-binding efficiency (Figure 3).
- Follow step 2.7 to analyze the column flow-through.
- Column wash
- Add 500 µL of the wash buffer to each column. Centrifuge at 4 °C for 3 min at 800 x g. Collect the flow-through wash (wash #1).
- Repeat step 3.3.1 for a total of three washes. Collect the flow-through washes #2 and #3. Do not pool the consecutive column flow-through washes.
- To further evaluate the column’s histone-binding efficiency, analyze the three column washes by following step 2.7 (Figure 4).
- Histone elution
- Transfer the column into a new labelled 1.5 mL tube.
- Add 50 µL of the histone elution buffer. Centrifuge at 4 °C for 3 min at 800 x g. Save the flow-through containing histone proteins.
- For an additional elution, repeat step 3.4.2. Do not combine the first and second flow-through eluates as they differ in histone quantity and purity.
4. Precipitation of Core Histones
- Add perchloric acid (PCA) to the purified histones to a final concentration of 4% PCA (e.g., add 3 µL of 70% PCA to 50 µL of purified histones from step 3.4.2.
- Centrifuge for 3 s to collect all residual liquid from the tube wall. Mix by pipetting up and down 6x.
- Place the tubes in a rack and incubate for 24 h at 4 °C.
- The next day, prechill a microcentrifuge to 4 °C and centrifuge the samples for 75 min at maximum speed at 4 °C.
- After the centrifugation is complete, a small white pellet containing precipitated histones will be visible on the bottom of the tube. Do not vortex the sample.
- Carefully aspirate the supernatant and, without disturbing the pellet, add 500 µL of ice-cold 4% PCA to the sample.
- Centrifuge at 4 °C for 10 min at maximum speed. Carefully aspirate the supernatant.
- Repeat step 4.7 2x.
- Without disturbing the pellet, add 500 µL of ice-cold acetone. Centrifuge at 4 °C for 10 min at maximum speed. Carefully aspirate the supernatant.
- Repeat step 4.9 2x.
- Carefully aspirate the supernatant, leave the tubes uncapped, and allow the sample to dry on ice for 30 min. Check if all residual acetone has evaporated.
- Leave the tubes uncapped and allow the sample to dry at room temperature (RT) for 5 min.
- Resuspend the pellet in 30 µL of sterile water. Do not pipet up and down. Flick the tube gently with a finger.
- Cap all tubes and allow the histones to reconstitute on ice for 30 - 50 min, depending on the pellet size. Check if the pellet is resuspended.
- Cap all tubes and allow the pellet to further resuspend at RT for 5 min.
NOTE: This solution (first and second elution from step 3.4.3) is constituted of purified and desalted histones and may be used for further quantification and histone acetylation analysis.
5. Quantification of Eluted Histone Proteins
- Use a spectrophotometer according to the manufacturer’s protocol to quantify the total histone proteins obtained after the final elution in step 4.15. Measure the absorbance at 230 nm. Record the A260/A280 ratio indicative of the sample contamination with nucleic acid.
- Use the following formula to calculate the histone concentration (x):

Here, OD is the optical density measured at A230 nm. - A histone concentration of ~1.5 mg/mL is considered an average yield for cell lines, while a histone concentration of ~5.0 mg/mL is considered an average yield for 30 mg of tissue.
6. Western Blot Analysis
- Adjust the purified and eluted histone from step 4.15 to ~10 µg of histone protein/sample.
- Add the appropriate volume of water and 4x Laemmli sample buffer to adjust the loading volumes.
- Denature the samples for 10 min at 99 °C. Cool them on ice. Centrifuge for 3 s to collect all residual liquid and condensation from the tube wall.
- Load the samples onto an SDS-PAGE gel and run the gel for 1 h at 100 V.
- To visualize total histone protein, stain the gel overnight with Coomassie Brilliant Blue R-250 staining solution and destain during three consecutive washes (1 h/wash) with Coomassie Brilliant Blue R-250 destaining solution.
NOTE: The first elution of histone proteins contains high-quality histones (Figure 5A) while the second elution contains low to no levels of histones (Figure 5B). - To quantify histone PTMs, use a transfer system (see Table of Materials) to transfer the histone protein from the SDS-PAGE gel (step 6.4) onto a PVDF membrane.
- To assemble the transfer sandwich, open the transfer system cassette and place the PVDF membrane stack (labelled as Bottom+) on the bottom of the cassette with the membrane facing up. Roll the membrane gently with a blot roller to remove any air from between the stack and the membrane.
- Lay the gel on top of the membrane, roll the gel gently with a blot roller to remove any air from between the membrane and the gel, and place the top stack on the gel. Roll gently again and place the cassette cover on top of the sandwich, press down firmly, and turn the nob clockwise to lock.
- Insert the cassette to the transfer system slot. On the apparatus screen, select Turbo Protocol. Use a 3 min protocol for a single mini gel or a 7 min protocol for more than two mini gels.
- Stain the membrane with Ponceau S stain for 5 min and visualize the total histone proteins.
- Wash them in 1x Tris-buffered saline (TBS) with 0.1% Tween 20 for 2 h and block them in 5% milk for 1 h. Incubate with primary and secondary antibody (overnight at 4 °C or 1 h at RT, respectively) or according to a previously optimized protocol.
NOTE: In the current protocol, antibodies against acetylated histones H4K12 (Figure 6 and Figure 7) and H3K27 (Figure 8) were used.
Results
To illustrate the progression of the histone purification protocol and composition of all analyzed fractions, we evaluated different histone extracts from human microglial BV2 cells. To demonstrate the quantification of histones H3 and H4 PTMs (i.e., acetylation), we used brain tissue lysates.
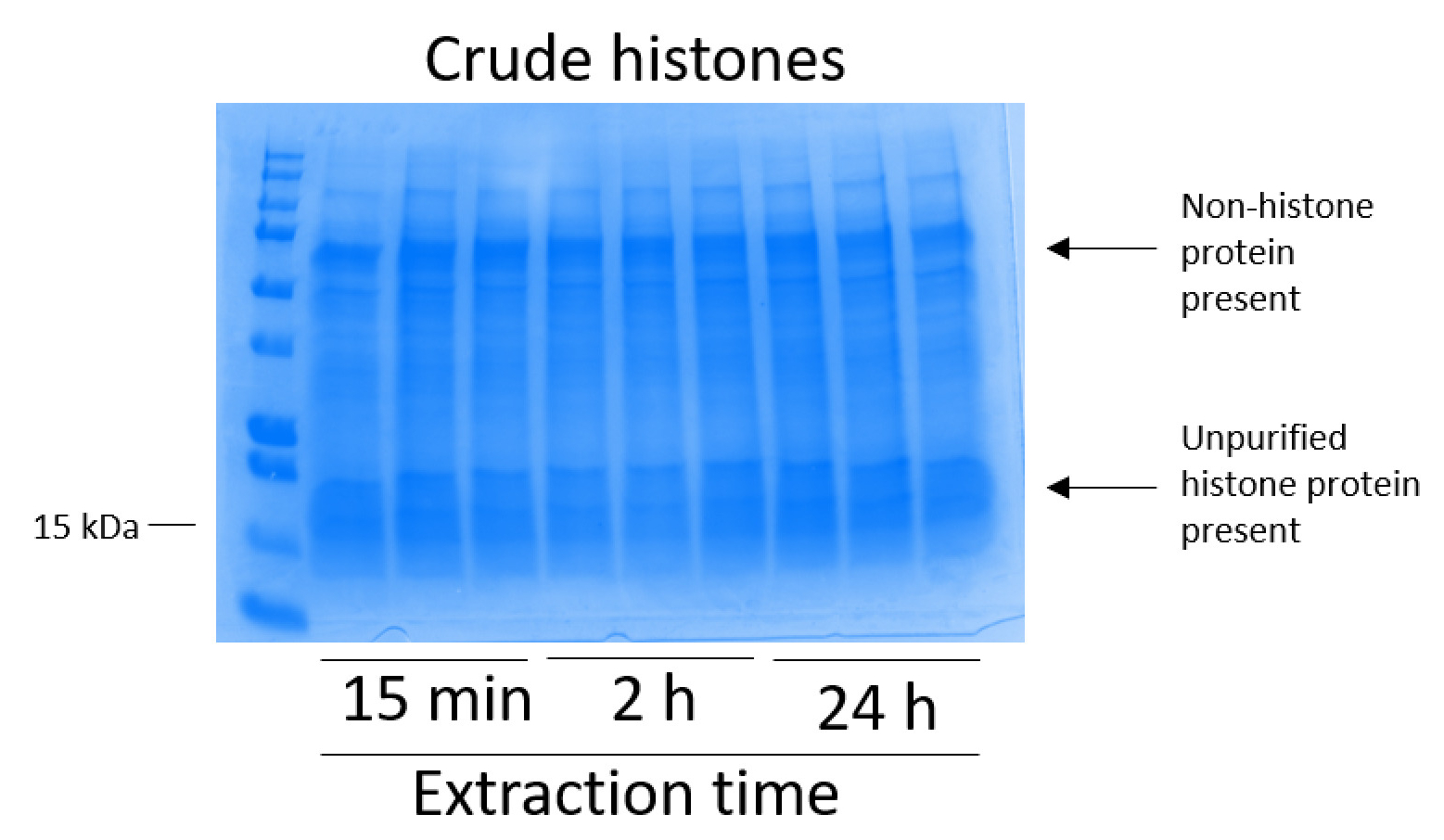
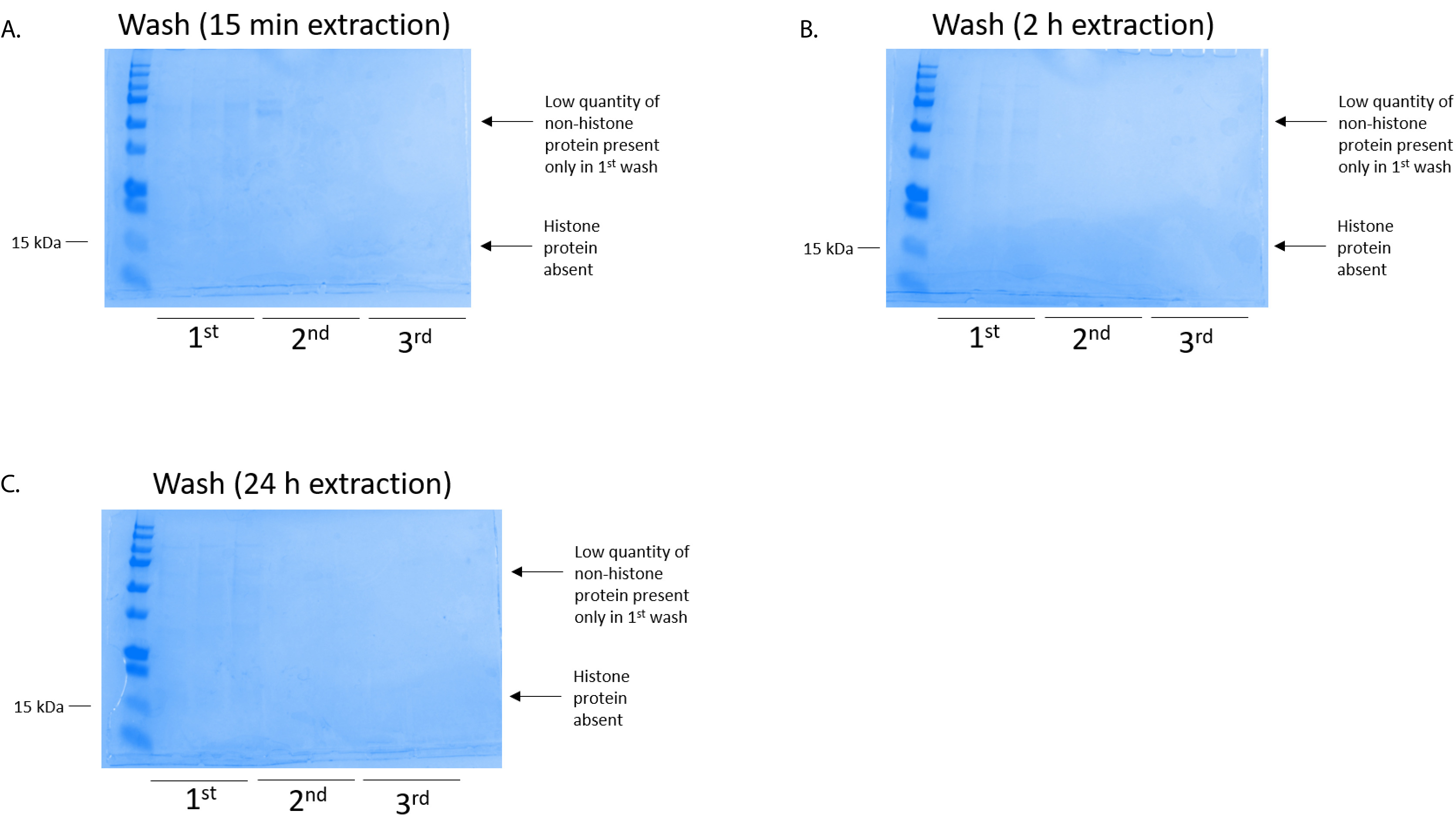
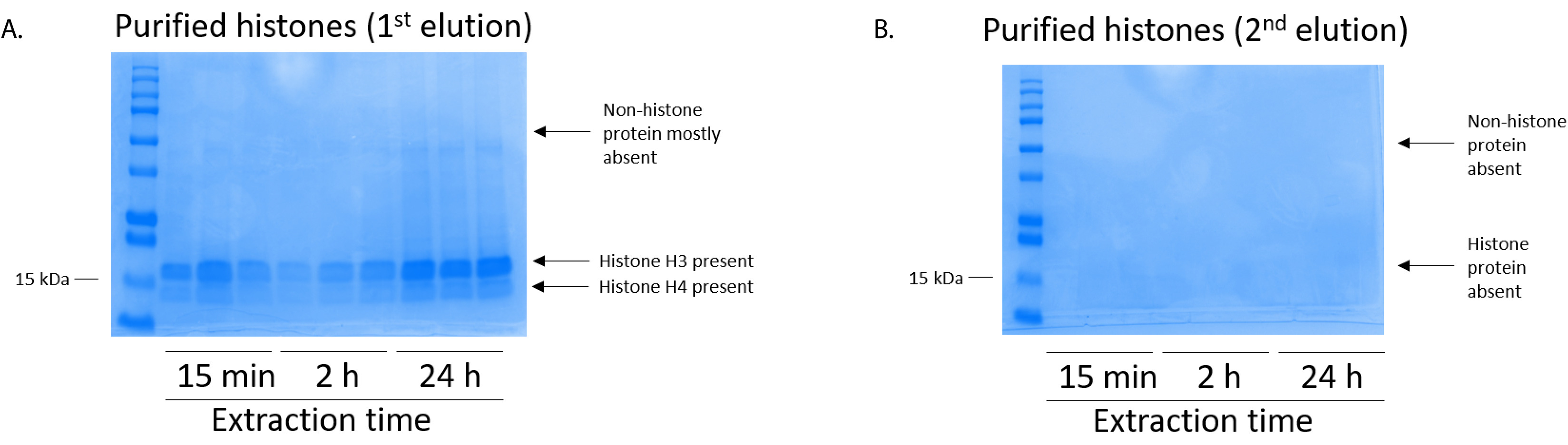
BV2 cells were plated at 5 x 106 cells per dish in 10 cm tissue culture-treated dishes and allowed to grow to confluency for 48 h. Cells were then collected, and histones were released from chromatin by an incubation in extraction buffer containing 0.4 M sulfuric acid, 1 mM KCl, 1 mM MgCl2, 50 mM Tris-HCl (pH 8.0), and 1x protease inhibitor. The extraction time, between 15 min and 24 h, did not affect the overall composition of crude histone extracts as determined by Coomassie Brilliant Blue staining (Figure 2). Next, crude histones were passed through the equilibrated histone columns and the flow-through was analyzed. High column-binding efficiency is determined by the absence of histone protein in the flow-through when analyzed by Coomassie Brilliant Blue staining. We determined the column-binding efficiency to be 100% as there were no detectable histone proteins present in the analyzed flow-through (Figure 3). All membranes with bound histones were then washed three times with wash buffer to remove any remaining impurities, leaving only histone proteins bound to the silica gel. We determined that, for all histone extraction times (i.e.,15 min, 2 h, and 24 h), the first membrane wash was the most important to remove nonhistone contaminations from the columns, while the second and third washes did not influence the sample purity. Thus, depending on the sample type, the last two washes might be omitted. Following the first elution of histone protein from the column (using elution buffer containing 1mM NaCl and EDTA), histones were precipitated overnight with 4% perchloric acid and then pelleted, washed, and analyzed for the enrichment of purified histones H3 and H4. We observed that 24 h of extraction time increases the amount of H3 and H4 histones in the purified fraction compared to 15 min and 2 h of extraction time (Figure 5A). The second elution from the column did not result in high-quality or high-quantity histones (Figure 5B).
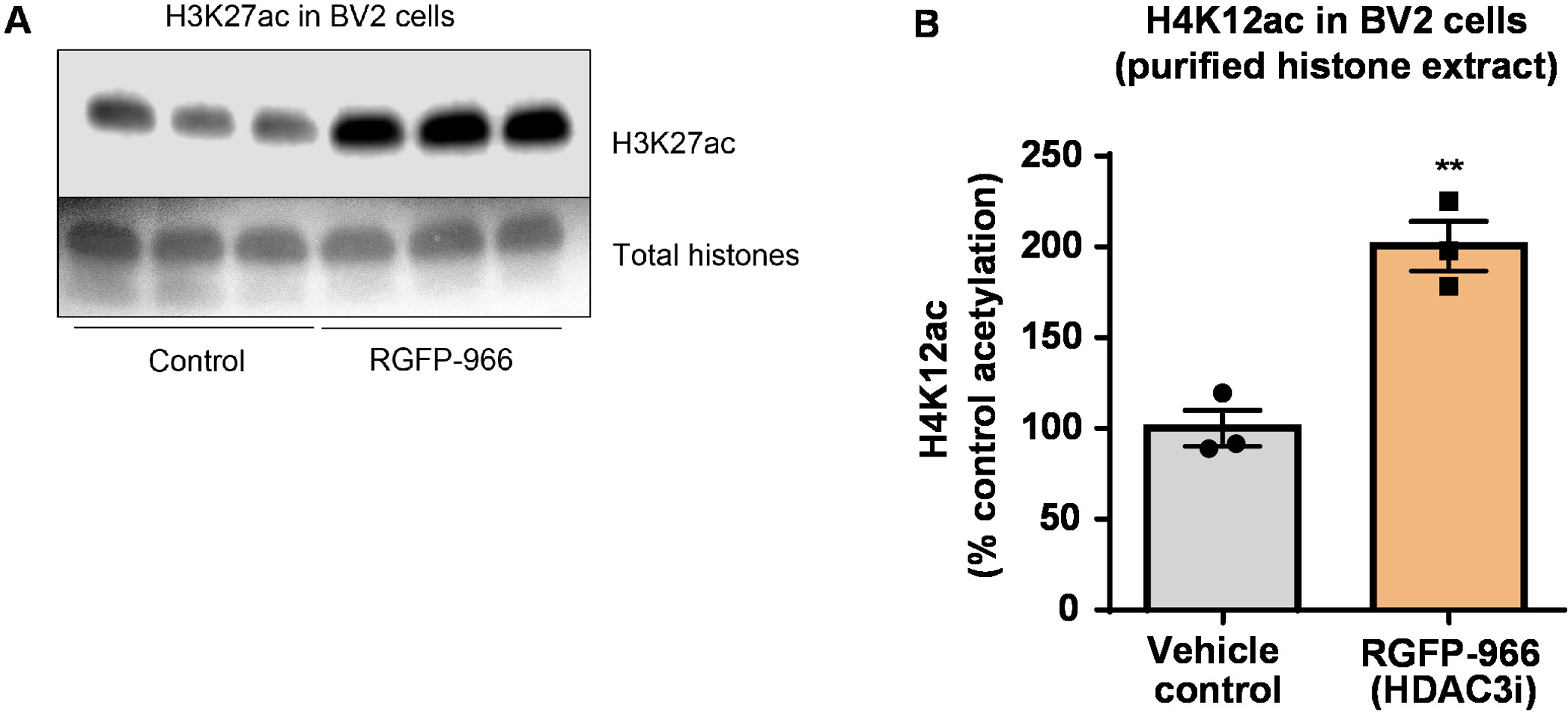
Next, we used brain tissue homogenates to quantify histone H3 and H4 PTMs, namely acetylation. Wild-type (C57BL6/J) male mice were administered a broadly acting HDAC inhibitor (tributyrin) at a dose of 5 g/kg orally for 3 d. Whole-brain tissue was collected on day 4 and crude histones were extracted according to the described protocol. Using an unpaired t-test, we determined that tributyrin increases the acetylation of histones in the crude extract (t(6) = 6.184, P = 0.0004); however, impurities are detected in the extract (the histone bands are not clearly defined). Thus, H4K12ac antibody does not have a high specificity (Figure 6). To further evaluate the applicability of the presented protocol to smaller tissue sections, we collected the prefrontal cortex from triple transgenic Alzheimer's disease (3 x Tg-AD) mice treated daily with 10 mg/kg M344, a class I and IIb HDAC inhibitor, for four months. Histone purification and precipitation was performed according to the herein described protocol. Using the purified histone H3 and H4 fraction, we determined that M344 increases H4K12 acetylation 2.4-fold (t(6) = 13.03, P < 0.0001), with a high specificity of H4K12ac antibody (Figure 7). Similarly, we observed an increase in histone H3 acetylation in BV2 cells in response to another HDAC inhibitor, namely the selective HDAC3 inhibitor, RGFP-966. 10 µM of RGFP-966 causes an approximately twofold increase of acetylation at histone H3K27 after 24 h of treatment. Student's unpaired t-test was used to compare control versus treated cells.

Figure 1: Timeline of the histone purification protocol. All steps for histone analyses are shown below along with the estimated time needed for each step. Figures depicting the outcome of particular steps and presented within the manuscript are referred to in parentheses. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative Coomassie Brilliant Blue-stained gel demonstrating crude histones extracted from BV2 cells. BV2 cells were cultured for 48 h before the histone extraction protocol began. Crude histones were extracted for 15 min, 2 h, and 24 h, with three replicates for each time point (this is also the case in Figure 3, Figure 4, and Figure 5). Both nonhistone and histone proteins are present in the crude histone extract. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative Coomassie Brilliant Blue-stained gel demonstrating column flow-through following histone purification steps from BV2 cells. Following crude histone passing through the histone binding column, only nonhistone proteins are present in the flow-through. Histone proteins are absent in this fraction. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Representative Coomassie Brilliant Blue-stained gel demonstrating a column wash following a histone purification step from BV2 cells. Regardless of the histone extraction times, which were (A) 15 min, (B) 2 h, or (C) 24 h, low quantities of nonhistone proteins were only present in the first-wash histonebinding column. Histone proteins were absent in all washes. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative Coomassie Brilliant Blue-stained gel demonstrating elutions following a histone purification step from BV2 cells. (A) High-quality purified and desalted histone H3 and H4 were detected after the first elution from the histone purification column. (B) The second elution from the histone purification column did not yield a high quality or quantity of histones H3 or H4. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: The broadly acting HDAC inhibitor, tributyrin, increases H4K12 acetylation in the whole brain of wild-type mice. (A) This panel shows a representative western blot depicting an increase of H4K12 acetylation in the crude histone extract collected from the whole brain of wild-type mice in response to the broadly acting HDAC inhibitor, tributyrin. (B) This panel shows the quantification of the increase in H4K12 acetylation in vivo. Unpaired t-test was used to compare groups (t(6) = 6.076, P = 0.0005). The bars represent the mean ± the standard error of the mean (SEM). N = 8. ***P < 0.0001. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: The class I and IIb HDAC inhibitor, M344, increases H4K12 acetylation in the prefrontal cortex of triple transgenic Alzheimer's disease (3 x Tg-AD) mice. (A) This panel shows a representative western blot depicting an increase of H4K12 acetylation in the purified and desalted histone extract collected from the prefrontal cortex of 3 x Tg-AD mice in response to the inhibition of HDACs by M344. (B) This panel shows the quantification of the increase of H4K12 acetylation in response to M344 administered at a 10 mg/kg daily dose for four months. Unpaired t-test was used to compare groups (t(6) = 13.30, P < 0.0001). The bars represent the mean ± the SEM. N = 8. ****P < 0.00001. Please click here to view a larger version of this figure.
{kind=link}

Figure 8: The selective HDAC3 inhibitor, RGFP-966, increases H3K27 acetylation in BV2 microglial cells. (A) This panel shows a representative western blot depicting an increase of H3K27 acetylation in the purified and desalted histone extract collected from BV2 cells in response to the HDAC3 inhibition by RGFP-966. (B) RGFP-966 causes an approximately twofold increase of acetylation at histone H3 and lysine (K) 27 after 24 h of treatment. Unpaired t-test was used to compare control versus treated cells (t(4) = 5.981, P = 0.002). The bars represent the mean ± the SEM. N = 6. **P < 0.01. Please click here to view a larger version of this figure.
{kind=link}
| Representative tissue (one hemisphere) * | Average tissue weight (mg) | Extraction Buffer (mL) | Number of strokes |
| Mouse cerebellum | 40 | 1 | 40 |
| Mouse frontal cortex | 30 | 0.3 | 20 |
| Mouse hippocampus | 27 | 0.3 | 18 |
| Mouse entorhinal cortex | 19 | 0.3 | 17 |
| * All experiments were performed on adult male mice. | |||
| Average age: 16 months. Average weight: 30 grams. | |||
Table 1: Optimized condition for brain tissue homogenization.
Discussion
In the current work, we demonstrated an optimized method to purify core histone proteins and quantify histone H3 and H4 PTMs (e.g., acetylation). The presented protocol is a comprehensive workflow that incorporates optimized procedures regarding cells and brain tissue preparation, crude histone purification, and detailed histone precipitation, elution, and quantification, which are followed by histone electrophoresis and robust histone PTM quantification. The large amount of details provided here allows for a replicable generation of high-quality data, despite the need for lengthy manipulations of the histone samples.
Many currently published protocols require the use of HPLC to isolate pure fractions of histones H3 and H419. Although HPLC is a powerful technique, its complexity and low-throughput deter most molecular biologists and nonexperts from its frequent use. Indeed, HPLC is not available to many labs, and highly-skilled personnel is required to operate the instrument. HPLC is oftentimes time-consuming, expensive, and potentially hazardous. Presented here is an inexpensive, medium-throughput strategy to achieve results of similar quality which bypasses HPLC. The reported strategy is also more practical and suitable for use in almost any lab as it uses a simple spin column approach that does not require specialized instrument operation skills. Additionally, histone H3/H4 tetramer is extracted as a single, pure, and abundant fraction, enabling a reliable quantification of preserved PTMs on each of the proteins.
PTMs are extremely sensitive to changes in oxidative stress and changes in pH20,21. Thus, in contrast to previously published methods18, we report an efficient strategy of rinsing the cells in serum-free media to ensure a minimum metabolic disturbance of the cells and to avoid the interference of the native PTMs with serum components. The current protocol not only bypasses the traditional nuclei isolation but also provides optimal times for cell lysis and the exact tissue homogenization procedure that allows for the preservation of the nuclear envelope while avoiding nuclear aggregation. Although the extraction time can be manipulated based on the number of cells, the cell type used, the tissue size, etc., extended lysis is not desirable as it might lead to the lysis of the nuclei and DNA release, making the sample difficult to handle. Importantly, multiple checkpoints within the protocol exist for the validation of successful histone purification (e.g., steps 2.7 and 3.2.3). This strategy also facilitates troubleshooting throughout the lengthy procedure.
Another important and unique feature of the presented protocol is its full compatibility with downstream western blot analysis tools and others if so desired. Histone proteins are detected at ~15 kDa13,22,23 and, similarly to other small molecular weight proteins, have been proven challenging to detect by standard immunoblotting techniques. The use of a high-performance and high-throughput transfer system in combination with optimal resolution protein gels allows for the maintenance of protein native confirmation (in the absence of SDS) and activity in the absence of SDS and high transfer efficiency of the low molecular weight histone proteins, thus assuring a reliable histone PTM quantification.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors express their gratitude to the Florida Department of Health Ed and Ethel Moore Alzheimer's Research Program (grants 6AZ08 and 7AZ26), the NIH-NIAAA (grant 5R01AA023781-03), and the American Heart Association (grant 17PRE33660831).
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mL microcentrifuge tubes | ThermoFiser Scientific | 05-408-129 | or equivalent from other sources |
| Sterile water | Gibco | 15-230-204 | or equivalent from other sources |
| 70% perchloric acid | Sigma Aldrich | 311421 | or equivalent from other sources |
| 100% acetone | Sigma Aldrich | 270725 | or equivalent from other sources |
| pH-indicator strips, non-bleeding | Milliipore Sigma | 1095310001 | |
| 4x SDS sample buffer | BIO-RAD | 161-0747 | |
| Benchtop rotor | Cole-Parmer | UX-04397-34 | or equivalent from other sources |
| 1.5 mL tube rack | ThermoFiser Scientific | 05-541 | or equivalent from other sources |
| Histone purification mini kit | Active Motif | 40026 | spin columns included in the kit |
| Protease Inhibitor Cocktail | ThermoFiser Scientific | 78430 | or equivalent from other sources |
| Nanodrop instrument | ThermoFiser Scientific | ND-2000 | |
| Tissue culture dishes | VWR | 10062-880 | required for histone extraction from cultured cells |
| Tissue culute media | varies based on cell line used | varies based on cell line used | required for histone extraction from cultured cells |
| Low-serum media | ThermoFiser Scientific | 51985091 | required for histone extraction from cultured cells |
| Plastic cell scraper | Falcon | 353086 | required for histone extraction from cultured cells |
| SDS-PAGE gradient gel | BIO-RAD | 456-9035 | required for histone extraction from cultured cells |
| Coomassie Brilliant Blue R-250 Staining Solution | BIO-RAD | 1610436 | required for histone extraction from cultured cells |
| Coomassie Brilliant Blue R-250 Destaining Solution | BIO-RAD | 1610438 | required for histone extraction from cultured cells |
| Trans-Blot Turbo Mini PVDF Transfer Packs | BIO-RAD | 1704156 | required for histone extraction from cultured cells |
| Trans-Blot Turbo Transfer System | RIO-RAD | 1704150 | required for histone extraction from cultured cells |
| Ponceau S stain | CellSignalling | 59803S | required for histone extraction from cultured cells |
| Dounce homogenizer (size/cap sc 7mL) with a small size clearance | Kimble Chase | 885302-0007 | required for histone extraction from tissues |
| 100% bleach | Clorox | 68973 | required for histone extraction from tissues |
| H4K12ac antibody | Active Motif | 39166 | required for PTMs quantification via WB |
| H3K27ac antibody | Active Motif | 39134 | required for PTMs quantification via WB |
References
- Holliday, R. Is there an Epigenetic Component in Long-term Memory? Journal of Theoretical Biology. 200, 339-341 (1999).
- DeWoskin, V. A., Million, R. P. The epigenetics pipeline. Nature Reviews Drug Discovery. 12, 661-662 (2013).
- Eberharter, A., Becker, P. B. Histone acetylation: a switch between repressive and permissive chromatin. EMBO Reports. 3, 224-229 (2002).
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature. 389, 349-352 (1997).
- Sartor, G. C., Powell, S. K., Brothers, S. P., Wahlestedt, C. Epigenetic Readers of Lysine Acetylation Regulate Cocaine-Induced Plasticity. The Journal of Neuroscience. 35, 15062-15072 (2015).
- Komatsu, N., et al. SAHA, a HDAC inhibitor, has profound anti-growth activity against non-small cell lung cancer cells. Oncology Reports. 15, 187-191 (2006).
- Bahari-Javan, S., Sananbenesi, F., Fischer, A. Histone-acetylation: a link between Alzheimer's disease and post-traumatic stress disorder. Frontiers in Neuroscience. 8, 160(2014).
- Roh, T. -Y., Cuddapah, S., Zhao, K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes & Development. 19, 542-552 (2005).
- Mutskov, V., Felsenfeld, G. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. The EMBO Journal. 23, 138-149 (2004).
- Howe, L., Brown, C. E., Lechner, T., Workman, J. L. Histone acetyltransferase complexes and their link to transcription. Critical Reviews in Eukaryotic Gene Expression. 9, 231-243 (1999).
- Jenuwein, T., Allis, C. D. Translating the histone code. Science. 293, 1074-1080 (2001).
- Bowman, G. D., Poirier, M. G. Post-Translational Modifications of Histones That Influence Nucleosome Dynamics. Chemical Reviews. 115, 2274-2295 (2015).
- Volmar, C. -H., Wahlestedt, C. Histone deacetylases (HDACs) and brain function. Neuroepigenetics. 1, 20-27 (2015).
- Plagg, B., Ehrlich, D., Kniewallner, K. M., Marksteiner, J., Humpel, C. Increased Acetylation of Histone H4 at Lysine 12 (H4K12) in Monocytes of Transgenic Alzheimer's Mice and in Human Patients. Current Alzheimer Research. 12, 752-760 (2015).
- Bhaskara, S., et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 18, 436-447 (2010).
- Mottamal, M., Zheng, S., Huang, T. L., Wang, G. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules. 20, 3898-3941 (2015).
- Ramakrishnan, S., et al. HDAC 1 and 6 modulate cell invasion and migration in clear cell renal cell carcinoma. BMC Cancer. 16, 617(2016).
- Wapenaar, H., Dekker, F. J. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clinical Epigenetics. 8, 59(2016).
- Klinker, H., Haas, C., Harrer, N., Becker, P. B., Mueller-Planitz, F. Rapid Purification of Recombinant Histones. PLoS ONE. 9, e104029(2014).
- Chen, K., et al. Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-translational Modification Landscapes. ACS Chemical Neuroscience. 9, 838-848 (2018).
- Simithy, J., Sidoli, S., Garcia, B. A. Integrating Proteomics and Targeted Metabolomics to Understand Global Changes in Histone Modifications. Proteomics. , e1700309 (2018).
- Volmar, C. -H., et al. An Epigenetic Approach for the Modulation of Amyloid Precursor Protein (APP) Processing and Improvement of Memory in Alzheimer's Disease. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 40, S470(2015).
- Volmar, C. -H., et al. M344 promotes nonamyloidogenic amyloid precursor protein processing while normalizing Alzheimer’s disease genes and improving memory. Proceedings of the National Academy of Sciences. 114 (43), E9135-E9144 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved