
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Traducción de la purificación de afinidad de ribosoma (TRAP) para investigar el desarrollo de la raíz de Arabidopsis thaliana a una escala específica de tipo de célula
En este artículo
Resumen
La traducción de la purificación de afinidad ribosoma (TRAP) ofrece la posibilidad de diseccionar programas de desarrollo con un procesamiento mínimo de órganos y tejidos. El protocolo produce ARN de alta calidad a partir de células dirigidas con una subunidad ribosomal etiquetada con proteína fluorescente verde (GFP). Las herramientas de análisis aguas abajo, como qRT-PCR o RNA-seq, revelan perfiles de expresión específicos del tejido y del tipo de célula.
Resumen
En este artículo, damos instrucciones prácticas para obtener datos de traducción de diferentes tipos de células raíz de Arabidopsis thaliana a través del método de traducción de purificación de afinidad ribosoma (TRAP) y la preparación de bibliotecas de baja entrada optimizadas consecutivas.
Como material de partida, empleamos líneas vegetales que expresan la proteína ribosomal etiquetada por GFP RPL18 de una manera específica del tipo de célula mediante el uso de promotores adecuados. Antes de la inmunopurificación y la extracción de ARN, el tejido se congela a presión, lo que preserva la integridad del tejido y simultáneamente permite la ejecución de estudios de series temporales con alta resolución temporal. En particular, las estructuras de las paredes celulares permanecen intactas, lo que es un inconveniente importante en procedimientos alternativos como enfoques basados en la clasificación celular activado por fluorescencia que se basan en el protoduro de tejidos para aislar poblaciones celulares distintas. Además, no es necesaria ninguna fijación tisular como en las técnicas basadas en microdisección de captura láser, lo que permite obtener ARN de alta calidad.
Sin embargo, el muestreo de subpoblaciones de células y solo el arn asociado al poliso limita gravemente los rendimientos del ARN. Por lo tanto, es necesario aplicar métodos de preparación de bibliotecas suficientemente sensibles para la adquisición exitosa de datos por ARN-seq.
TRAP ofrece una herramienta ideal para la investigación de plantas, ya que muchos procesos de desarrollo implican vías de señalización mecánica y relacionadas con la pared celular. El uso de promotores para dirigirse a poblaciones celulares específicas está cerrando la brecha entre el nivel de órgano y de una sola célula que a su vez sufren de poca resolución o costos muy altos. Aquí, aplicamos TRAP para estudiar la comunicación célula-célula en la formación de raíces laterales.
Introducción
Impulsada por la creciente aplicación de técnicas de secuenciación de próxima generación, se podría aumentar la resolución espacial en la biología del desarrollo. Los estudios contemporáneos tienen como objetivo disuajar tejidos hasta tipos de células especializadas, si no de un solo nivel de células1,,2,3,4. Para ello, en los últimos cincuenta años se ha ideado una plétora de métodos diferentes (véase la Figura 1A)5,6,7,8,9,10,11,12,13,14,15.
Muchas herramientas en la ciencia vegetal han sido adaptaciones de técnicas que fueron pioneras en la investigación animal. Este no es el caso del método que estamos introduciendo en detalle aquí. En 2005, equipado con una sólida experiencia en traducción de proteínas, el Bailey-Serres Lab se propuso diseñar proteínas ribosomales para la posterior purificación de afinidad16. Por lo tanto, podrían evitar el perfilado de polisomas que consume mucho tiempo y requiere mucha mano de obra, que se basa en la ultracentrifugación con un gradiente de sacarosa y se utilizó para evaluar la traducción de ribosomas desde la década de 196017,18. Desde entonces, el método se ha denominado purificación de afinidad ribosoma traslacional (TRAP)16. Después de estudios de traducción exitosos en plantas, Heiman et al. adaptó TRAP para animales19 y otros ampliaron su aplicación a la levadura20, Drosophila21, Xenopus22 y zebrafish23,,24.
Aunque la modificación genética del sistema modelo es un requisito previo para trap, que limita su aplicación a especies susceptibles a la transformación genética, se puede aprovechar simultáneamente esta objeción a los subconjuntos objetivo de células que son de especial interés y, por lo tanto, extremadamente difíciles de aislar del tejido/órgano intacto25 (por ejemplo, células dendríticas altamente ramificadas en un cerebro de ratón o hifas fúngicas en el tejido vegetal infectado). En las plantas, todas las células se mantienen en su lugar a través de paredes celulares que forman la base del esqueleto hidrostático26. Para liberar una célula vegetal de esta matriz, los científicos han cortado físicamente la célula de su tejido circundante a través de la microdisección de captura láser (LCM)27 o han realizado digestión enzimática de las paredes celulares28. Entre estas últimas células, las llamadas protoplastias, la población de interés está etiquetada fluorescentemente y se puede separar mediante la clasificación celular activada por fluorescencia (FACS)7. LCM generalmente requiere que una muestra sea fija e incrustada en la cera, lo que en última instancia deteriora la calidad de su ARN29. Los métodos basados en FACS producen ARN de alta calidad, pero el proceso de protoplasting en sí mismo introduce diferencias en la expresión génica30 y los tejidos con paredes celulares secundarias modificadas y gruesas son notoriamente difíciles de tratar. Además, se supone que muchos procesos de desarrollo en las plantas dependen de señales transmitidas mecánicamente y, por lo tanto, la integridad de la pared celular es de suma importancia31. Dos métodos, que utilizan un acceso directo para eludir el aislamiento celular operando en el nivel de nucleii, son la clasificación nuclear activada por fluorescencia (FANS) y el aislamiento de núcleos etiquetados en tipos de células específicos (INTACT). Al igual que en TRAP, utilizan promotores específicos del tipo de celda para marcar los núcleos, que posteriormente se enriquecen a través de la clasificación o el tirón hacia abajo, respectivamente8,15. Un desafío importante para todos estos enfoques es obtener suficiente material de ARN de subconjuntos de células en un tejido. Como TRAP captura solo una fracción de los ARN celulares, la recolección de muestras es un cuello de botella considerable. Por lo tanto, se necesitan protocolos de preparación de bibliotecas especialmente sensibles para producir datos de alta calidad a partir de cantidades de entrada bajas.
Desde su creación, TRAP se ha utilizado en combinación con microarrays de ADN o, como los costos de secuenciación disminuyeron significativamente en los últimos años, ARN-seq10,32,33. Una multitud de preguntas de investigación ya se ha aclarado como se revisó en Sablok et al.34. Estamos convencidos de que seguirán más informes en los próximos años, ya que la técnica es muy versátil a la hora de combinar diferentes promotores para apuntar a tipos de células específicas. Eventualmente, esto se hará incluso de una manera inducible, y puede combinarse con el sondeo de la reacción de la planta a muchos factores de estrés biótico y abiótico. Además, cuando no se dispone de líneas transgénicas estables, los sistemas de expresión de raíces peludas también se han utilizado con éxito para realizar TRAP en tomate y medicago35,,36.

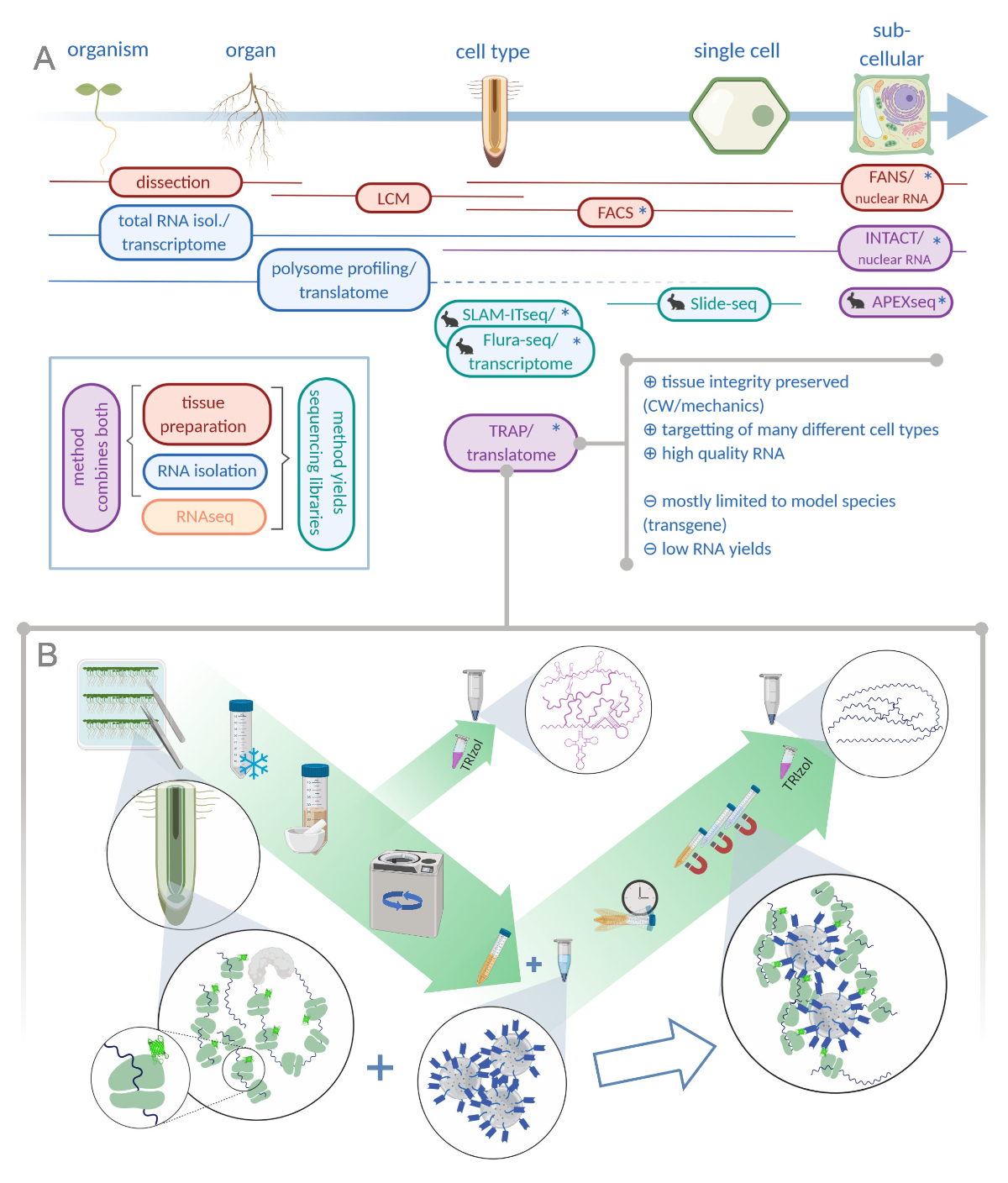
Figura 1: La traducción de la purificación de afinidad ribosoma (TRAP) complementa la cartera de análisis "omics". A. El aumento de los niveles de precisión analítica, hasta una sola célula o incluso la resolución subcelular se puede lograr mediante una plétora de métodos o combinaciones de los mismos. El esquema ofrece una visión general de las herramientas disponibles actualmente en el campo vegetal y animal. La recolección de tejidos a resolución celular se puede lograr mediante protocolos como LCM o FACS, que luego se acoplan al transcriptoma estándar o al análisis de perfiles/traducción de polisoma. TRAP e INTACT integran la captura de tejido y el aislamiento de ARN, ya que se basan en el etiquetado de epítopos. Sin embargo, INTACT sólo toma muestras de núcleos celulares y constituye, por lo tanto, un caso especial de análisis de transcriptoma. Un pequeño icono de conejo marca los métodos recientemente desarrollados en el campo animal: Mientras que SLAM-ITseq y Flura-seq se basan en el objetivo metabólico de ARN nacientes con bases de uracilo modificadas en células que expresan la enzima permisiva, Slide-seq hace uso de un portaobjetos de vidrio recubierto con códigos de barras de ADN que proporcionan información posicional en el rango celular. Se sigue un enfoque de etiquetado de proximidad en APEX-seq para muestrear ARN en compartimentos subcelulares específicos. En particular, el aumento de la resolución a menudo requiere la generación de material transgénico (asteriscos) y, por lo tanto, estos métodos se utilizan predominantemente para las especies modelo. TRAP es especialmente adecuado para estudios de ciencias vegetales que involucran pared celular (CW) o señalización mecánica, así como especies celulares que son difíciles de liberar de su matriz CW. B. Pasos detallados de laboratorio húmedo del procedimiento TRAP: Las plántulas que expresan proteína ribosomal etiquetada con GFP en distintos tipos de células (por ejemplo, endodermis de raíz) se cultivan en platos De Petri durante siete días y el material radicular cosechado por congelación rápida. Se recoge una muestra de control total de ARN del extracto crudo homogeneizado antes de peletizar los escombros a través de la centrifugación. Las perlas magnéticas anti-GFP se añaden al extracto transparente para realizar la inmunoprecipitación. Después de la incubación y tres pasos de lavado, el ARN asociado al polisoma (ARN TRAP/polisoma) se obtiene directamente a través de la extracción de fenol-cloroformo. LCM: microdisección de captura láser, FACS/FANS: clasificación celular/nuclear activada por fluorescencia, APEX-seq: método basado en ascorbación peroxidasa diseñada, INTACT: aislamiento de núcleos etiquetados en tipos de células específicas, alquilación vinculada a SLAM-ITseq: thiol(SH) para la secuenciación metabólica de ARN en tejido, Flura-seq: secuenciación de ARN con etiqueta de fluorouracilo (Creado con Biorender.com) Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
El objetivo de este artículo es proporcionar una descripción detallada del método TRAP, resaltar los pasos críticos y proporcionar orientación para un posible método de preparación de biblioteca.
Un experimento trap genérico consistirá esencialmente en los siguientes pasos (véase también la figura 1B):(1) Preparación de material vegetal, incluida la clonación de la construcción de etiquetado de ribosoma, producción y selección de líneas transgénicas, cultivo y abultamiento de semillas, esterilización y chapado, y aplicación/tratamiento de tensión (opcional) y recolección de tejidos; 2) inmunopurificación, incluida la homogeneización tisular y la limpieza del extracto crudo, el lavado de cuentas y la inmunopurificación, y las etapas de lavado; 3) extracción de ARN y evaluación de la calidad; y (4) la preparación de la biblioteca.
La raíz de Arabidopsis ha sido un sistema modelo para estudiar el desarrollo de la planta desde su introducción como planta modelo37,,38. Aquí, la aplicación de TRAP se muestra en el contexto del desarrollo de la raíz lateral de la planta. En las plantas, la acumulación de todo el sistema radicular se basa en la ejecución de este programa y por lo tanto es muy importante para la supervivencia del organismo39. En Arabidopsis,las raíces laterales se originan a partir de tejido periciclo que reside junto a vasos xilemas y, por lo tanto, se denomina periciclo de polo xilem (XPP; véase la figura 2C)40. Algunas células XPP, que se encuentran en lo profundo de la raíz, adquieren una identidad celular fundadora y, tras un desencadenante hormonal local, comienzan a proliferar por hinchazón y división anticlinalmente41. Sin embargo, debido a la presencia de una matriz de pared celular rígida, este proceso ejerce tensión mecánica en los tejidos circundantes. En particular, la endodermis superpuesta se ve afectada, ya que se encuentra en el camino del eje de crecimiento de la raíz lateral42,,43,44. De hecho, el primordium recién formado tendrá que crecer a través de la célula de endodermis superpuesta(Figura 2C2),mientras que las células de corteza y epidermis simplemente se apartan para que el primordium finalmente surja45,46. El trabajo reciente en nuestro laboratorio ha demostrado que la endodermis está contribuyendo activamente a acomodar la proliferación en el periciclo. El bloqueo dirigido de la señalización hormonal endodérmica es suficiente para inhibir incluso la primera división en las células XPP47. Por lo tanto, la comunicación periciclo-endodermis constituye un punto de control muy temprano para el desarrollo de la raíz lateral en Arabidopsis. Sin embargo, no se sabe cómo se realiza esta charla cruzada. Para desentrañar este misterio, elegimos el enfoque TRAP-seq para apuntar a las células XPP y endodeérmicas. Para enriquecer las células en el programa de raíz lateral, imitamos el desencadenante hormonal aplicando exógenamente un análogo de auxina (ácido 1-naftalencia, NAA)48,que al mismo tiempo permitió resolver temporalmente la fase inicial de la formación de la raíz lateral.
Protocolo
1. Clonación de transgénicos, producción de líneas transgénicas y selección
- Clonar el promotor de elección en el vector de entrada adecuado. Utilice un método de clonación basado en recombinación (Tabla de materiales) y recombine los promotores en pDONRP4-P1r. Clonar RPL18 (con etiqueta de afinidad o proteína fluorescente de elección) utilizando clonación basada en recombinación en pDONRP1-P249.
- Combine el vector de entrada que contiene RPL18 con el vector de entrada que contiene el promotor en una reacción de recombinación de dos fragmentos en el vector de destino adecuado con el cassette de selección FAST-rojo50 para facilitar la selección directa de semillas transgénicas.
- Verifique el vector recombinado secuenciando y transformarlo en agrobacterias adecuadas y competentes. Sumergir las plantas de Arabidopsis y después de 3-4 semanas de cosecha y seleccionar semillas T151.
- Utilice la microscopía para identificar líneas bien que expresan y verificar los patrones de expresión de acuerdo con la actividad del promotor reportada en varias líneas independientes. Seleccione líneas que muestren un patrón de expresión representativo con una sola inserción de ADN en T. Esto podría ayudar a minimizar el silenciamiento y será ventajoso para los cruces genéticos.
- Seleccione la descendencia T3 que sea homocigota para el gen marcador.
2. Propagación y esterilización
- TRAP específico del tipo de celda aísla el ARN de un número limitado de células diana por raíz. Para generar el material de partida necesario, propague líneas homocigotas. Con este fin, utilice condiciones de crecimiento estándar con un enfoque especial en el control del crecimiento de hongos.
NOTA: Si no se pueden obtener líneas de inserción individuales, aumente los lotes en grandes poblaciones a lo largo de pocas generaciones para evitar el silenciamiento transgeneracional inducido por El ADN T. - Esterilice grandes cantidades de semillas de Arabidopsis con una ronda de gas cloro y una ronda de 70% EtOH.
- Esparza las semillas uniformemente en platos Petri cuadrados de 12 cm x 12 cm (menos de 0,3 ml de semillas/placa) y apiló las en un desecador u otro recipiente adecuado. Evite la formación de amos o montones, ya que las semillas deben ser accesibles al gas. Realizar la esterilización de gas durante la noche con los volúmenes de lejía y HCl según se informó52: 100 ml de lejía (13%) con 6 ml de conc. HCl en un desecador de 60 L. Desfumigar durante al menos 1 h antes de recoger las semillas en un recipiente estéril.
ADVERTENCIA: 37% HCl es altamente corrosivo y requiere un manejo cuidadoso. El gas cloro es tóxico, usa una campana de humos. - Tomar 0,1 ml de semillas secas esterilizadas con gas por placa y mezclarlas con solución de esterilización (70% EtOH, 0,01% Tween) a temperatura ambiente. Incubar durante 20 min, decantar EtOH y lavar las semillas 3-4 veces con estéril H2O.
- Transfiera las semillas empapadas en tubos de 50 ml y diluya con agar estéril 0,1% para obtener 1 ml de lodo de semillas imbibe por placa (0,1 ml de semilla/1 ml de lodo).
NOTA: Debido a eventos de integración transgénica, las líneas de plantas pueden ser susceptibles a diferentes técnicas de esterilización; especialmente el tiempo de incubación de EtOH fue crítico. En nuestras manos, los pasos de esterilización dual eran necesarios para evitar la contaminación por hongos durante los experimentos. Esto es especialmente importante cuando se realizan series temporales, ya que la contaminación de un único punto de tiempo dificulta todo el experimento. Bien podría ser que la esterilización dual no siempre es necesaria, dependiendo de las condiciones de cultivo locales.
- Esparza las semillas uniformemente en platos Petri cuadrados de 12 cm x 12 cm (menos de 0,3 ml de semillas/placa) y apiló las en un desecador u otro recipiente adecuado. Evite la formación de amos o montones, ya que las semillas deben ser accesibles al gas. Realizar la esterilización de gas durante la noche con los volúmenes de lejía y HCl según se informó52: 100 ml de lejía (13%) con 6 ml de conc. HCl en un desecador de 60 L. Desfumigar durante al menos 1 h antes de recoger las semillas en un recipiente estéril.
3. Placado
- Prepare estos pasos con antelación. Vierta 1/2 Placas MS (pH 5.8) con 1% de agar en las cantidades necesarias para el experimento (20-30 por muestra/punto de tiempo). Corte las puntas de la pipeta de 1 ml para agrandar el diámetro de la punta a unos 3-4 mm con una cuchilla de afeitar. Autoclave las puntas. Cree un soporte de plantilla para enchapar tres filas de semillas por plato con tapas cuadradas de plato Petri. Prepare una campana de flujo laminar para proporcionar un ambiente de trabajo estéril y etiquete las placas a procesar.
NOTA: Si se procesan muchas placas al mismo tiempo, las etiquetas de color pueden acelerar el etiquetado. - Coloque las placas de agar vacías en el soporte de la plantilla y distribuya 1 ml de semillas impregnadas uniformemente en tres filas. Coloque las placas procesadas en pilas en el flujo laminar hasta que las semillas estén secas (es decir, se adhieran a la superficie del agar). No deje las placas más tiempo, ya que el agar también se secará.
- Una vez que las semillas estén suficientemente secas, cierre las tapas y selle cada placa con cinta adhesiva de microporo. Estratificar las semillas durante dos días a 4oC en la oscuridad y luego colocarlas en una cámara de crecimiento.
4. Tratamiento de tejidos (opcional)
NOTA: En este protocolo, delineamos el tratamiento exógeno de las raíces de Arabidopsis con la variante de auxina sintética NAA. Dependiendo de la pregunta experimental en cuestión, esta parte necesita ser ajustada o puede ser omitida por completo.
- Preparar tiras de papel tisú de 1,5 - 2 cm de altura y 10 cm de longitud. Los tiempos de incubación prolongados requieren que el tejido sea autoclavedo antes de su uso.
- Retire la cinta de microporos de todas las placas que tienen que someterse al tratamiento hormonal. Diluir 1 ml de NAA de 10 mM (disuelto en DMSO) en 1 L de solución líquida, autoclave 1/2 MS (pH 5.8) y remojar el papel tisú en la solución (10 M NAA).
- Use pinzas para aplicar una tira de papel tisú en cada fila de raíces. Utilice suavemente los dedos para eliminar las burbujas de aire. Vacíe el exceso de líquido de la placa, cierre la tapa y etiquete la placa con el tiempo. Para tiempos de incubación prolongados, vuelva a colocar las placas en la cámara de crecimiento.
5. Cosecha
- Recuperar placas para cada réplica biológica/ punto de tiempo / tratamiento. Recoja nitrógeno líquido en un recipiente limpio de Dewar y tubos de etiquetas (15 o 50 ml) para las diferentes muestras de tejido. Prepare un soporte de espuma de poliestireno.
ADVERTENCIA: Familiarícese con los procedimientos de manipulación de nitrógeno líquido (aireación, congelación, tubos potencialmente explosivos). - Abra la placa y retire el papel tisú con fórceps, teniendo cuidado de no separar las raíces de la superficie del agar. Con una cuchilla quirúrgica, corte una vez por fila a lo largo de la unión de raíz de brote en un solo trazo determinado. Limpie las cuchillas entre las muestras e intercambie con frecuencia para garantizar la nitidez.
- Con las pinzas, desliza el dedo a lo largo de las raíces de cada fila para recogerlas en tres paquetes. Agarre las raíces y vacíelas en un tubo de 50 ml lleno de nitrógeno líquido para congelarlas.
NOTA: No intente ensamblar las raíces en estructuras densas (como bolas) ya que son difíciles de moler en el siguiente paso. - Proceder con todas las placas que constituyen una muestra (en el orden de los tiempos de incubación) y verter el exceso de nitrógeno líquido. Utilice la tapa del tubo para evitar que las raíces se derramen. A continuación, cierre la tapa y recoger todos los tubos en el recipiente Dewar. Almacene el tejido radicular a -80 oC.
6. Inmunopurificación
NOTA: Este paso tiene como objetivo obtener ARN TRAP/polisoma de alta calidad. Por lo tanto, siga estrictamente los consejos de buenas prácticas para el manejo del ARN. Realice todos los pasos de esta sección en un banco estéril y limpie todos los equipos y utensilios de laboratorio con una solución de eliminación de RNase(Tabla de materiales). Use guantes y cámbielos inmediatamente cuando estén contaminados con muestras, hielo u otras fuentes que no se hayan limpiado. Dado que este es un aspecto muy crucial, se incluye una sección sobre la reutilización de equipos junto con consejos de eliminación de residuos.
- Preparación del búfer
- Prepare las soluciones de stock de acuerdo con la Tabla 1 y el autoclave (A) o el esterilizado por filtros. A menos que se especifique lo contrario, el disolvente es agua libre de RNase.
- Disolver y alícuota ditiothreitol (TDT), fluoruro de fenimetilsulfonil (PMSF), cicloheximida (CHX) y cloramphenicol (CAM) en sus respectivos disolventes, tal como se indica en la Tabla 1, y almacenarlos a -20oC. Todas las demás existencias pueden permanecer a temperatura ambiente.
- Premezcle las existencias - con ingredientes 1-4 para tampón de lavado (WB) y 1-6 para tampón de extracción de polisoma (PEB) - para evitar la mezcla de tampón que consume mucho tiempo antes de cada extracción. Por lo tanto, sólo añadir agua y los ingredientes congelados (7-10) el día de la extracción. Mantener las existencias premezcladas y el agua libre de RNase a 4oC.
NOTA: La concentración de TDT es 1/5 de la concentración notificada de Zanetti et al. 2005, ya que la interacción de nanobody con el GFP es sensible a las altas concentraciones de TDT.
| Ingredientes | Concentración de stock | Añadir volumen en ml para 50 ml de WB* | Añadir volumen en ml para 50 ml de PEB* | ||
| 1 | Tris, pH 9 | Un | 2 M | 5 | 5 |
| 2 | Kcl | Un | 2 M | 5 | 5 |
| 3 | Egta | Un | 0.5m | 2.5 | 2.5 |
| 4 | MgCl2 | Un | 1 M | 1.75 | 1.75 |
| 5 | Pte | Un | 20% (v/v) | 0 | 2.5 |
| 6 | mezcla de detergente | Un | 0 | 2.5 | |
| Tween 20 | 20% (v/v) | ||||
| Tritón-X 100 | 20% (v/v) | ||||
| Brij-35 | 20% (p/v) | ||||
| Igepal | 20% (v/v) | ||||
| 7 | Tdt | ₳ | 0.5m | 0.1 | 0.1 |
| 8 | Pmsf | ₳ | 0,1 M (isopropanol) | 0.5 | 0.5 |
| 9 | Cicloheximida | ₳ | 25 mg/ml (EtOH) | 0.1 | 0.1 |
| 10 | Cloranfenicol | ₳ | 50 mg/ml (EtOH) | 0.05 | 0.05 |
Tabla 1: Asesoramiento sobre la composición del búfer y la mezcla. Los ingredientes con las concentraciones de stock dadas mezcladas en las cantidades dadas producen 50 ml de WB o PEB. Tris: tris-(hidroximetil)-aminometano, EGTA: etilenglicol-bis(éter de aminoetilo)-N,N,N',N'-ácido tetraacético, PTE: Polioxietileno-(10)-tridecyl éter, A: autoclave, . *Llenar hasta 50 ml con agua libre de RNase.
- Homogeneización/molienda de tejidos
- Enfríe la centrífuga y coloque homogeneizadores y tubos de centrífuga en hielo. Descongelar alícuotas de TDT, PMSF, CHX y CAM. Mezclar PEB y WB de las soluciones de stock en tubos de 50 ml de acuerdo con los requisitos del día (a de muestras) y enfriar sobre hielo.
NOTA: Añadir PMSF sólo poco antes de su uso, ya que la vida media de PMSF en agua es de sólo 30 minutos. - Preparar mucho nitrógeno líquido en un vaso de Dewar y recuperar muestras de tejido de un almacenamiento de -80 oC. Use guantes de algodón debajo de los guantes de laboratorio estándar para evitar quemaduras por morteros fríos. Vierta nitrógeno líquido en morteros y plagas hasta que estén lo suficientemente fríos como para permitir la molienda. Se recomienda idear un sistema para distinguir morteros (etiquetar o mantener en un cierto orden).
- Vacíe la muestra de tejido en un mortero y muele cuidadosamente hasta que todo el material sea un polvo blanco. Si es necesario, agregue nitrógeno líquido para mantener el tejido congelado o para facilitar una mejor molienda.
- Agregue 5 ml de PEB a la muestra y mezcle rápidamente con el polvo antes de que el búfer se congele. Mientras que esta muestra se descongela (mezcla de vez en cuando) procesar otra muestra.
- Tan pronto como la mezcla pueda ser transferida, vacíe la suspensión en un homogeneizador de vidrio y manténgala en hielo. Con 2 ml adicionales de PEB, enjuague el mortero y el pestillo y agréguelo a la muestra en el homogeneizador.
NOTA: Evite una muestra completamente líquida, ya que esto permite la degradación del ARN. - Moler la suspensión manualmente hasta que el extracto sea homogéneo. Recomendamos un mínimo de 4 a 5 sueldos.
NOTA: Puede requerir un poco de tiempo de espera adicional para permitir que la suspensión se descontezcan aún más. El manejo de homogeneizadores requiere cierta diligencia. No aplique fuerza bruta y tenga cuidado con las fuerzas de succión. Si no se tiene en cuenta, esto conducirá a derrames, contaminación o destrucción del homogeneizador. - Vierta el extracto de raíz bruta en un tubo de centrífuga de 50 ml (mantener en hielo).
NOTA: Por lo general, varias muestras se pueden moler antes de la transferencia. Se requiere un manejo paralelo de molienda, transferencia y homogeneización. Trate de trabajar rápidamente, pero no se apresure; mantener la calma. Mantenga siempre las muestras homogeneizadas en hielo.
- Enfríe la centrífuga y coloque homogeneizadores y tubos de centrífuga en hielo. Descongelar alícuotas de TDT, PMSF, CHX y CAM. Mezclar PEB y WB de las soluciones de stock en tubos de 50 ml de acuerdo con los requisitos del día (a de muestras) y enfriar sobre hielo.
- Colección total de muestras de ARN
- Transfiera las alícuotas de 200 l de cada muestra bruta a un tubo de microcentrífuga limpio (etiquetado y enfriado en hielo de antemano).
- Proceda con la extracción de ARN como se detalla para las muestras de TRAP en los puntos 7.1 y 7.2. Realice estos pasos mientras las muestras se limpian en la centrífuga.
- Realizar un tratamiento DNase con el ARN total resuspendido para eliminar la contaminación del ADN y limpiar la reacción utilizando un kit comercial(Tabla de materiales).
NOTA: Las extracciones totales de ARN generalmente producen altas concentraciones y las muestras deben diluirse considerablemente. Recomendamos medir la concentración después de la dilución por el protocolo sensible Qubit.
- Borrar el extracto crudo
- Tome el cubo de hielo con muestras de 6.2.7 y centrifugarlas durante 15 minutos a 16.000 x g y 4oC.
NOTA: Para equilibrar la centrífuga, empareje las muestras en consecuencia. En caso de que esto no sea del todo posible, ajuste una muestra añadiendo PEB. - Vierta el sobrenadante en un tubo de centrífuga fresco (enfriado en hielo de antemano) y repita la centrifugación (15 min a 16.000 x g y 4 oC). Esta transferencia se puede realizar rápidamente junto a la centrífuga.
- Mientras el extracto crudo se está limpiando, inicie el lavado de perlas GFP para el paso 6.6.
NOTA: Guarde este cubo de hielo para balancearse en la coctelera, pero no vuelva a colocarlo en el banco estéril, ya que podría estar contaminado.
- Tome el cubo de hielo con muestras de 6.2.7 y centrifugarlas durante 15 minutos a 16.000 x g y 4oC.
- Lavado de cuentas
- Perlas magnéticas GFP de Aliquot (#samples x 60 l, mesa de materiales)en un tubo de 1,5 ml. Colocar en el soporte magnético. Una vez que las cuentas hayan recogido, retire el sobrenadante.
- Añadir 1 ml de WB frío, resuspender las cuentas y recogerlas de nuevo. Deseche el tampón de lavado y repita una vez más con 1 ml de WB.
- En última instancia, resuspenda las perlas en WB al volumen inicial utilizado en el paso 6.5.1.
- Inmunopurificación (IP)
- Inmediatamente después de la centrifugación, vierta el sobrenadante despejado en tubos etiquetados de 15 ml y agregue 60 ml de perlas lavadas por muestra.
- Coloque todas las muestras horizontalmente en el cubo de hielo y colóquelas en una coctelera. Dejar que la mezcla se incuba durante 2 h para unir los polisomas etiquetados con GFP a las perlas.
- Recoger las perlas en el soporte magnético para tubos de 15 ml (sobre hielo) y añadir PMSF al PEB restante. Descarta el sobrenadante. Vierta aproximadamente 5 ml de PEB en las perlas y resuspenderlas inclinando. Agitar las muestras durante 15 minutos en la misma configuración que en la sección 6.6.2.
- Repita los lavados con WB a un total de 3 lavados (1 x PEB, 2 x WB). Antes de cada intercambio de búfer, agregue PMSF.
- Recoger las perlas en 1 ml de WB y transferirlas a un tubo de 1,5 ml. Finalmente, recoja las perlas una vez más en el soporte magnético y retire todo el líquido. Cierre el tubo y manténgalo en hielo hasta que se procesen todas las muestras.
- Transporte las muestras a una campana de humos para la extracción de ARN.
- Eliminación y reacondicionamiento de residuos de suministros de laboratorio.
- Si se realiza de acuerdo con las buenas prácticas de laboratorio (ver sección 2.2.1), el procedimiento de esterilización produce una solución acuosa de NaCl. Deje el gas de cloro, así como el HCl residual y la lejía, para desfumigar en la campana de humos.
- Eliminación de PEB y WB: A medida que CHX se descompone a un pH alto, recoger todos los líquidos y llevar al pH>9. Deseche los residuos líquidos en los residuos químicos halogenados. Todos los sólidos (tejidos, pipetas serológicas, guantes, etc.) deben eliminarse como residuos químicos.
- Recoger líquidos que contienen fenol por separado, así como material contaminado con fenol (puntas, tubos y guantes).
- Lavar a mano morteros, plagas y homogeneizadores (esponjas y cepillos) con jabón y enjuagar bien. Posteriormente, hornee el material a >220 oC durante la noche. Envuelva en papel de aluminio antes del tratamiento o colóquelo en un recipiente cubierto a prueba de calor.
- Cepille los tubos de centrífuga limpios con detergente y luego el troto de dietalpirocarbonato (DEPC) en la campana de humos. Para ello, añadir DEPC líquido al agua desionizada (1 ml de DEPC a 1 L de H2O) y mezclar mediante el temblor. Coloque los tubos centrífugos en una bandeja autoclavable que capture el agua DEPC derramada. Vierta la suspensión en los tubos y déjela durante 3 h o durante la noche. DEPC se descompone en el proceso de autoclave posterior.
ADVERTENCIA: DEPC es altamente tóxico.
7. Extracción de ARN y QC
- Extracción de ARN
- Enfríe la centrífuga de la mesa a 4 oC.
- Añadir 1 ml de reactivo a base de ácido-guanidinio-fenol(Tabla de Materiales)a cada muestra, invertir para resuspender las cuentas o la suspensión total de ARN e incubar durante 5 minutos sobre hielo. ¡No vórtice!
- Añadir 200 l de cloroformo e incubar durante 3 minutos sobre hielo. A continuación, vórtice a fondo las muestras.
- Para ayudar a la separación de fases, centrífuga a máx. velocidad de 10-15 min, 4oC.
- Etiquetar tubos de baja retención de 1,5 ml(Tabla de materiales)y alícuota de 650 ml de isopropanol en cada uno.
- Tomar cuidadosamente la fase acuosa superior (ca. 650 oL) y transferir a los tubos preparados con isopropanol. Evite tocar la fase orgánica rosa.
- Precipitar el ARN durante la noche a -20 oC.
NOTA: Se recomienda almacenar las muestras en isopropanol a -20 oC o -80 oC y solo solubilizar en agua cuando sea necesario. El ARN acuoso se degrada incluso a -80 oC cuando se almacena durante semanas/meses.
- Precipitación de ARN
- Enfríe la centrífuga de la mesa a 4 oC.
- Preparar fresco 80% EtOH con agua libre de RNase y enfriar a -20 oC (5 min a -80 oC ayuda a acelerar el proceso).
- Centrifugar las muestras a máxima velocidad (ca. 13.000 x g)durante 30 min y desechar el sobrenadante. El pellet no será visible, tan cuidadosamente pipetear como si estuviera allí. Añadir 1 ml de frío 80% EtOH e invertir el tubo una o dos veces.
- Centrifugar de nuevo durante 30 minutos a la velocidad máxima y repetir el lavado a un total de dos lavados.
- Gire hacia abajo durante 2 minutos y retire todo EtOH residual con una punta de 10 l. Deje que el pellet se seque durante 3-5 minutos (no más) a temperatura ambiente y resuspenda en agua libre de RNase de 20 ol.
- Mantenga las muestras en hielo y realice un control de calidad lo antes posible. Proceda a almacenar las muestras a -80 oC. Evite los ciclos de congelación-descongelación.
- Control de calidad utilizando equipos dedicados(Tabla de Materiales)de acuerdo con las recomendaciones del fabricante.
8. Preparación de la biblioteca
- síntesis y amplificación de ADNc con el kit de ARN de entrada ultrabaja SMARTer v4
- Calcular la dilución de cada muestra para tener 1,5 ng de ARN-TRAMPA o ARN total en un volumen de 4,75 l. Realizar todas las reacciones en tubos PCR y diluir muestras con alícuotas frescas de agua libre de RNase.
- Realice todos los pasos de acuerdo con las recomendaciones del fabricante con 1/2 los volúmenes de reacción. Amplifique el ADNc con 12-13 ciclos de PCR.
- Limpie el PCR añadiendo 0,5 l de tampón de lisis 10x y 25 l de perlas SPRI(Tabla de materiales). Si se procesan muchas muestras, el búfer de lisis y los perlas se pueden mezclar previamente. Asegúrese de que las perlas estén uniformemente dispersas antes de pipetear.
- Continúe con el protocolo en volúmenes de reacción completos (17 l de búfer de elución). No deje que las cuentas se sequen durante más de 3 minutos. Las muestras sobrescadas pueden ser rescatadas potencialmente por tiempos prolongados de incubación.
- Mida las concentraciones de muestra con el kit de ADN Qubit HS.
NOTA: El kit SMARTer v4 puede tolerar hasta 200 pg de entrada. Obtuvimos bibliotecas en casos donde no se pudieron determinar valores de Qubit (por debajo de 250 pg, límite de detección) con un PCR de 16 ciclos. Sin embargo, el material de entrada limitado también puede producir bibliotecas menos complejas.
- Fragmentación y ligadura de adaptadores PCR con el kit de preparación de la biblioteca de ADN Nextera XT
- Diluir el ADNc con agua libre de RNase para obtener una concentración de 200 pg/l y pipeta de 1,25 l en un tubo PCR.
- Realice todos los pasos según el fabricante con 1/4 de los volúmenes de reacción. Amplifique el ADNc con 12 ciclos de PCR y adaptadores compatibles para las muestras que pertenecen a una agrupación de secuenciación. Con los kits de índice A y D de Illumina se pueden multiplexar hasta 384 muestras.
- Para la limpieza de PCR, agregue 12,5 l de tampón de resuspensión y 22,5 l de perlas SPRI (relación de 0,9x). Elule la muestra con 22 ml de tampón de elución.
NOTA: El control de calidad y la agrupación fueron realizados por la empresa de secuenciación(Tabla de materiales)y, por lo tanto, no se necesitaba una normalización basada en cuentas. La reacción de fragmentación enzimática (tagmentation) es muy sensible a la entrada de material, ya que cada enzima solo corta una vez. Por lo tanto, no exceda la recomendación de concentración.
Resultados
Para la evaluación de la calidad, el procedimiento antes mencionado debe sondearse en varios pasos intermedios: validación del patrón de expresión en planta, control de calidad del ARN polisómico aislado, así como de las bibliotecas finales. QRT-PCR utilizando genes marcadores conocidos se puede, además, realizar para confirmar la respuesta a la condición de tratamiento o para afinar las condiciones experimentales.
Discusión
Verificación del patrón de localización RPL18
El patrón de expresión adecuado de la subunidad ribosomal etiquetada es fundamental para evitar la interpretación errónea de los datos de cualquier experimento TRAP. Por lo tanto, la incorporación de GFP como etiqueta de epítope a RPL18 permite muy elegantemente la verificación del patrón de expresión deseado y, consecutivamente, la extracción de la fracción de polisoma del mismo tejido. Los enfoques más invasivos para asegurar patrones de p...
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Nos gustaría dar las gracias a Jean-Claude Walser, del Centro de Diversidad Genética de Zúrich, por su asesoramiento experto crucial en la fase inicial de este proyecto. El trabajo en el laboratorio de Vermeer fue apoyado por una beca de cátedra SNF (PP00P3_157524) y una subvención de equipo R'EQUIP (316030_164086) de la Fundación Nacional Suiza de Ciencias (SNSF) otorgada a JEMV.
Materiales
| Name | Company | Catalog Number | Comments |
| Sterilization | |||
| bleach, 13% | Sigma | 71696 | |
| beaker | VWR | 214-1172/74/75 | |
| desiccator with porcelaine plate (DURAN) | Sigma/Merck | Z317454-1EA/Z317594-1EA | |
| EtOH, p.a. | Honeywell | 02860-1L | |
| HCl, 37% | Roth | 4625.1 | |
| Tween 20 | Sigma | P9416 | |
| Plate growth + harvesting | |||
| MS salts, basal salt mixture, incl. MES buffer | Duchefa | M0254 | |
| agar plant for cell culture | Applichem/Panreac | A2111.1000 | |
| DMSO | Sigma | D4540 | |
| forcepts | Rubis Switzerland | 5-SA model | |
| KOH | Fluka | 60370 | |
| micropore/surgical tape | 3M | 1530-0 | |
| NAA | Duchefa | N0903 | |
| petri dishes 120x120 mm | Greiner bio-one | 688102 | |
| scalpel | VWR/Swann-Morton | 233-5454 | |
| tissues, neutral, two-layered | any supplier of your choice | ||
| Immunoprecipitation | |||
| GFP-beads: gtma-100 GFP-Trap_MA | Chromotek | e.g. gtma-100 | |
| Brij-35 | Sigma | P1254-500G | |
| centrifuge tubes (in accordance with centrifuge) | Beckman Coulter | 357001 | |
| Chloramphenicol | Applichem | C0378-25G | |
| cotton gloves | VWR | 113-7355 | |
| Cycloheximide, HPLC grade | Sigma | 01810-1G | |
| DEPC | VWR | E174 | might have long delivery times |
| DTT | Fluka | 43815 | |
| EGTA | Sigma | 3054.3 | |
| homogenizers DUALL 23 | KONTES GLASS CO (via VWR) | SCERSP885450-0023 (set) | SCERSP885451-0023 pestle only - SCERSP885452-0023 cylinder only; long delivery times |
| Igepal CA-360 | Sigma | I3021-100ml | |
| KCl | Sigma | 60130 | |
| MgCl2 hexahydrat | Roth | 2189.2 | |
| mortar and pestle | VWR | 470148-960 & 470019-978 | |
| PMSF | Roche | 10 837 091 001 | |
| Polyoxyethylene-(10)-tridecylether/PTE | Sigma | P2393-500G | |
| RNase-free water | Roth | T143.3 | |
| RNAZap | Thermo Fisher | AM9780/AM9782 | for cleaning surfaces |
| Tris, >99.3% | Roth | AE15.3 | |
| Triton X-100 | Fluka | T8787-250ml | |
| Tween 20 | Sigma | P9416-100ml | |
| RNA extraction | |||
| 2-Propanol, p.a. | Sigma | 33539-1L-GL-R | |
| Chloroform, HPLC grade | Scharlau | CL02181000 | |
| EtOH, p.a. | Honeywell | 02860-1L | |
| low-retention microcentrifuge tubes, 1.5 ml | Eppendorf/Sigma | Z666548-250EA | LoBind |
| RNase-free DNase set | Qiagen | 79254 | |
| RNeasy MiniElute Cleanup Kit | Qiagen | 74204 | |
| TRIzol reagent | ThermoFisher/Ambion | 15596018 | |
| Library preparation | |||
| 15/50 mL Tube Magnetic Separator | Abraxis | PN 472250 | |
| AMPure beads | Beckman Coulter | A63881 | |
| Index Kit A | Illumina | FC-131-2001 | |
| Index Kit D | Illumina | FC-131-2004 | |
| neodymium magnets | Amazon/other | 6 x 1.5 mm range: N42 (NdFeB) | |
| Nextera XT kit | Illumina | FC-131-1024/1096 | https://emea.support.illumina.com/ |
| PCR strips | ThermoScientific | AB-0266 | |
| SMARTer v4 kit | Takara Bioscience | 634892 | https://www.takarabio.com/ |
| Bioanalyzer | Agilent | 2100 Bioanalyzer Instrument | specialized equipment for RNA/DNA quality control |
| Tapestation | Agilent | 4200 Tapestation Instrument | specialized equipment for RNA/DNA quality control |
| Fragment Analyzer | Agilent | 5400 Fragment Analyzer System | specialized equipment for RNA/DNA quality control (high throughput) |
| LabChip | PerkinElmer | LabChip GX Touch Nucleic Acid Analyzer | specialized equipment for RNA/DNA quality control (high throughput) |
| Qubit 4 Fluorometer | ThermoFisher | Q33239 | specialized equipment for RNA/DNA concentration determination |
| qRT-PCR | |||
| GATA23 | Microsynth | fwd: AGTGAGAATGAA AGAAGAGAAGGG; rev: GTGGCTGCGAAT AATATGAATACC | |
| GH3.3 | Microsynth | fwd: CAAACCAATCCT CCAAATGAC; rev: ACTTATCCGCAA CCCGACT | |
| LBD29 | Microsynth | fwd: TCTCCAACAACA GGTTGTGAAT; rev: AAGGAGCCTTAG TAGTGTCTCCA | |
| UBC21 | Microsynth | fwd: TGCGACTCAGGG AATCTTCT; rev: TCATCCTTTCTT AGGCATAGCG | |
| SsoAdvanced Universal SYBR Green | Bio-Rad | #172-5270 | |
| iScript Adv cDNA Kit | Bio-Rad | #172-5038 | |
| miscellaneous | |||
| Falcon tubes 15 ml, Cellstar | Greiner bio-one | 188261 | |
| Falcon tubes 50 ml, Cellstar | Greiner bio-one | 210261 | |
| filter tips 1 ml | Axygen | TF-1000-R-S | |
| filter tips 10 µl | Axygen | TF-10-R-S | |
| filter tips 100 µl | Axygen | TF-100-R-S | |
| filter tips 20 µl | Axygen | TF-20-R-S | |
| filter tips 200 µl | Axygen | TF-200-R-S | |
| microcentrifuge tubes 1.5 ml | SARSTEDT | 72.690.001 | |
| Propidium iodide | Sigma | P4170-100MG | |
| sequencing company | Novogene | en.novogene.com |
Referencias
- Van Verk, M. C., Hickman, R., Corné, M. J., Pieterse, M., Van Wees, S. C. RNA-Seq: Revelation Of The Messengers. Trends In Plant Science. 18 (4), 175-179 (2013).
- Libault, M., Pingault, L., Zogli, P., Schiefelbein, J. Plant Systems Biology At The Single-Cell Level. Trends In Plant Science. 22 (11), 949-960 (2017).
- Mustroph, A., et al. Profiling Translatomes Of Discrete Cell Populations Resolves Altered Cellular Priorities During Hypoxia In Arabidopsis. Proceedings Of The National Academy Of Sciences Of The United States Of America. 106 (44), 18843-18848 (2009).
- Karve, R., Iyer-Pascuzzi, A. S. Digging Deeper: High-Resolution Genome-Scale Data Yields New Insights Into Root Biology. Current Opinion In Plant Biology. 24, 24-30 (2015).
- Warner, J. R., Knopf, P. M., Rich, A. A Multiple Ribosomal Structure In Protein Synthesis. Proceedings of The National Academy of Sciences of The United States of America. 49 (1), 122-129 (1963).
- Gautam, V., Sarkar, A. K. Laser Assisted Microdissection, An Efficient Technique To Understand Tissue Specific Gene Expression Patterns And Functional Genomics In Plants. Molecular Biotechnology. 57 (4), 299-308 (2015).
- Bargmann, B. O. R., Birnbaum, K. D. Fluorescence Activated Cell Sorting Of Plant Protoplasts. Journal of Visualized Experiments. (36), e1673 (2010).
- Deal, R. B., Henikoff, S. The Intact Method For Cell Type-Specific Gene Expression And Chromatin Profiling In Arabidopsis Thaliana. Nature Protocols. 6 (1), 56-68 (2011).
- Dougherty, J. D. The Expanding Toolkit Of Translating Ribosome Affinity Purification. The Journal of Neuroscience: The Official Journal Of The Society For Neuroscience. 37 (50), 12079-12087 (2017).
- Mustroph, A., Juntawong, P., Bailey-Serres, J. Isolation Of Plant Polysomal mRNA By Differential Centrifugation And Ribosome Immunopurification Methods. Methods in Molecular Biology. 553, 109-126 (2009).
- Matsushima, W., et al. SLAM-ITseq: Sequencing Cell Type-Specific Transcriptomes Without Cell Sorting. Development. 145 (13), (2018).
- Basnet, H., et al. Flura-Seq Identifies Organ-Specific Metabolic Adaptations During Early Metastatic Colonization. Elife. 8, (2019).
- Rodriques, S. G., et al. Slide-Seq: A Scalable Technology For Measuring Genome-Wide Expression At High Spatial Resolution. Science. 363 (6434), 1463-1467 (2019).
- Fazal, F. M., et al. Atlas Of Subcellular RNA Localization Revealed By Apex-Seq. Cell. 178 (2), 473-490 (2019).
- Slane, D., Bayer, M., Kaufmann, K., Mueller-Roeber, B. Cell Type-Specific Gene Expression Profiling Using Fluorescence-Activated Nuclear Sorting. Plant Gene Regulatory Networks: Methods And Protocols. , 27-35 (2017).
- Zanetti, M. E., Chang, I. F., Gong, F., Galbraith, D. W., Bailey-Serres, J. Immunopurification Of Polyribosomal Complexes Of Arabidopsis For Global Analysis Of Gene Expression. Plant Physiology. 138 (2), 624-635 (2005).
- King, H. A., Gerber, A. P. Translatome Profiling: Methods For Genome-Scale Analysis Of mRNA Translation. Briefings In Functional Genomics. 15 (1), 22-31 (2016).
- Mašek, T., Valášek, L., Pospíšek, M., Nielsen, H. Polysome Analysis And RNA Purification From Sucrose Gradients. RNA: Methods And Protocols. , 293-309 (2011).
- Heiman, M., et al. A Translational Profiling Approach For The Molecular Characterization Of Cns Cell Types. Cell. 135 (4), 738-748 (2008).
- Halbeisen, R. E., Scherrer, T., Gerber, A. P. Affinity Purification Of Ribosomes To Access The Translatome. Methods. 48 (3), 306-310 (2009).
- Thomas, A., et al. A Versatile Method For Cell-Specific Profiling Of Translated mRNAs In Drosophila. Plos One. 7 (7), e40276 (2012).
- Watson, F. L., et al. Cell Type-Specific Translational Profiling In The Xenopus Laevis Retina. Developmental Dynamics. 241 (12), 1960-1972 (2012).
- Lam, P. Y., Harvie, E. A., Huttenlocher, A. Heat Shock Modulates Neutrophil Motility In Zebrafish. Plos One. 8 (12), e84436 (2013).
- Fang, Y., et al. Translational Profiling Of Cardiomyocytes Identifies An Early Jak1/Stat3 Injury Response Required For Zebrafish Heart Regeneration. Proceedings Of The National Academy Of Sciences Of The United States Of America. 110 (33), 13416-13421 (2013).
- Mustroph, A., Zanetti, M. E., Girke, T., Bailey-Serres, J. Isolation And Analysis Of mRNAs From Specific Cell Types Of Plants By Ribosome Immunopurification. Methods In Molecular Biology. 959, 277-302 (2013).
- Monshausen, G. B., Gilroy, S. Feeling Green: Mechanosensing In Plants. Trends In Cell Biology. 19 (5), 228-235 (2009).
- Day, R. C., Grossniklaus, U., Macknight, R. C. Be More Specific! Laser-Assisted Microdissection Of Plant Cells. Trends In Plant Science. 10 (8), 397-406 (2005).
- Sheen, J. Signal Transduction In Maize And Arabidopsis Mesophyll Protoplasts. Plant Physiology. 127 (4), 1466-1475 (2001).
- Datta, S., et al. Laser Capture Microdissection: Big Data From Small Samples. Histology And Histopathology. 30 (11), 1255-1269 (2015).
- Birnbaum, K., et al. A Gene Expression Map Of The Arabidopsis Root. Science. 302 (5652), 1956 (2003).
- Hamant, O., Haswell, E. S. Life Behind The Wall: Sensing Mechanical Cues In Plants. BMC Biology. 15 (1), 1354 (2017).
- Vragović, K., et al. Translatome Analyses Capture Of Opposing Tissue-Specific Brassinosteroid Signals Orchestrating Root Meristem Differentiation. Proceedings of The National Academy of Sciences of the United States of America. 112 (3), 923-928 (2015).
- Wang, Y., Jiao, Y. Translating Ribosome Affinity Purification (Trap) For Cell-Specific Translation Profiling In Developing Flowers. Methods In Molecular Biology. 1110, 323-328 (2014).
- Sablok, G., Powell, J. J., Kazan, K. Emerging Roles And Landscape Of Translating mRNAs In Plants. Frontiers in Plant Science. 8, 1443 (2017).
- Ron, M., et al. Hairy Root Transformation Using Agrobacterium Rhizogenes As A Tool For Exploring Cell Type-Specific Gene Expression And Function Using Tomato As A Model. Plant Physiology. 166 (2), 455-469 (2014).
- Reynoso, M. A., et al. Evolutionary Flexibility In Flooding Response Circuitry In Angiosperms. Science. 365 (6459), 1291-1295 (2019).
- Dolan, L., et al. Cellular Organisation Of The Arabidopsis Thaliana Root. Development. 119 (1), 71 (1993).
- Ristova, D., Barbez, E. . Root Development. , (2018).
- Shekhar, V., Stӧckle, D., Thellmann, M., Vermeer, J. E. M. The Role Of Plant Root Systems In Evolutionary Adaptation. Current Topics in Developmental Biology. 131, 55-80 (2019).
- Malamy, J. E., Benfey, P. N. Down And Out In Arabidopsis: The Formation Of Lateral Roots. Trends in Plant Science. 2 (10), 390-396 (1997).
- de Smet, I., et al. Bimodular Auxin Response Controls Organogenesis In Arabidopsis. Proceedings of the National Academy of Sciences of The United States of America. 107 (6), 2705-2710 (2010).
- Péret, B., et al. Arabidopsis Lateral Root Development: An Emerging Story. Trends In Plant Science. 14 (7), 399-408 (2009).
- Vilches-Barro, A., Maizel, A. Talking Through Walls: Mechanisms Of Lateral Root Emergence In Arabidopsis Thaliana. Current Opinion in Plant Biology. 23, 31-38 (2015).
- Porco, S., et al. Lateral Root Emergence In Arabidopsis Is Dependent On Transcription Factor Lbd29 Regulation Of Auxin Influx Carrier Lax3. Development. 143 (18), 3340-3349 (2016).
- Stoeckle, D., Thellmann, M., Vermeer, J. E. Breakout-Lateral Root Emergence In Arabidopsis Thaliana. Current Opinion in Plant Biology. 41, 67-72 (2018).
- Banda, J., et al. Lateral Root Formation In Arabidopsis: A Well-Ordered Lrexit. Trends in Plant Science. 24 (9), 826-839 (2019).
- Vermeer, J. E. M., et al. A Spatial Accommodation By Neighboring Cells Is Required For Organ Initiation In Arabidopsis. Science. 343 (6167), 178-183 (2014).
- Vanneste, S., et al. Cell Cycle Progression In The Pericycle Is Not Sufficient For Solitary Root/Iaa14-Mediated Lateral Root Initiation In Arabidopsis Thaliana. The Plant Cell. 17 (11), 3035-3050 (2005).
- Marques-Bueno, M. M., et al. A Versatile Multisite Gateway-Compatible Promoter And Transgenic Line Collection For Cell Type-Specific Functional Genomics In Arabidopsis. The Plant Journal : For Cell and Molecular Biology. 85 (2), 320-333 (2016).
- Shimada, T. L., Shimada, T., Hara-Nishimura, I. A Rapid And Non-Destructive Screenable Marker, Fast, For Identifying Transformed Seeds Of Arabidopsis Thaliana. The Plant Journal : For Cell and Molecular Biology. 61 (3), 519-528 (2010).
- Clough, S. J., Bent, A. F. Floral Dip: A Simplified Method For Agrobacterium Mediated Transformation Of Arabidopsis Thaliana. The Plant Journal. 16 (6), 735-743 (1998).
- Lindsey, B. E., Rivero, L., Calhoun, C. S., Grotewold, E., Brkljacic, J. Standardized Method For High-Throughput Sterilization Of Arabidopsis Seeds. Journal Of Visualized Experiments. (128), e56587 (2017).
- Andersen, T. G., et al. Diffusible Repression Of Cytokinin Signalling Produces Endodermal Symmetry And Passage Cells. Nature. 555, 529-533 (2018).
- Schroeder, A., et al. The Rin: An Rna Integrity Number For Assigning Integrity Values To Rna Measurements. BMC Molecular Biology. 7, 3 (2006).
- Vragović, K., Bartom, E., Savaldi-Goldstein, S. Quantitation Of Cell Type-Specific Responses To Brassinosteroid By Deep Sequencing Of Polysome-Associated Polyadenylated RNA. Methods in Molecular Biology. 1564, 81-102 (2017).
- Bertin, B., Renaud, Y., Aradhya, R., Jagla, K., Junion, G. Trap-Rc, Translating Ribosome Affinity Purification From Rare Cell Populations Of Drosophila Embryos. Journal Of Visualized Experiments. (103), e52985 (2015).
- Livak, K. J., Schmittgen, T. D. Analysis Of Relative Gene Expression Data Using Real-Time Quantitative PCR And The 2(-Delta Delta C(T)) Method. Methods. 25 (4), 402-408 (2001).
- Jiao, Y., Meyerowitz, E. M. Cell-Type Specific Analysis Of Translating Rnas In Developing Flowers Reveals New Levels Of Control. Molecular Systems Biology. 6, 419 (2010).
- Tian, C., et al. A Gene Expression Map Of Shoot Domains Reveals Regulatory Mechanisms. Nature Communications. 10 (1), 141 (2019).
- Townsley, B. T., Covington, M. F., Ichihashi, Y., Zumstein, K., Sinha, N. R. Brad-Seq: Breath Adapter Directional Sequencing: A Streamlined, Ultra-Simple And Fast Library Preparation Protocol For Strand Specific mRNA Library Construction. Frontiers in Plant Science. 6, 366 (2015).
- Song, Y., et al. A Comparative Analysis Of Library Prep Approaches For Sequencing Low Input Translatome Samples. BMC Genomics. 19 (1), 696 (2018).
- Basu, D., Haswell, E. S. Plant Mechanosensitive Ion Channels: An Ocean Of Possibilities. Current Opinion in Plant Biology. 40, 43-48 (2017).
- Brady, S. M., et al. A High-Resolution Root Spatiotemporal Map Reveals Dominant Expression Patterns. Science. 318 (5851), 801 (2007).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados