
Method Article
Real-time Live-cell Flow Cytometry to Investigate Calcium Influx, Pore Formation, and Phagocytosis by P2X7 Receptors in Adult Neural Progenitor Cells
* These authors contributed equally
In This Article
Summary
Providing single-cell sensitivity, real-time flow cytometry is uniquely suited to quantify multimodal receptor functions of live cultures. Using adult neural progenitor cells, the P2X7 receptor function was assessed via calcium influx detected by calcium indicator dye, transmembrane pore formation by ethidium bromide uptake, and phagocytosis using fluorescent latex beads.
Abstract
Live-cell flow cytometry is increasingly used among cell biologists to quantify biological processes in a living cell culture. This protocol describes a method whereby live-cell flow cytometry is extended upon to analyze the multiple functions of P2X7 receptor activation in real-time. Using a time module installed on a flow cytometer, live-cell functionality can be assessed and plotted over a given time period to explore the kinetics of calcium influx, transmembrane pore formation, and phagocytosis. This simple method is advantageous as all three canonical functions of the P2X7 receptor can be assessed using one machine, and the gathered data plotted over time provides information on the entire live-cell population rather than single-cell recordings often obtained using technically challenging patch-clamp methods. Calcium influx experiments use a calcium indicator dye, while P2X7 pore formation assays rely on ethidium bromide being allowed to pass through the transmembrane pore formed upon high agonist concentrations. Yellow-green (YG) latex beads are utilized to measure phagocytosis. Specific agonists and antagonists are applied to investigate the effects of P2X7 receptor activity. Individually, these methods can be modified to provide quantitative data on any number of calcium channels and purinergic and scavenger receptors. Taken together, they highlight how the use of real-time live-cell flow cytometry is a rapid, cost-effective, reproducible, and quantifiable method to investigate P2X7 receptor function.
Introduction
The study of purinergic signaling is broad and multifaceted, involving cellular physiology, biochemistry, and pharmacology. Purinergic signaling is involved in an infinite number of cellular and molecular processes, from cancer and cell cycle regulation to cell-cell communications and stem cell biology; as such, there often exists a potential to modulate purinergic signaling for a therapeutic benefit. Of the purinergic receptors, P2X7 receptor has received significant attention in recent years due to its potential as a therapeutic target for numerous inflammatory conditions1. Methods to study the receptor have evolved and been adapted over the years to facilitate this research2,3,4,5. Here, we describe a live-cell flow cytometry method to investigate the multiple functions of P2X7 receptors in adult neural progenitor cells derived from the subventricular zone (SVZ) and the hippocampal dentate gyrus.
The P2X7 receptor was first described as the P2Z receptor, or the ‘cell death’ receptor, as its activation with high concentrations of adenosine triphosphate (ATP) results in the formation of a large transmembrane pore permeable to molecules up to 900 Da6. This leads to cytoskeletal rearrangement, transmembrane pore formation, and, potentially, apoptosis and/or necrosis7. Traditionally, this function of P2X7 is quantified by the uptake of large molecular weight dyes such as YO-PRO-1 or ethidium bromide, which fluoresce when intercalated with DNA3,8. Plate reader methods, which are rapid and allow for upscaling, generally do not allow for the observation of kinetics. The method described here is based on ethidium uptake and allows the fluorescence increase to be observed over time, providing a greater depth of information with regard to the speed of pore formation. P2X7 receptors have since been shown to facilitate a number of nonimmune functions, with distinct responses depending on exposure time and agonist concentration9,10. Brief activation by lower concentrations of ATP results in cation influx for the purposes of neurotransmitter and signal transduction11. Using flow cytometry to measure calcium influx overcomes the problems associated with cumbersome and technically challenging methods—particularly, patch clamping to measure inward currents which provide invaluable details as to the change in potential across a cell membrane but do not allow for population analysis2. The third function of P2X7 receptors occurs in the absence of ATP, where P2X7 receptors have been demonstrated to facilitate phagocytosis in both the immune system and the nervous system9,12,13. Advancements in microscopy techniques have allowed the visualization of cytoskeletal rearrangements during the uptake process, although quantification and population analysis can still present a challenge.
The live-cell flow cytometry method detailed here allows for the investigation of all three main functions of P2X7 receptors in real-time. The inclusion of a time module device on the flow cytometer allows temperature control and continual stirring of cells in suspension. Agonist and antagonist stimuli can be delivered within a second, allowing the near uninterrupted measurement of the cellular response. This presents a rapid and simple method to reproducibly quantify function while avoiding the use of multiple assay systems. It is important to note that this protocol may easily be adapted to suit any cell type and could be used to examine other receptor subtypes given the inclusion of specific agonists or inhibitors, depending on their properties.
Protocol
Animals were treated in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and approved for use by the Griffith University Animal Ethics committee.
1. Neurosphere Culture of Neural Progenitor Cells from the Adult SVZ and Hippocampus
NOTE: The dissection protocol presented here is based on work by Walker and Kempermann, and a detailed protocol for the dissection of neural progenitor cells from adult mice is available elsewhere14. Culture conditions have been modified from Babu and colleagues15. Adult female C57BL/6 mice aged 8–12 weeks were used.
- In a biological safety cabinet, prepare the culture medium (neural basal medium) supplemented with neural stem cell proliferation supplement, 2 mM glutamine, 20 ng/mL epidermal growth factor (EGF), 10 ng/mL basic fibroblast growth factor (bFGF), and 2 µg/mL heparin.
- Euthanize two mice by CO2 inhalation. Alternatively, anesthetize the mice according to institutional guidelines and immediately euthanize them by cervical dislocation. Spray their heads with 70% ethanol and decapitate them. Transfer each head to a sterile tube containing Hank’s balanced salt solution (HBSS) with 1x penicillin/streptomycin solution (P/S).
- Perform dissections in a laminar flow hood under a dissection microscope.
- Using forceps and dissection scissors, remove tissue and bone to expose the brain, and transfer it to a sterile dish containing HBSS with P/S.
- Position the brain ventral side up and use a scalpel blade to make a complete coronal incision across the optic chiasm. Hold the brain steady with the forceps and use one swift, clean, downward motion. Avoid sawing to minimize cell death and to help maintain tissue structure.
NOTE: If not held securely, the brain may tilt forward during the cut, compromising the number of progenitor cells obtained from the SVZ. - Isolate the SVZ from the rostral half of the brain.
- Locate both ventricles, separated by the septum, with the corpus callosum forming a white bridge above them.
- Use forceps to remove the septum separating the two ventricles and isolate the lateral walls of the anterior lateral ventricles. Do this by cutting away above, below, to the sides, and at the front with fine dissection scissors. There will remain just a small cup-like shape.
- Prepare a clean Petri dish lid with a few drops of HBSS in the middle and transfer the SVZ to the liquid. Do not allow the tissue to dry or to touch a dry surface. Stand to the side while dissecting the hippocampi.
- Isolate the hippocampi from the caudal half of the brain.
- Make a midline cut between the hemispheres to sever the corpus callosum. Use the lateral ventricles as a guide and gently unfold the cortex to expose the hippocampus. Once the cortex has been unrolled, the hippocampus can be seen as a dense, white, curved structure.
- Use fine dissection scissors or forceps to isolate the hippocampus from the neighboring tissue.
- With forceps, remove excess white matter, blood vessels, and any membranous tissue covering the hippocampus.
- Prepare a clean Petri dish lid prepared with a few drops of HBSS and transfer the hippocampus to the liquid. Repeat for the other hemisphere.
- Once the hippocampus and the SVZ from two mice have been isolated and transferred to their respective Petri dish lids, mechanically dice the tissue using a scalpel blade. Chop the tissue in one Petri dish lid for approximately 1 min, rotating every 10 s until the tissue appears smooth and only fine pieces remain. Take a clean scalpel blade and repeat for the other dish, dicing the tissue for 1 min.
- Using a 1 mL pipette, transfer all the tissue from each Petri dish lid to separate tubes containing 1 mL of 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA). Use one tube for the SVZ and one tube for the hippocampus.
- Do this by first pipetting about half of the trypsin-EDTA to the tissue in the dish to minimize air bubbles and to prevent the tissue coming into contact with the dry pipette tip as it is transferred to the tube. Rinse the dish and the scalpel blade with the rest of the trypsin-EDTA to collect as much tissue as possible and add it to the tube.
- Incubate the tissue with the trypsin in a 37 °C water bath for 30 min, agitating the tube every 10 min to properly dissociate the tissue.
- Triturate the tissue using a fire-polished glass Pasteur pipette to create a single cell suspension. Take care not to overtriturate as this will cause excessive cell lysis. This is a critical step for optimal tissue dissociation.
- Observe the tube contents during the trituration process and stop at the earliest signs that the majority of tissue clumps have been removed. The trypsin solution should become slightly cloudy when the cells have gone into single-cell suspension, although some clumps may remain.
NOTE: As an indication, passing the suspension up and down 10x–15x is adequate. - Add culture medium up to 5 mL to neutralize the trypsin, and spin at 300 x g for 3 min. Wash a further 2x with HBSS and pass the medium through a 70 µm cell strainer to remove any tissue clumps.
- Transfer all the cells in 15 mL of medium to a T75 cell culture flask.
NOTE: Spheres should form in the passage zero (P0) SVZ culture after 7–10 days and after 15–20 days in the hippocampal culture. Refrain from washing or feeding the cells during this time to maximize the number of neural progenitor cells in culture. If more progenitors are required, or to decrease the P0 incubation time, it is possible to increase the number of mice sacrificed. - Maintain cultures at 37 °C with 5% CO2 and, following the initial P0 culture phase, passage every 7–10 days as necessary, using a biological safety cabinet and standard tissue culture practices.
NOTE: The P0 hippocampal culture should generate enough spheres to passage into a T25 flask; the SVZ culture will generate many more spheres and can be passaged into a T75. If the P0 hippocampal spheres have adhered, use a 200 µL pipette to gently lift the sphere. - Passage the spheres when they reach 150 to 200 µm in diameter. Collect the spheres and medium in a 15 mL tube and allow the spheres to settle by gravity for approximately 5 min. Alternatively, spin at a low speed (100 x g) for 2 min.
- Remove the medium and dissociate the cells with dissociation reagent for 7–10 min, depending on the size of the spheres.
NOTE: The dissociation reagent used will have significant effects on the outcome of the culture, so please refer to the Table of Materials for specifics. - Wash the cells by adding 5 mL of HBSS to the cell solution and centrifuge at 300 x g for 3 min. Remove the supernatant, resuspend the cells in culture medium, and seed at approximately 150,000 cells in 5 mL of medium in a T25 cell culture flask or equivalent. Maintain the cultures at 37 °C and 5% CO2.
- Confirm neural progenitor cell status by identifying the expression of stem cell markers such as Sox2 and ASCL1 before proceeding to any downstream protocol9.
NOTE: This can be done by the researcher’s preferred method (e.g., immunochemistry by microscopy, flow cytometry or western blot, or by using quantitative polymerase chain reaction (qPCR)).
2. Preparation of a Single-cell Suspension for Analysis by Flow Cytometry
- Prepare the required media. These include the following.
- Prepare Na+ medium containing 145 mM NaCl, 5 mM KOH, 10 mM HEPES, 5 mM D-glucose, 0.1% bovine serum albumin (BSA), and 0.1 mM CaCl2. This medium is also used in a calcium-free form, with 0.1 mM CaCl2 omitted.
- Prepare K+ medium containing 145 mM KCl, 5 mM KOH, 10 mM HEPES, 5 mM D-glucose, and 0.1% BSA.
NOTE: These media were extensively optimized and are detailed in previous publications4,5. - Prepare Stock ATP by weighing enough ATP powder for around 20 mL of a 100 mM stock. Dissolve the powder in 17 mL of KCl buffer (145 mM KCl, 5 mM KOH, and 10 mM HEPES, pH 7.5) and slowly, while stirring, add 2 mL of 18% (w/v) tetramethylammonium hydroxide (TMA) to the solution to bring the pH to 6.8–7.0.
- Adjust the final volume to 20 mL. Do not overshoot the pH past 7.0. Store the stock at -80 °C; note that the ATP aliquots are stable for at least 6 months.
NOTE: The free molecular weight of anhydrous ATP is 551.14 g/mol, and this does not include the molecular weight of the water and disodium molecules, which may vary from batch to batch and should be taken into consideration for the calculations. TMA is toxic, and care must be taken when handling. Read the safety data sheet and prepare the stocks in a fume cabinet. - Prepare stock BzATP by dissolving the BzATP in ultrapure H2O for a final stock concentration of 10 mM.
- Create a single-cell suspension as described in steps 1.16 and 1.17. Count the cells using a hemocytometer or automatic cell counter. Resuspend the cells in the required medium (e.g., Na+ medium, culture medium) for the experiment being conducted (as described below) and place the cells on ice in the meantime.
- For each experiment, ensure there is enough sample to include controls for forward and side scatter, as well as for the calibration of voltage and compensation settings. Note, as an indication, that spheres grown in a T75 flask typically yield around 8 x 106 cells per flask.
- Set the flow cytometer settings using control samples.
- Use forward and side scatter to selectively gate the living cells.
NOTE: Forward scatter provides information regarding cell size based on light diffraction, while side scatter provides a measure of internal complexity or granularity. Flow events with small forward and side scatter may be considered as dead cells. - Adjust the voltage and gain of the flow cytometer according to the manufacturer’s instructions. Run a trial sample to ensure the capture of data at the maximum fluorescence intensity. No compensation is required for single channel acquisitions.
- Use forward and side scatter to selectively gate the living cells.
3. Measuring Calcium Influx by Live-cell flow Cytometry
- Following the preparation of a single-cell suspension, resuspend the cells in 1 mL of calcium-free Na+ medium and load them with 2 ng/mL of calcium indicator dye according to the manufacturer’s protocol (refer to the Table of Materials) with 10 µL of 5% pluronic acid. Incubate the cells for 30 min at 37 °C.
- Wash the cells by adding 3–5 mL of calcium-free Na+ medium and centrifuging gently (200 x g for 4 min). Remove the supernatant and resuspend the cells in calcium-free Na+ medium, washing a second time.
- Resuspend the cells in 1 mL of calcium-free Na+ medium, place them on ice, and allow them to de-esterify for 30 min.
- Wash 1x more by adding 3–5 mL of K+ medium and centrifuging (200 x g for 4 min); then, resuspend the cells in K+ medium and aliquot them into fluorescence assisted cell sorting (FACS) tubes at a concentration of 1 x 106 cells per 500 µL per FACS tube.
NOTE: The number of FACS tubes per sample will depend on the number of treatments and repeats that are included. - Place the FACS tubes on ice until the cells are ready to be analyzed. Do not leave the cells on ice for an extended period but begin the assay as soon as possible.
- For some samples, preincubate the cells with P2X7 receptor-specific inhibitor AZ10606120 (1 µM for 10–15 min) or A438079 (10 µM for 30 min) at 37 °C prior to analysis.
- A few minutes prior to running the first sample, add CaCl2 (to a final concentration of 1 mM in the FACS tube) and place the tube in a 37 °C water bath to recover.
- Drop a clean, small magnetic stirrer into the FACS tube and position the tube in the time module linked to a circulating 37 °C water bath to control the sample temperature. Select a low stirring speed to ensure movement of the sample without introducing a vortex effect. Place the water jacket tube adapter onto the sample platform and close the lever arm of the FACS machine.
- Initiate sample acquisition and run the sample for 3 min at around 1,000 events per second.
- At the 40 s mark, quickly remove the tube and add the P2X7 agonist, either 1 mM ATP or 300 µM BzATP, and replace the tube to continue the acquisition.
- While the first sample is recording, prepare the second sample with CaCl2 and place it in 37 °C to allow sufficient time for the cells to warm up prior to analysis. Once the first sample has finished, clean the intake by running a water sample, and then the acquisition of the second sample can begin as described in steps 3.8 and 3.9. Always clean the intake between samples.
4. Measuring Pore Formation by Live-cell Flow Cytometry
- Create a single-cell suspension as described in steps 1.16 and 1.17. Save a few milliliters of the old medium, use it to resuspend the cells at a concentration of 1 x 106 cells per 100 µL per FACS tube, and place it on ice until ready.
- Prior to running the assay, add 900 µL of K+ medium for a final volume of 1 mL and place the tubes in a 37 °C water bath for 10 min to recover.
- If applicable, preincubate the cells with treatments, including the P2X7-specific inhibitors AZ10606120 (1 µM for 10–15 min) or A438079 (10 µM for 30 min).
- Immediately prior to running the assay, add 25 µM ethidium bromide to the FACS tube; then, add the magnetic stirrer, place it on the FACS machine according to step 3.7, and begin the acquisition.
NOTE: Ethidium bromide is toxic, and care must be taken when handling it. Dispose of used FACS tubes appropriately. - To induce the formation of pores in the cell membrane, add 1 mM ATP or 100 µM BzATP 40 s after the start of the acquisition.
- Run the samples at around 1,000 events per second for 6 min.
- While the first sample is running, take the second sample from the ice and place it in the 37 °C water bath to allow sufficient time for the cells to recover prior to the analysis. Once the first sample has finished with the acquisition, clean the intake by running a water sample; then, the second sample can be placed on the machine to begin recording as described in steps 3.8 and 3.9.
5. Measuring Phagocytosis by Live-cell Flow Cytometry
- Create a single-cell suspension as described in steps 1.16 and 1.17. Resuspend the cells in conditioned medium and aliquot them into FACS tubes at a concentration of at least 1 x 106 cells per 100 µL per FACS tube. Dilute the cells to a final concentration of 1 x 106 cells/mL with Na+ medium (e.g., add 900 µL of Na+ medium) and place the cells on ice until the analysis is performed.
- Use 1 µm of YG latex beads (microspheres) as phagocytic targets for real-time phagocytosis assays.
NOTE: Other colors may also be substituted, but differently sized beads were found to be inadequate targets of phagocytosis4. - Prior to running the first sample, transfer the cells to a 37 °C water bath and incubate them for approximately 7–10 min to allow the cells to recover.
- Add any treatments requiring preincubation to their respective tubes, including 1 mM ATP for 15 min, 300 µM oxidized ATP (oxATP) for 40 min, 20 µM cytochalasin D for 20 min, and 4% paraformaldehyde (PFA) for 20 min.
- No preincubation is required for 5% human serum. If treatments are added at approximately the same time, the samples can be run in reverse order while the others continue to incubate. For example, run the controls and serum first, then the ATP-treated sample, followed by cytochalasin D and PFA, and oxATP last.
- Place the sample on the cytometer with the magnetic stirrer as described in steps 3.8 and 3.9 and, then, initiate sample acquisition.
- Remove the sample tube from the machine 15–20 s after the start of the acquisition, and add 5 µL of undiluted YG beads. Return the sample FACS tube and continue the acquisition. Run the samples for 7–8 min at around 1,000 events/s.
- While the first sample is running, take the second sample from the ice and place it in a 37 °C water bath to allow sufficient time for the cells to recover prior to analysis. Once the first sample is finished, clean the intake by running a water sample and, then, begin acquisition on the second sample as described in steps 3.8 and 3.9.
6. Data Analysis
- Export the data to a spreadsheet. The data analysis will depend on the experimental question.
NOTE: Be aware that different runs may have different baseline intensities, so it is important to run the assay for a designated time at the start (around 40 s) prior to adding any agonists and to normalize the data by calculating the change in fluorescence (the fluorescence at any given time point [F] divided by the fluorescence at time point zero [F0], or F/F0). - To quantify the rate or kinetics of the P2X7 function in question, calculate the area under the curve or the sum of the trapezoids created under the curve for each 10 s time period16.
- To determine the effects of treatments, average the fluorescence intensity over the final 10–20 s of recording and compare the treatments. Determine the significance by t-test or analysis of variance.
Results
Neural progenitor cell cultures
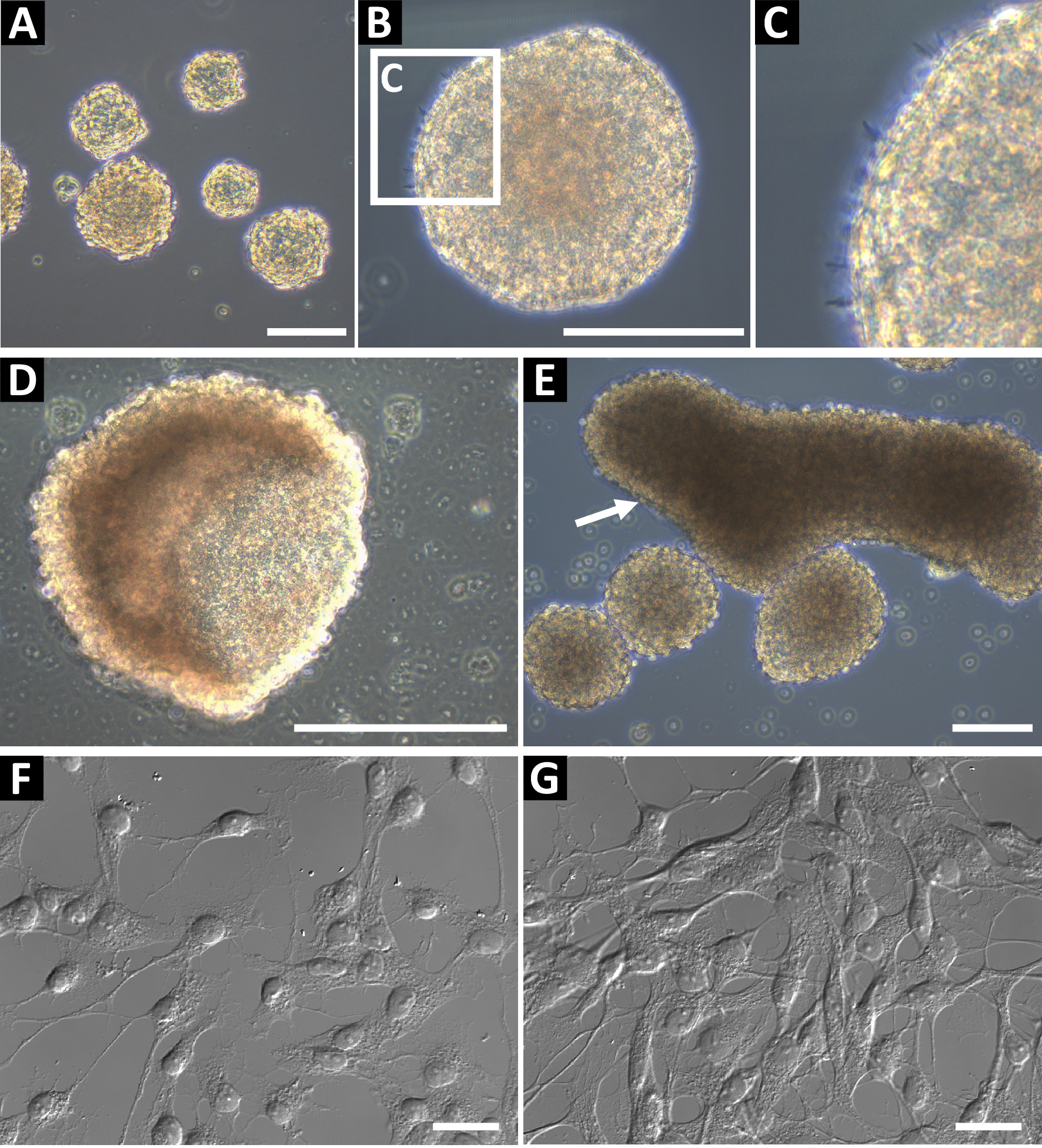
Neural progenitor sphere cultures derived using this method should be phase bright and have a smooth round edge (Figure 1A,B). In healthy cultures, small microspikes can be observed on the edges (Figure 1C). At late passages, or if fed inadequately, spheres can form a hollow cup shape (Figure 1D) or large oblong shapes (Figure 1E, indicated by arrow). These cultures should not be used for flow cytometry or any other downstream applications, as these features it may be indicative of differentiation. To confirm the neural progenitor status, the cells were plated on glass coverslips coated with poly-L-ornithine and laminin for immunocytochemistry (Figure 1F and, at higher confluency, Figure 1G). Cells were stained for GFAP, nestin, Sox2, vimentin, ASCL1, BLBP, Prox1, and DCX to identify the cells as Type 2 progenitor cells (hippocampus) or Type C progenitor cells (SVZ)9. Cells should have a well-defined nucleus and extended processes.

Figure 1: Representative hippocampal neural progenitor cell culture. (A) Hippocampal neural progenitor cells are isolated from adult mice and cultured as neurospheres until approximately 100 to 150 µm in diameter. (B) Neurospheres should have a smooth periphery, (C) and small microspikes may be observed on their surface. When spheres are too long in culture, they can form (D) cup or (E) oblong shapes. These cultures should not be used for experiments. To confirm the neural progenitor status of the cells, seed them as a single-cell suspension on poly-L-ornithine (PLO) and laminin-coated glass coverslips for immunochemistry. Cells should have a small soma and branching processes, (F) at low confluency and (G) ready for immunochemistry. Scale bars = 100 µm. Please click here to view a larger version of this figure.
{kind=link}
Calcium influx by live-cell flow cytometry
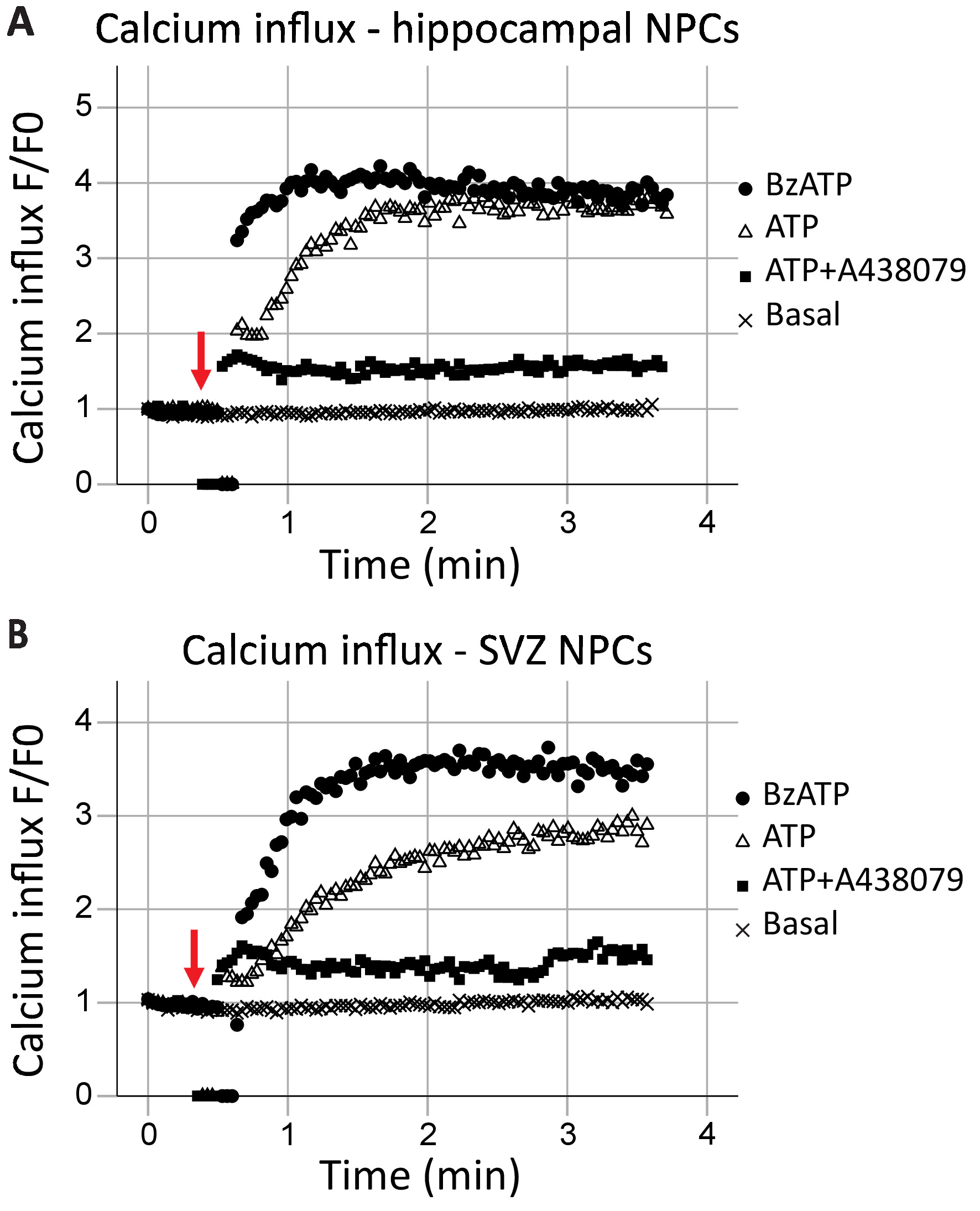
This protocol allows for the analysis of P2X7 receptor function as a calcium channel in real-time. The kinetics of receptor function, as well as the effects of different agonists and antagonists, can also be assessed. When plotted over time, calcium influx in the hippocampal and SVZ neural progenitor cells was generally similar (Figure 2A and Figure 2B, respectively). Agonists (either ATP or BzATP) were added at the 40 s mark, as indicated by the red arrow. For a brief moment, the tube is removed from the recording point to add the agonist, resulting in data points of zero. This will allow for the identification of the time when the agonist was added. BzATP rapidly activates P2X7 receptors, opening the ion channel and allowing calcium influx, which binds to Fluo-8 and fluoresces. ATP application generally results in a more gradual calcium influx. It has a lower affinity to P2X7 when compared to BzATP and will also result in G-protein-coupled receptor activation, a slower signaling pathway which releases calcium from the endoplasmic reticulum. The inclusion of P2X7 antagonists A438079 and AZ10606120 (data not shown) reduced the calcium influx in response to agonist application.

Figure 2: Live-cell calcium influx in neural progenitor cells from the hippocampus and SVZ. P2X7 receptor calcium channel function was demonstrated in (A) hippocampal and (B) SVZ-derived progenitor cells by changes in Fluo-8 fluorescence. Application of the general P2X agonist ATP and the P2X7 agonist BzATP result in P2X7 ion channels opening, allowing calcium influx. The influx was blocked with the P2X7-specific inhibitors A438079 or AZ10606120 (data not shown). F = fluorescence; F0 = fluorescence at time point zero. Please click here to view a larger version of this figure.
{kind=link}
Pore formation by live-cell flow cytometry
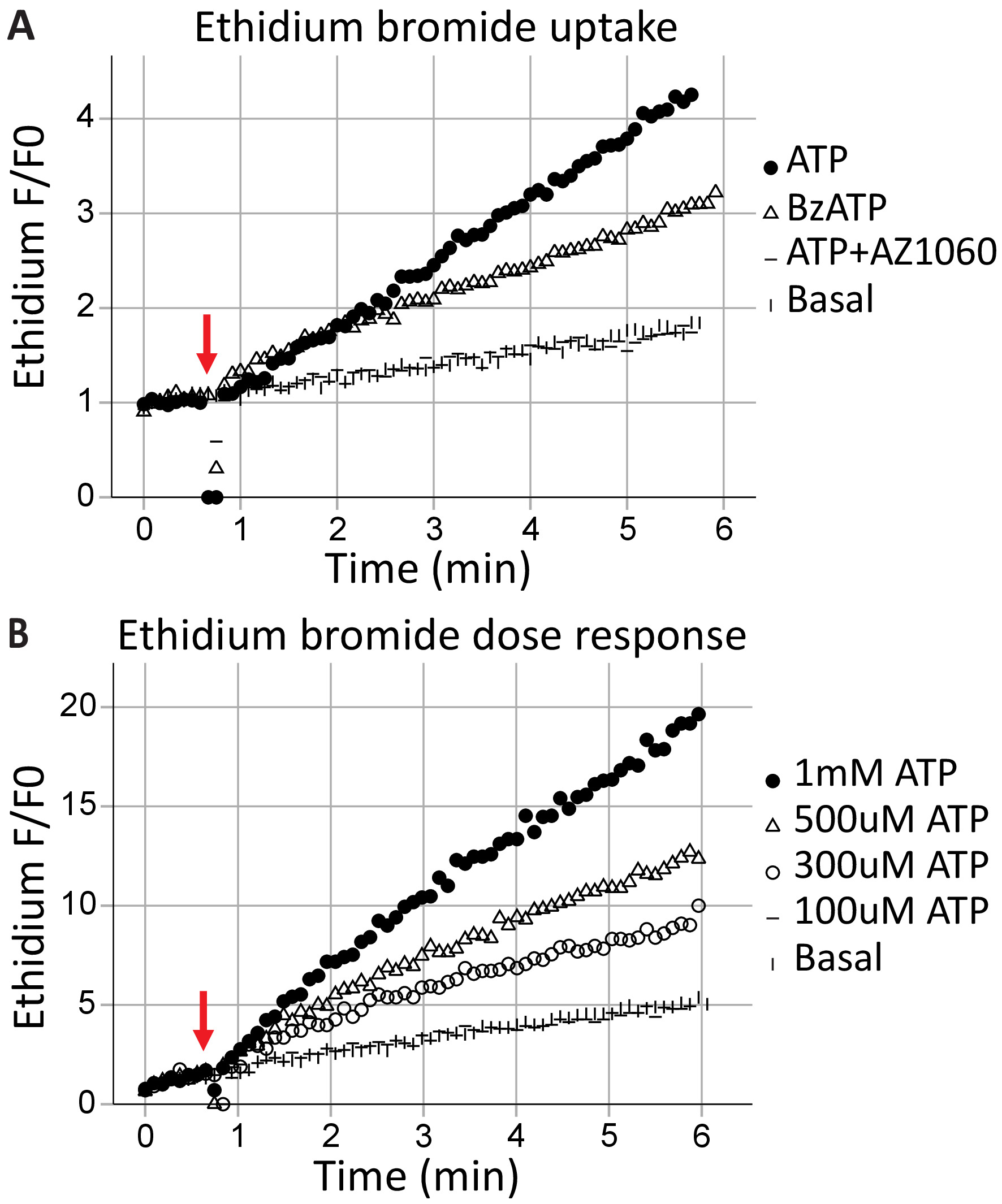
Transmembrane pore formation is a canonical feature of P2X7 receptors, results in macromolecule exchange, and can lead to cell death. Ethidium+ is a large molecule (314 Da) excluded from healthy cells; its uptake and subsequent intercalation with DNA results in fluorescent emissions and can be used to assess the ability of P2X7 receptors to form transmembrane pores. Following the application of the agonists ATP (1 mM ATP) and BzATP (100 μM) at the 40 s (indicated by the arrow), time-resolved flow cytometry captures the ethidium bromide entering the cells in real-time (Figure 3A). This effect was attenuated by the P2X7-specific inhibitor AZ10606120. The ethidium bromide uptake assay demonstrates a functional P2X7 receptor C-terminus17 and is good evidence for full-length P2X7 receptor expression. ATP concentration-response assays illustrate the effects of agonist concentration on P2X7 pore formation, using change in ethidium bromide fluorescence over time (Figure 3B). Agonist dose concentration curves together with receptor-specific inhibitors provide strong evidence for receptor activation.

Figure 3: P2X7 transmembrane pore formation measured by ethidium uptake. The addition of ethidium bromide moments before the start of acquisition is used to measure the formation of P2X7 transmembrane pores. High concentrations of ATP and BzATP result in (A) P2X7 receptor pore formation, allowing ethidium bromide to enter the cell. The P2X7 inhibitor AZ10606120 attenuates this phenomenon and provides evidence for functional P2X7 receptors. (B) ATP concentration-response assays demonstrated significant pore formation at 500 μM and 1 mM but not at lower concentrations. Please click here to view a larger version of this figure.
{kind=link}
Phagocytosis by live-cell flow cytometry
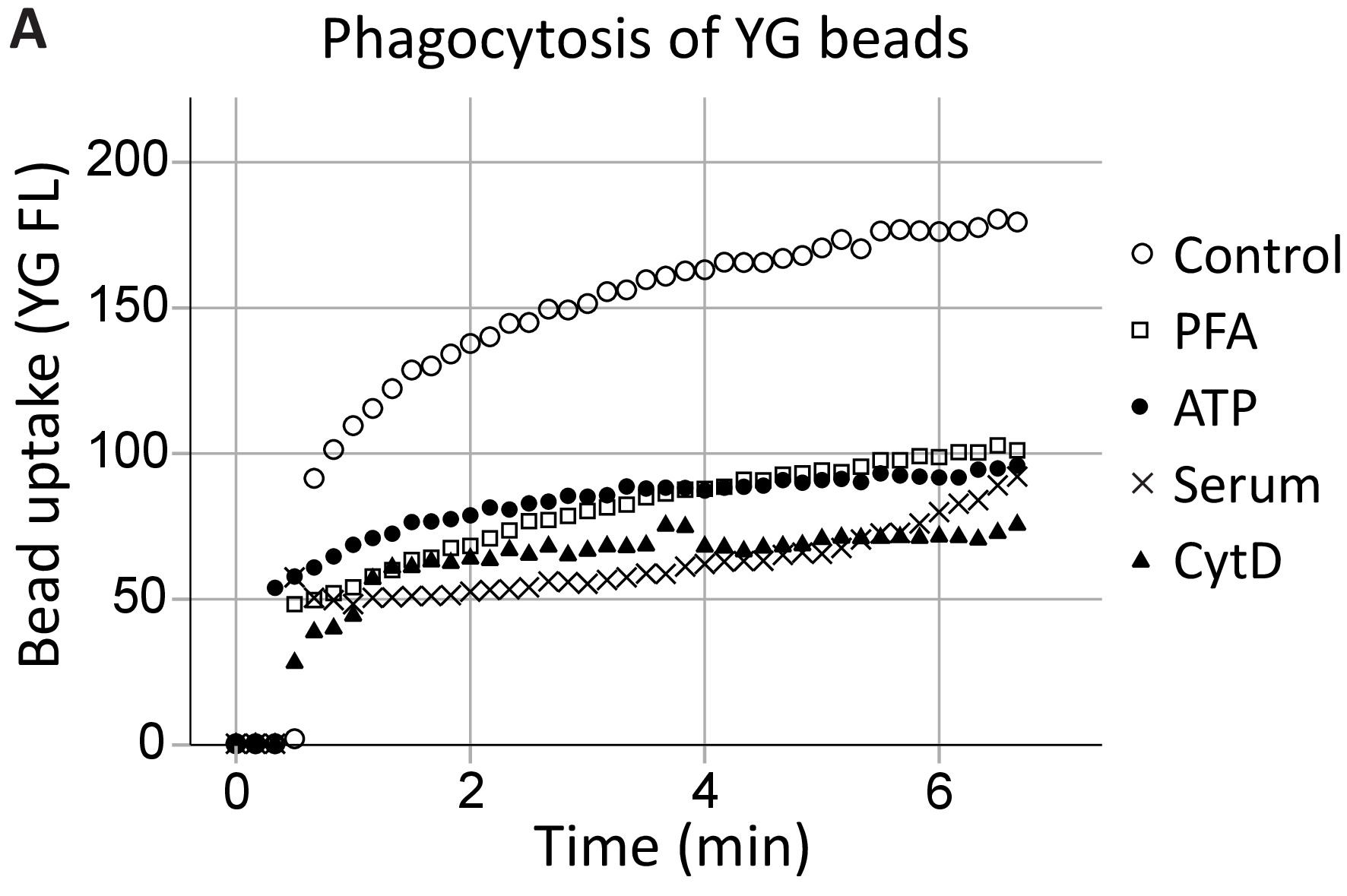
Our group has previously demonstrated that extracellular ATP inhibits P2X7-mediated phagocytosis by dissociating the P2X7 C-terminus from the cytoskeleton, specifically, nonmuscle myosin IIA18,19. This method expands on these findings to demonstrate P2X7 receptor involvement in phagocytosis by hippocampal and SVZ neural progenitor cells in real-time (Figure 4, an example of hippocampal phagocytosis). Uninhibited phagocytosis (control) levels of 1 µm YG latex beads were established as the positive control. ATP inhibited the phagocytosis of YG beads to the same extent as the nonspecific inhibitors, namely PFA fixation and the actin polymerization inhibitor cytochalasin D, while 5% serum abolished all innate phagocytosis20.

Figure 4: YG bead uptake demonstrating the phagocytic capacity of neural progenitors via P2X7 receptors. YG bead uptake by neural progenitor cells is observable using live-cell flow cytometry in real-time. Control levels of phagocytosis are established initially, and if the number of cells allows, reconfirmed at the end of the run. Involvement of P2X7 receptors is indicated by the inhibition of phagocytosis in the presence of ATP, as this dissociates the C-terminus from the membrane cytoskeleton, preventing P2X7-mediated cytoskeletal rearrangements. The application of ATP blocked phagocytosis to the same extent as the use of nonspecific inhibitors of phagocytosis, including paraformaldehyde (PFA) and cytochalasin D (CytD). Please click here to view a larger version of this figure.
{kind=link}
Discussion
This paper presents a detailed protocol for the analysis of P2X7 receptor function in neural progenitor cell cultures derived from the adult neurogenic niches. The potential applications for adult neural progenitor cells range from research to therapeutic purposes, and so the method of culture must be robust and reproducible. There are a number of key aspects to this protocol that may impact the quality of the endpoint culture. Once removed from the skull, the brain should not be allowed to dry and the dissection should be performed as quickly as possible. Particularly with the hippocampus, extra care taken to remove any blood vessels or membranous tissue will result in superior progenitor cell yields. The dissociation and trituration process can heavily impact the number of spheres obtained in a culture; agitating the tissue during the incubation with trypsin-EDTA will result in a more homogeneous solution. The use of a fire-polished glass pasture pipette over a P1000 plastic pipette tip is highly recommended to reduce cell death and improve the resulting culture. Avoid overtriturating. Despite these precautions, the procedure can create a lot of debris in the P0 culture, and to avoid losing progenitor cells, washing or feeding the culture should be avoided until spheres have formed.
A number of differences between the hippocampal and SVZ cultures will be obvious at P0. Hippocampal cultures yield fewer spheres, and these generally adhere. Use a pipette tip to gently lift off the spheres for the initial passage. Adherent spheres were not observed in subsequent passages. Different brands of tissue culture flasks may cause the spheres, particularly hippocampal spheres, to adhere and grow as colonies on the bottom of the dish. This was not found to alter any downstream results for this protocol but should be monitored, and consistency should be maintained where possible.
Previous methods used to measure P2X7 receptor function, such as patch clamping to record calcium influx, are time-consuming and laborious and may only provide information on a single cell. This protocol presents a rapid and reproducible method to analyze all three main functions of P2X7 receptors using one machine. Time-resolved live-cell flow cytometry allows for whole population analysis and provides the researcher with information regarding the kinetics of calcium influx, pore formation, and/or phagocytic function. In addition to this, flow cytometry can be easily used as a method for assessing marker expression patterns and population analysis based on cell size or protein expression levels.
When conducting these experiments, differences in maximal calcium influx, ethidium uptake, or phagocytosis rates may be observed between repeats. To minimize this, the sphere size, culture conditions, and feeding regime must be consistent as the health of the cells will have a significant impact on the results obtained. The time on ice can also influence the data, so ensure everything is prepared ahead of time so that the time on ice is minimal. Ensure that the calcium indicator dye loading time is consistent. Another factor that may lead to large inconsistencies in the maximal calcium recordings is the variation between ATP batches. The preparation of ATP stocks is crucial, and the use of different batches for the same experiments should be avoided. Comparing old and new batches to ensure the ATP is consistent is also recommended. The effectiveness of P2X7 antagonists may also be cell-line- and batch-dependent, so optimization of incubation times and concentrations may be required.
It is worth noting that calcium influx/efflux is one of the most fundamental and complex cellular functions and can be mediated by many receptors. The ATP-induced calcium influx, as a classic measurement for P2X7 channel/pore function, may not accurately reflect the true function of P2X7 receptors, as ATP may also activate P2Y receptors to release intracellular calcium. In this case, barium may be a better cation to use instead of calcium as its influx is unidirectional16. To differentiate the contribution from P2Y receptors in calcium influx, conditions where 1 mM EDTA or ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) is added to the K+ medium instead of CaCl2 may be used in this assay.
This protocol may also be easily adapted to suit other cell types and can be useful for investigating the functionality of alternate ion channel receptors or receptors that participate in phagocytosis. This method may also be adapted to a flow cytometry machine without a time module. As an example, phagocytosis assays may be performed where YG beads are added 7–8 min prior to the analysis by conventional flow cytometry. Keep the cells at 37 °C and continuously swirl them. This will not provide real-time information but differences in the mean final fluorescence will still inform the researcher regarding the functionality of P2X7 receptors.
Interest in P2X7 receptors as a drug target21,22 or even as a drug delivery route23,24 is growing rapidly, and so methods to study this enigmatic receptor must be continuously adapted and improved upon to facilitate these studies. This protocol outlines methodologies that may be used to explore P2X7 function in adult neural progenitor cells, and it is hoped that achieving a greater understanding of P2X7 receptors in the neurogenic niches may lead to advancements in the treatment of stroke and other ischemic injuries.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank Maria Kasherman and Xin Huang for their contributions to this research. This work was supported by grants from the Rebecca L. Cooper Medical Research Foundation to M.W., T.C.L., and M.L., and to T.C.L. from the National Health and Medical Research Council (NHMRC) of Australia (571100 and 1048082) and the Baxter Charitable Foundation (Sydney, Australia). B.G. was supported by the Australian Research Council (ARC) Future Fellowship (FT120100581), NHMRC Project Grants (1048082, 1061419, and 1120095) and the Victorian Government’s Operational Infrastructure Support Grant to the Florey Institute. M.L. was supported by a Charles D. Kelman, M.D. Postdoctoral Award (2010) from the International Retinal Research Foundation (USA).
Materials
| Name | Company | Catalog Number | Comments |
| A438079 | Tocris | 2972/10 | |
| ATP | Sigma-Aldrich | A2383 | |
| AZ10606120 | Tocris | 3323/10 | |
| bzATP | Sigma-Aldrich | B6396 | |
| Cytochalasin D | Sigma-Aldrich | C8273 | |
| FACSCalibur | Becton Dickinson | ||
| Fluo-8AM | AAT-Bioquest | 21080 | Fluo-4AM and Fura-Red AM have also been used successfully |
| Fluoresbrite YG Microspheres | Polysciences Inc | 17154-10 | 1.00 µm, yellow-green |
| Glutamine | ThermoFisher Scientific | 25030081 | 200 mM |
| HBSS | ThermoFisher Scientific | 14170112 | |
| Heparin | Sigma-Aldrich | H3149 | |
| NeuroCult Basal Medium | Stemcell Technologies | 5700 | Mouse and rat |
| NeuroCult Proliferation Supplement | Stemcell Technologies | 5701 | Mouse and rat |
| Oxidized ATP | Sigma-Aldrich | A6779 | |
| Pluronic F-127 | Sigma-Aldrich | P2443 | pluronic acid |
| Recombinant Murine EGF | Peprotech | 315-09 | |
| Recombinant Murine FGF-basic | Peprotech | 450-33 | |
| Tetramethylammonium Hydroxide | Sigma-Aldrich | T7505 | |
| Time Zero Module | Cytek Biosciences | ||
| Tissue culture flasks | BD Falcon (Corning) | 353108 (T25), 353136 (T75) | Blue vented screw cap |
| TrypLE Express | Gibco | 12604013 | |
| Trypsin-EDTA (0.25%) | ThermoFisher Scientific | 25200056 | with phenol red |
| UltraPure Ethidium Bromide | ThermoFisher Scientific | 15585011 | 10 mg/mL |
References
- Sperlagh, B., Illes, P. P2X7 receptor: an emerging target in central nervous system diseases. Trends in Pharmacological Sciences. 35 (10), 537-547 (2014).
- Liang, X., et al. Quantifying Ca2+ current and permeability in ATP-gated P2X7 receptors. Journal of Biological Chemistry. 290 (12), 7930-7942 (2015).
- Rat, P., Olivier, E., Tanter, C., Wakx, A., Dutot, M. A fast and reproducible cell- and 96-well plate-based method for the evaluation of P2X7 receptor activation using YO-PRO-1 fluorescent dye. Journal of Biological Methods. 4 (1), 64(2017).
- Gu, B. J., et al. A quantitative method for measuring innate phagocytosis by human monocytes using real-time flow cytometry. Journal of Quantitative Cell Science: Cytometry Part A. 85 (4), 313-321 (2014).
- Jursik, C., et al. A quantitative method for routine measurement of cell surface P2X7 receptor function in leucocyte subsets by two-colour time-resolved flow cytometry. Journal of Immunological Methods. 325 (1-2), 67-77 (2007).
- Surprenant, A., Rassendren, F., Kawashima, E., North, R. A., Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science. 272 (5262), 735-738 (1996).
- Delarasse, C., et al. Neural progenitor cell death is induced by extracellular ATP via ligation of P2X7 receptor. Journal of Neurochemistry. 109 (3), 846-857 (2009).
- North, R. A. Molecular physiology of P2X receptors. Physiological Reviews. 82 (4), 1013-1067 (2002).
- Leeson, H. C., et al. P2X7 Receptors Regulate Phagocytosis and Proliferation in Adult Hippocampal and SVZ Neural Progenitor Cells: Implications for Inflammation in Neurogenesis. Stem Cells. , (2018).
- Glaser, T., et al. Modulation of mouse embryonic stem cell proliferation and neural differentiation by the P2X7 receptor. PLoS One. 9 (5), e96281(2014).
- Papp, L., Vizi, E. S., Sperlagh, B. Lack of ATP-evoked GABA and glutamate release in the hippocampus of P2X7 receptor-/- mice. Neuroreport. 15 (15), 2387-2391 (2004).
- Wiley, J. S., Gu, B. J. A new role for the P2X7 receptor: a scavenger receptor for bacteria and apoptotic cells in the absence of serum and extracellular ATP. Purinergic Signalling. 8 (3), 579-586 (2012).
- Lovelace, M. D., et al. P2X7 receptors mediate innate phagocytosis by human neural precursor cells and neuroblasts. Stem Cells. 33 (2), 526-541 (2015).
- Walker, T. L., Kempermann, G. One mouse, two cultures: isolation and culture of adult neural stem cells from the two neurogenic zones of individual mice. Journal of Visualized Experiments. (84), e51225(2014).
- Babu, H., et al. A protocol for isolation and enriched monolayer cultivation of neural precursor cells from mouse dentate gyrus. Frontiers in Neuroscience. 5, 89(2011).
- Gu, B. J., Wiley, J. S. Broad applications of mulit-colour time-resolved flow cytometry. Flow Cytometry - Recent Perspectives. , (2012).
- Cheewatrakoolpong, B., Gilchrest, H., Anthes, J. C., Greenfeder, S. Identification and characterization of splice variants of the human P2X7 ATP channel. Biochemical and Biophysical Research Communications. 332 (1), 17-27 (2005).
- Gu, B. J., Saunders, B. M., Jursik, C., Wiley, J. S. The P2X7-nonmuscle myosin membrane complex regulates phagocytosis of nonopsonized particles and bacteria by a pathway attenuated by extracellular ATP. Blood. 115 (8), 1621-1631 (2010).
- Gu, B. J., Rathsam, C., Stokes, L., McGeachie, A. B., Wiley, J. S. Extracellular ATP dissociates nonmuscle myosin from P2X(7) complex: this dissociation regulates P2X(7) pore formation. American Journal of Physiology-Cell Physiology. 297 (2), C430-C439 (2009).
- Gu, B. J., et al. P2X7 receptor-mediated scavenger activity of mononuclear phagocytes toward non-opsonized particles and apoptotic cells is inhibited by serum glycoproteins but remains active in cerebrospinal fluid. Journal of Biological Chemistry. 287 (21), 17318-17330 (2012).
- Bhattacharya, A. Recent Advances in CNS P2X7 Physiology and Pharmacology: Focus on Neuropsychiatric Disorders. Frontiers in Pharmacology. 9 (30), (2018).
- Burnstock, G., Knight, G. E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signalling. 14 (1), 1-18 (2018).
- Alves, L. A., et al. Pore forming channels as a drug delivery system for photodynamic therapy in cancer associated with nanoscintillators. Oncotarget. 9 (38), 25342-25354 (2018).
- Pacheco, P. A., et al. P2X7 receptor as a novel drug delivery system to increase the entrance of hydrophilic drugs into cells during photodynamic therapy. Journal of Bioenergetics and Biomembranes. 48 (4), 397-411 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved