Method Article
Detection of Retrotransposition Activity of Hot LINE-1s by Long-Distance Inverse PCR
In This Article
Summary
This article outlines a simple PCR-based assay to monitor the activity of an active LINE-1 retrotransposon and to map de novo retrotranspositions in a given genome. Using the MCF7 cell line, we demonstrate herein how this method can be applied to detect activity of a LINE-1 located at 22q12.1.
Abstract
Long interspersed nuclear elements 1 (LINE-1s) are the only family of mobile genetic elements in the human genome that can move autonomously. They do so by a process called retrotransposition wherein they transcribe to form an mRNA intermediate which is then consequently inserted into the genome by reverse transcription. Despite being silent in normal cells, LINE-1s are highly active in different epithelial tumors. De novo LINE-1 insertions can potentially drive tumorigenesis, and hence it is important to systematically study LINE-1 retrotransposition in cancer. Out of ~150 retrotransposition-competent LINE-1s present in the human genome, only a handful of LINE-1 loci, also referred to as "hot" LINE-1s, account for the majority of de novo LINE-1 insertion in different cancer types. We have developed a simple polymerase chain reaction (PCR)-based method to monitor retrotransposition activity of these hot LINE-1s. This method, based on long-distance inverse (LDI)-PCR, takes advantage of 3´ transduction, a mechanism by which a LINE-1 mobilizes its flanking non-repetitive region, which can subsequently be used to identify de novo LINE-1 3´ transduction events stemming from a particular hot LINE-1.
Introduction
Long interspersed nuclear elements (LINE-1s) are a family of mobile genetic elements called retrotransposons that can independently move from one place to another via a copy-and-paste mechanism called retrotransposition. Over evolutionary time, the human genome has accumulated more than 500,000 copies of LINE-1 repeats1. However, most of the LINE-1 copies present in the genome are mutated and hence cannot move via retrotransposition; only ~150 copies have intact copy of DNA sequence necessary for them to move2. In normal somatic cells, the mobility of these LINE-1s is restricted by different host factors3. These restrictions are relieved in different epithelial tumors, causing LINE-1s to be derepressed and resulting in many de novo insertions in the tumor genome4. Some of these tumor-associated de novo insertions have been shown to cause insertional mutagenesis in genes, hence driving tumor progression5,6. Therefore, it is important to be able to map novel insertions in the tumor genome.
Existing methods to detect de novo LINE-1 insertions use (1) whole-genome sequencing approach4,7,8, where different computational algorithms are used to find de novo LINE-1 insertions from WGS data, or (2) next-generation sequencing that targets the 3´ end of young, potentially active LINE-1s9,10,11,12,13. However, finding novel insertions among several thousand near-identical copies with these methods is far from trivial, and the challenge is further aggravated by tumor heterogeneity and genomic alterations associated with LINE-1 insertion4.
Studies using these existing methods showed that just a few LINE-1s account for the majority of de novo LINE-1 insertions observed in tumors7,8. Therefore, to answer whether or not a particular tumor sample displays LINE-1 activity, it suffices to map retrotransposition events caused by this handful of highly active LINE-1 loci. In this article, we describe a simple polymerase chain reaction (PCR)-based method14 that can be used to monitor the activity of a particular LINE-1 locus in the first intron of the TTC28 gene at 22q12.1 that is highly active in colorectal cancer7,8. This LINE-1 locus will be referred to as TTC28-LINE-1 throughout the article. This assay specifically identifies de novo LINE-1 retrotransposition events that mobilize non-repetitive sequence on the 3´ flanking region of the source LINE-1 by a mechanism called 3´ transduction15. 3´ transduction occurs due to the weak LINE-1 polyadenylation signal (PAS) that causes the transcriptional machinery to skip it and to instead terminate transcription at the stronger PAS downstream, thus capturing the flanking non-repetitive sequence (henceforth referred to as the "unique tag") which is then inserted into the target location alongside the LINE-1 sequence. Philippe et al.16 recently showed that different cell types can express different LINE-1 loci. In light of this finding, this method can be applied to monitor the activity of the most highly expressed LINE-1 that mobilize their unique tag in the cancer type of interest.
The first step in LDI-PCR is digestion of genomic DNA with a restriction enzyme that generates a restriction fragment containing the LINE-1 being assayed (here, TTC28-LINE-1) and its unique tag (Figure 1). Digested DNA is then circularized by self-ligation and PCR amplified using inverse primers located within the unique tag. By doing so, the full-length source LINE-1 at its "native" location is always amplified and alongside it, offspring LINE-1 insertions at different target loci containing the unique tag will also be amplified (Figure 1), thus reporting retrotransposition activity of the LINE-1 in question.
Protocol
This research was approved by the Institutional Review Board and Ethics committee of Helsinki University Hospital. Signed informed consent was obtained from the subject for the blood sample used to demonstrate this protocol.
1. Designing inverse primers and selecting restriction enzymes (bioinformatics)
- Determining a LINE-1 associated unique tag
- Download the TTC28-LINE-1 sequence in FASTA format from a LINE-1 database such as L1Base17. The L1base ID for TTC28-LINE-1 is 135.
- Include 5 kb sequence flanking both 5´ and 3´ ends of the LINE-1 sequence and annotate it in a word processor.
NOTE: Here the LINE-1 flanking sequence is annotated in brown font, and LINE-1 sequence is in grey font (Supplemental File). - Enter 1 kb sequence downstream of selected LINE-1's cognate PAS into a PAS prediction tool such as polyadq18 or Dragon PolyA spotter19 and annotate all the polyadenylation signal in this 1 kb window.
NOTE: If there is no PAS in the 1 kb window, search for PAS in next 1 kb window downstream. TTC28-LINE-1's own weak PAS is highlighted in pink and all the other PAS in 1 kb window downstream is highlighted in red (Supplemental File). - Annotate the sequence between the end of LINE-1's cognate PAS and the strongest PAS downstream as "unique tag".
NOTE: The "unique tag" of TTC28-LINE-1 is highlighted in yellow (Supplemental File).
- Designing inverse primers
- Design inverse PCR primers by entering the "unique tag" sequence into a web-based primer-designing tool like Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/) or NCBI’s primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). Since primer pairs designed by these tools face each other facilitating conventional PCR, use the reverse-complement function for the primer pair to perform inverse PCR.
NOTE: As LINE-1 transductions are heavily truncated at their 5´ end and the size of the transduced region is highly variable, aim to keep the distance between the two inverse primers minimal, by setting the "PCR product length" parameter in NCBI primer-BLAST to the minimum. In the case of multiple PAS within the unique tag, design several primer pairs that correspond to different PASs. Design primers close to the PAS, as LINE-1 insertions initiate from the 3´end of the RNA intermediate and the 5´ end is variably truncated. Here three primer pairs were designed, highlighted in teal and green, corresponding to three strong polyadenylation signals in the unique tag of TTC28 LINE-1 (Supplemental File).
- Design inverse PCR primers by entering the "unique tag" sequence into a web-based primer-designing tool like Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/) or NCBI’s primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). Since primer pairs designed by these tools face each other facilitating conventional PCR, use the reverse-complement function for the primer pair to perform inverse PCR.
- Selecting restriction enzymes
- Digest the LINE-1 sequence along with its 5 kb upstream and downstream flanks in silico using web-based tool such as RestrictionMapper. This will give a comprehensive list of restriction enzymes that digests this region, generating different restriction fragments.
- Select restriction enzymes that cut the native locus of LINE-1 as follows: at the 5´ end, either upstream of the LINE-1´s 5´end or far 5´end of the LINE-1 itself, and at the 3´ end, downstream of the LINE-1's unique tag.
NOTE: The selected restriction enzyme should be insensitive to DNA methylation, should be heat-inactivatable, and should generate staggered "sticky" ends that are complementary to each other. In order to demonstrate LDI-PCR, SacI restriction enzyme that cuts DNA at GAGCTC sites, highlighted here in light green, is used (Supplemental File). - Take note of the restriction fragment size made by selected restriction enzymes. This should not be longer than 12 kb as it might not be efficiently amplified by PCR.
2. Making circular DNA templates for long-distance inverse PCR
- DNA extraction and quality assessment
- Extract genomic DNA from samples (tumor or blood) using commercially available DNA extraction kits that can extract good quality, high molecular weight DNA necessary for LDI-PCR according to the manufacturer's instructions. Alternatively, high molecular weight DNA can also be extracted by phenol:chloroform20.
- Measure the DNA concentration using a fluorometer according to the manufacturer's instructions, and run 100 ng of DNA on 1% (w/v) agarose gel containing ethidium bromide (0.5 µg/mL) in 1x Tris-acetate-EDTA (TAE) buffer at 4.5 V/cm21 alongside λ-HindIII DNA molecular weight markers to check DNA quality and quantity.
- Digesting genomic DNA
- Make a digestion reaction mix (final volume of 50 µL) by adding 20 units SacI restriction enzyme, 5 µL of 10x reaction buffer (Table of Materials), 100 ng of DNA (up to 44 µL) in a 0.2 mL PCR tube on ice (1 µL of restriction enzyme from most manufacturers is sufficient to completely digest 100 ng of genomic DNA). Mix the solution by flicking the tube, and centrifuge briefly.
- Use a thermal cycler to incubate the reaction mix at 37 °C for 1 h, followed by heat inactivation at 65 °C for 5 min.
- Self-ligating the digested genomic DNA
- To the 50 µL digestion mix (after step 2.2.2), add 8 µL of 10x T4 DNA ligase buffer, 1 µL (5 units) of T4 DNA ligase and 21 µL of ultrapure water to make a final reaction volume of 80 µL. Mix the solution by flicking the tubes, and centrifuge briefly.
- Incubate in a thermal cycler at 22 °C for 10 min, terminating with a heat inactivation step at 65 °C for 10 min.
3. Long-distance inverse PCR
- Determining primer annealing temperature by gradient PCR
- Set an annealing temperature gradient of (A-4) °C, (A-2)°C, A, (A+2)°C, (A+4) °C where A is the theoretical annealing temperature of the primer pair calculated using the commercial vendor's web-based tool.
NOTE: Thermal cyclers from some manufacturers may not allow setting the temperature gradient manually. In that case, the automatic gradient setting can be used with a temperature range of (A-4) °C to (A+4) °C. - Prepare a master mix for the PCR by combining and mixing the following components in a 1.5 mL microcentrifuge tube: 4 µL of 5x reaction buffer, 0.4 µL of 10 mM dNTP, 5 µL of 2 µM PCR primer (forward and reverse, designed in step 1.3.1), 0.2 µL (0.1 U) of DNA polymerase per reaction. Set up one reaction for each annealing temperature in the gradient.
- For each reaction, aliquot 19 µL of the master mix into 0.2 mL PCR tubes and add 1 µL (1.25 ng) of circular DNA template made in section 2.
NOTE: Use circular self-ligated DNA generated from normal blood DNA as template so as to avoid consuming potentially precious tumor DNA for this optimization step. - Run gradient PCR program on a thermal cycler as described below: (i) one cycle of 3 min at 98 °C (denaturation); (ii) 35 cycles of (10 s at 98 °C [denaturation], 20 s at the temperature gradient of [A-4] to [A+4] °C [Annealing] and 1−6 min [30 s per kilobase of expected PCR product] at 72 °C [extension]); (iii) one cycle of 10 min at 72 °C (final extension).
- Run 6 µL of the PCR product in 1% agarose gel21 prepared in 1x TAE buffer at 4.5 V/cm and analyze the PCR products yielded at different annealing temperatures.
- Select the annealing temperature that yields a PCR product that corresponds to the expected size.
- Set an annealing temperature gradient of (A-4) °C, (A-2)°C, A, (A+2)°C, (A+4) °C where A is the theoretical annealing temperature of the primer pair calculated using the commercial vendor's web-based tool.
- Detecting de novo LINE-1 retrotransposition activity in the tumor genome
- Perform LDI-PCR using the inverse PCR primer pair on circular DNA templates generated from tumor samples (section 2). Follow same instructions as for gradient PCR (section 3.1), but this time replacing the temperature gradient with the optimal annealing temperature.
- Analyze the PCR products by agarose gel electrophoresis as done in step 3.1.5. PCR product of known size or the "native" PCR product corresponding to the LINE-1 at its native locus should be visible for each reaction. De novo LINE-1 3´ transduction in the tumor sample assayed is detectable as PCR products of different sizes, along with the native PCR product in the agarose gel.
4. Sequencing LDI-PCR products to reveal the identity of target sites for LINE-1 3´ transduction
- Perform single-molecule long read sequencing of all the PCR amplicons generated in each LDI-PCR reaction to identify the target integration sites of these LINE-1 3´ transduction events.
NOTE: Cloning and Sanger sequencing of the LDI-PCR products is also a possible, albeit cumbersome, approach. - Align the reads produced by single-molecule long read sequencing platforms to the reference genome using standard sequence alignment pipelines. Analyze the aligned reads using LDI-PCR software14 to identify de novo LINE-1 insertions and its target sites.
Results
In case of TTC28-LINE-1, there is more than one PAS within a 1 kb window downstream of its cognate PAS, hence the region between the TTC28-LINE-1 PAS and strongest PAS at 811 bp downstream was considered as the unique tag for TTC28-LINE-1. Three inverse PCR primer pairs were designed at this unique tag that correspond to different PASs present14. We selected three restriction enzymes: (i) NsiI that cuts 5´ upstream of TTC28-LINE-1 and 3´ outside the unique tag, and (ii) SacI and (iii) PstI that cut 5´ within the LINE-1 in its far 5´ end, and 3´ outside the unique tag. These generate restriction fragments of 10,288 bp, 5,699 bp, and 6,305 bp respectively.
In order to demonstrate this method, we performed LDI-PCR on DNA extracted from MCF7 cell line. This breast cancer cell line has been previously reported to display TTC28-LINE-1 activity16. For simplicity, we made a circular DNA template using one restriction enzyme, SacI, out of three and performed an LDI-PCR using one primer pair out of three (Table 1) to detect de novo LINE-1 insertions stemming from TTC28-LINE-1.
Good quality of DNA extracted from MCF7 cell line was ensured by agarose gel electrophoresis (Figure 2). Intact high molecular weight DNA shows that the genomic DNA is of optimum quality for this assay. If a smear is visible instead, this indicates poor quality of the extracted DNA, which in turn will hamper downstream procedures.
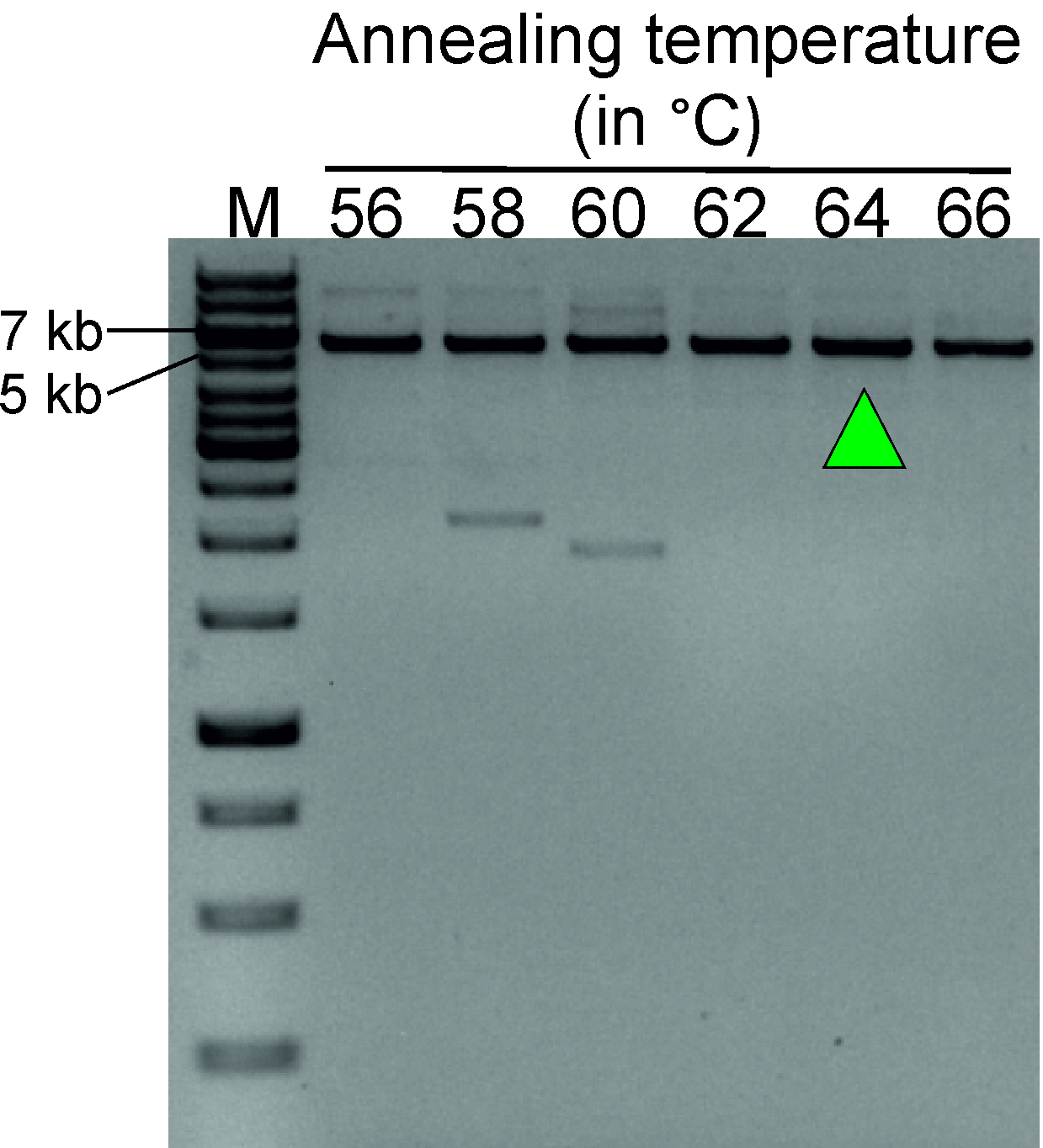
Figure 3 shows a representative result of a gradient PCR experiment, aimed at determining the optimal annealing temperature of the TTC28-LINE-1 inverse primer pair. Blood genomic DNA digested with SacI, followed by self-ligation to form a circular DNA template, was used for this reaction. A highly specific PCR product of expected size (5,649 bp) at 62, 64 and 66 °C shows that the optimum annealing temperature for this primer pair lies within the range of 62−66 °C.
We generated a circular DNA template by digesting MCF7 genomic DNA with the SacI restriction enzyme followed by self-ligation. Figure 4 shows that TTC28-LINE-1 3´ transduction occurs in MCF7 cell lines: de novo insertions can be detected as LDI-PCR products of varying sizes along with a native PCR product of known size (5,649 bp). To identify genomic coordinates of the de novo target sites, PCR amplicons can be sequenced (see protocol section 4).

Figure 1: Overview of LDI-PCR to detect LINE-1 3´ transduction. A circular DNA template is generated by first digesting (I) it with a restriction enzyme and self-ligating (II) it. This step is followed by inverse PCR (III) with inverse PCR primers targeted to the unique tag of the LINE-1 of interest (sequence between LINE-1's own weaker PAS, in pink, and stronger PAS downstream, in red). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Quality assessment of extracted DNA. 100 ng of DNA extracted from the MCF7 cell line and blood from a normal individual, which will be used as a control sample, were run alongside 1 µL and 2 µL of λ DNA/HindIII marker labelled as M. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Gradient PCR to determine optimal annealing temperature for inverse PCR primers (Table 1). LDI-PCR using inverse primer pairs at annealing temperature ranging from 56 to 66 °C shows a distinct PCR product at 62−66 °C. Green arrow indicates the selected annealing temperature for future experiments. Circular DNA template generated by digesting blood genomic DNA from a normal individual with SacI followed by self-ligation was used for this optimization step. M, marker (1 kb plus DNA ladder). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: LDI-PCR to identify LINE-1 3´ transduction stemming from TTC28-LINE-1. Circular DNA templates generated by digesting MCF7 and blood (from normal individual) genomic DNA with SacI followed by self-ligation were amplified by inverse primers in optimum annealing temperature. "Native" PCR product, marked with asterisk, of expected size (5,649 bp) was detected in both MCF7 DNA and normal blood DNA, while MCF7 also produced additional PCR products of varying sizes, indicating de novo LINE-1 retrotransposition. M, marker (1 kb DNA ladder). Please click here to view a larger version of this figure.
{kind=link}
| Primer name | Sequence (5´→ 3´) |
| L1_001 (rev) | TTCACTAAGCATGTATGTGGAAAAC |
| L1_002 (fwd) | CCCAAAATATACCCAATTACTGGCA |
Table 1: Inverse PCR primer pair designed for the unique tag of TTC28-LINE-114.
Supplemental File. Please click here to download this file.
Discussion
Here we describe a method that can be used to identify de novo LINE-1 insertions stemming from any active LINE-1 of interest. We have optimized this method for a highly active LINE-1, located at 22q12.1, and previously demonstrated it to be highly sensitive in detecting sub-clonal insertions in colorectal cancer14.
Success of LDI-PCR depends on the quality of genomic DNA. Therefore, we have included an additional quality control step to ensure that at the start of the protocol, high-molecular weight DNA is present (step 2.1.2). We recommend storing genomic DNA at -20 °C for long term storage, and to prepare aliquots in order to avoid cycles of freezing and thawing. Using genomic DNA from blood or patient-matched normal tissue is highly recommended to distinguish whether the LINE-1 retrotransposition detected is a germline or a somatic event. Since cut sites for restriction enzymes are stochastic in the genome, it is possible that a particular de novo LINE-1 insertion site might not harbor any cut sites for the restriction enzyme being used in its vicinity. Hence to increase the likelihood of detecting the majority of de novo LINE-1 insertions in tumor DNA, more than one restriction enzyme should be used in separate reactions to generate different libraries of circular DNA template. Furthermore, if the unique tag of the LINE-1 of interest has more than one PAS, then using primer pairs adjacent to each PAS improves the chances of detecting heavily truncated transductions.
Although elegant methods for genome-wide detection of de novo LINE-1 insertions exist, they can be overwhelming if the aim is to probe the retrotransposition competence of a particular LINE-1 in a specific cellular context. For this purpose, LDI-PCR can be an inexpensive and simple yet robust approach to visualize LINE-1 retrotransposition events. The targeting approach used in this method is similar to TS-ATLAS22; however, LDI-PCR avoids using linker oligonucleotides and can amplify both 5´ and 3´ junctions of de novo LINE-1 insertion simultaneously. Information regarding both 5´ and 3´ junctions of the LINE-1 insertion, the target site of integration, polyA tail and target-site modifications, all of which are hallmarks of LINE-1 retrotransposition, can be obtained by coupling LDI-PCR with single-molecule long-read sequencing technologies. Long reads thus generated contain the inserted LINE-1 sequence, its unique tag and the target sequences in one single read, circumventing difficulties of mapping short reads in the repetitive region.
There are two major limitations to using LDI-PCR for detection of LINE-1 activity. The first is inherent to PCR: it can only reliably amplify fragments up to 10 kb in size. This should be considered while selecting restriction enzyme(s), as the native fragment should not exceed this limit. Secondly, this method can only detect retrotransposition events that mobilize the LINE-1's 3´ flanking region by 3´ transductions. Hence, activity of those LINE-1s that do not exhibit 3´ transduction will not be detected using this method. Additionally, despite of being amplified by LDI-PCR, some LINE-1 retrotransposition events that (a) generate a PCR target of similar size as the "native" location or other retrotranspositions or (b) are rare or subclonal, may not be detected by agarose gel electrophoresis. Such LINE-1 retrotransposition events can be captured by sequencing the LDI-PCR product using single molecule long-read sequencing technologies14.
The workflow described here can be easily modified to detect the activity of other "hot" LINE-1s by using a suitable restriction enzyme and by designing inverse primers targeting these LINE-1s. In addition to detection of LINE-1 mediated 3´ transduction, this method can be adapted to detect less frequent LINE-1 mediated 5´ transductions23. Similar method have been used to identify the integration site of LINE-1 reporters in cell-based assays24 and proviral integration sites in cancer25. Besides LINE-1 insertions, this method can also be utilized to detect other genomic aberrations, such as DNA rearrangements, where information regarding the rearrangement-prone region pre-exists26.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to thank all our co-authors in the article where this method was first described14, especially Tatiana Cajuso, Kimmo Palin, Outi Kilpivaara and Esa Pitkänen for valuable discussions while developing the method. L.K. is funded by the Academy of Finland (grant numbers 25996, 292789, 306026 and 314394), the Sigrid Juselius Foundation, and the Finnish Cancer Society. B.P. is recipient of University of Helsinki Research Foundation PhD studentship, a Finnish Cancer Society dissertation grant, and a doctoral research grant from Ida Montinin Säätiö. We also thank Teemu Masalin (University of Helsinki) and Kul Shrestha from L.K.'s research group for assisting us with the video production.
Materials
| Name | Company | Catalog Number | Comments |
| 1 Kb DNA Ladder | New England Biolabs | N3232L | |
| 10 mM dNTP | ThermoFisher Scientific | 18427013 | |
| Acetic acid | ThermoFisher Scientific | 64-19-7 | Used to make TAE buffer |
| Agarose | BioNordika | BN-50004 | |
| Blood sample (frozen) | Blood sample from a healthy individual for control PCR | ||
| ChemiDoc XRS+ System | Bio-rad | 1708265 | |
| DNA Gel Loading Dye (6X) | ThermoFisher Scientific | R0611 | |
| DNeasy Blood & Tissue Kits | Qiagen | 69504 | |
| Ethidium Bromide | Bio-rad | 161-0433 | |
| Ethylenediaminetetraacetic acid (EDTA) | ThermoFisher Scientific | 25102-12-9 | Used to make TAE buffer |
| FastDigest buffer | ThermoFisher Scientific | B64 | |
| FastDigest SacI | ThermoFisher Scientific | FD1133 | |
| Generuler 1 Kb plus DNA Ladder | ThermoFisher Scientific | SM1331 | |
| Generuler Lambda DNA/HindIII Marker, 2 | ThermoFisher Scientific | SM0103 | |
| Mini-Sub Cell GT Cell | Bio-rad | 1704406 | |
| Phusion Green Hot Start II High-Fidelity DNA Polymerase | ThermoFisher Scientific | F537L | |
| PowerPac Basic Power Supply | Bio-rad | 1645050 | |
| Quantus Fluorometer | Promega | E6150 | |
| T4 DNA Ligase | ThermoFisher Scientific | EL0011 | |
| Tear-A-Way 96/8, 96 Well PCR Plate | 4titude | 4ti-0750/TA | |
| Tris(hydroxymethyl)aminomethane | ThermoFisher Scientific | 77-86-1 | Used to make TAE buffer |
| Veriti Thermal Cycler | Applied Bioscience | 4375786 |
References
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Brouha, B., et al. Hot LINE-1s account for the bulk of retrotransposition in the human population. Proceedings of the National Academy of Sciences of the United States of America. 100 (9), 5280-5285 (2003).
- Goodier, J. L. Restricting retrotransposons: a review. Mobile DNA. 7, 16 (2016).
- Lee, E., et al. Landscape of somatic retrotransposition in human cancers. Science. 337 (6097), 967-971 (2012).
- Miki, Y., et al. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Research. 52 (3), 643-645 (1992).
- Scott, E. C., et al. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Research. 26 (6), 745-755 (2016).
- Pitkanen, E., et al. Frequent L1 retrotranspositions originating from TTC28 in colorectal cancer. Oncotarget. 5 (3), 853-859 (2014).
- Tubio, J. M., et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 345 (6196), 1251343 (2014).
- Badge, R. M., Alisch, R. S., Moran, J. V. ATLAS: a system to selectively identify human-specific L1 insertions. American Journal of Human Genetics. 72 (4), 823-838 (2003).
- Rodic, N., et al. Retrotransposon insertions in the clonal evolution of pancreatic ductal adenocarcinoma. Nature Medicine. 21 (9), 1060-1064 (2015).
- Ewing, A. D., Kazazian, H. H. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Research. 20 (9), 1262-1270 (2010).
- Sanchez-Luque, F. J., Richardson, S. R., Faulkner, G. J. Retrotransposon Capture Sequencing (RC-Seq): A Targeted, High-Throughput Approach to Resolve Somatic L1 Retrotransposition in Humans. Methods in Molecular Biology. 1400, 47-77 (2016).
- Zhao, B., et al. Somatic LINE-1 retrotransposition in cortical neurons and non-brain tissues of Rett patients and healthy individuals. PLOS Genetics. 15 (4), e1008043 (2019).
- Pradhan, B., et al. Detection of subclonal L1 transductions in colorectal cancer by long-distance inverse-PCR and Nanopore sequencing. Scientific Reports. 7 (1), 14521 (2017).
- Moran, J. V., DeBerardinis, R. J., Kazazian, H. H. Exon shuffling by L1 retrotransposition. Science. 283 (5407), 1530-1534 (1999).
- Philippe, C., et al. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. Elife. 5, (2016).
- Penzkofer, T., et al. L1Base 2: more retrotransposition-active LINE-1s, more mammalian genomes. Nucleic Acids Research. 45 (D1), D68-D73 (2017).
- Tabaska, J. E., Zhang, M. Q. Detection of polyadenylation signals in human DNA sequences. Gene. 231 (1-2), 77-86 (1999).
- Kalkatawi, M., et al. Dragon PolyA Spotter: predictor of poly(A) motifs within human genomic DNA sequences. Bioinformatics. 29 (11), 1484 (2013).
- Sambrook, J., Russell, D. W. Purification of nucleic acids by extraction with phenol:chloroform. Cold Spring Harbor Protocols. 2006 (1), (2006).
- Green, M. R., Sambrook, J. Agarose Gel Electrophoresis. Cold Spring Harbor Protocols. 2019 (1), (2019).
- Macfarlane, C. M., et al. Transduction-specific ATLAS reveals a cohort of highly active L1 retrotransposons in human populations. Human Mutation. 34 (7), 974-985 (2013).
- Beck, C. R., Garcia-Perez, J. L., Badge, R. M., Moran, J. V. LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics. 12, 187-215 (2011).
- Morrish, T. A., et al. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nature Genetics. 31 (2), 159-165 (2002).
- Li, J., et al. Leukaemia disease genes: large-scale cloning and pathway predictions. Nature Genetics. 23 (3), 348-353 (1999).
- Pradhan, B., et al. Detection and screening of chromosomal rearrangements in uterine leiomyomas by long-distance inverse PCR. Genes, Chromosomes and Cancer. 55 (3), 215-226 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved