
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Tradurre la purificazione dell'affinità del ribosoma (TRAP) per studiare lo sviluppo della radice di Arabidopsis thaliana su una scala specifica del tipo di cella
In questo articolo
Riepilogo
Tradurre la purificazione dell'affinità di ribosomi (TRAP) offre la possibilità di sezionare programmi di sviluppo con una minima elaborazione di organi e tessuti. Il protocollo produce RNA di alta qualità da cellule mirate con una sottounità ribosomica denominata GFP (Proteina fluorescente verde). Gli strumenti di analisi a valle, come qRT-PCR o RNA-seq, rivelano profili di espressione specifici del tessuto e del tipo di cellula.
Abstract
In questo articolo, diamo istruzioni pratiche per ottenere dati translatomi da diversi tipi di cellule della radice di Arabidopsis thaliana tramite il metodo di purificazione dell'affinità del ribosoma di traduzione (TRAP) e la preparazione consecutiva della libreria a basso input.
Come materiale di partenza, impieghiamo linee vegetali che esprimono RPL18 di proteine ribosomiche marcate tramite GFP in modo specifico del tipo di cellula mediante l'uso di promotori adeguati. Prima dell'immunopurificazione e dell'estrazione dell'RNA, il tessuto viene congelato allo snap, che conserva l'integrità dei tessuti e contemporaneamente consente l'esecuzione di studi di serie temporali ad alta risoluzione temporale. In particolare, le strutture delle pareti cellulari rimangono intatte, il che è uno dei principali inconvenienti nelle procedure alternative come gli approcci basati sullo smistamento delle cellule attivate dalla fluorescenza che si basano sulla protonodurata dei tessuti per isolare popolazioni cellulari distinte. Inoltre, non è necessaria alcuna fissazione dei tessuti come nelle tecniche basate sulla microdissezione di cattura laser, che consente di ottenere RNA di alta qualità.
Tuttavia, il campionamento dalle sottopopolazioni di cellule e solo l'isolamento dell'RNA associato al polisomo limita gravemente la resa dell'RNA. È quindi necessario applicare metodi di preparazione della libreria sufficientemente sensibili per una corretta acquisizione dei dati da parte dell'RNA-seq.
TRAP offre uno strumento ideale per la ricerca sulle piante, poiché molti processi di sviluppo coinvolgono percorsi di segnalazione meccanica e correlati alle pareti cellulari. L'uso di promotori per colpire specifiche popolazioni di cellule sta colmando il divario tra il livello di organo e a livello di singola cellula che a sua volta soffrono di poca risoluzione o di costi molto elevati. Qui, applichiamo il TRAP per studiare la comunicazione cellulare nella formazione laterale della radice.
Introduzione
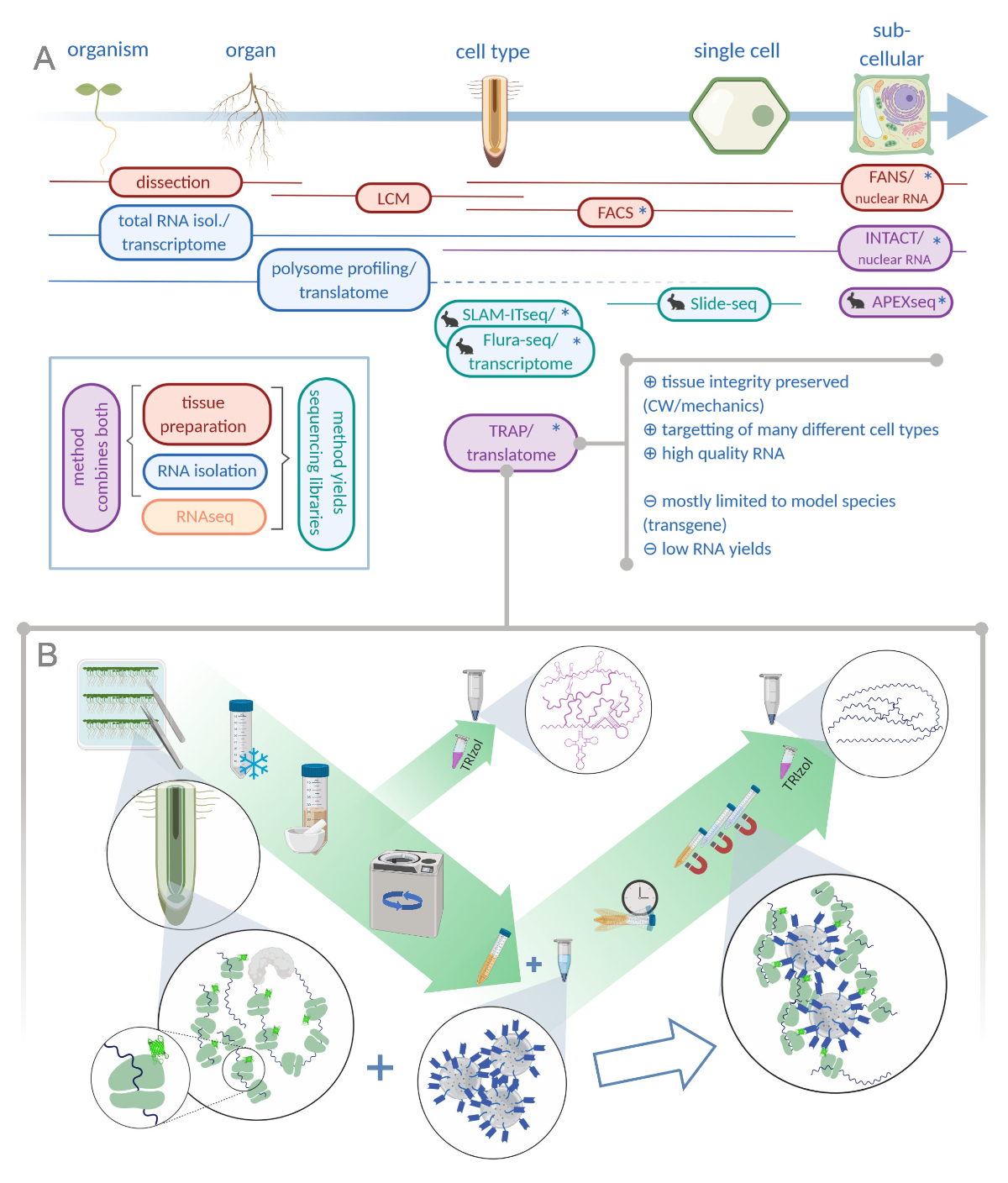
Spinto dalla crescente applicazione delle tecniche di sequenziamento di prossima generazione, la risoluzione spaziale nella biologia dello sviluppo potrebbe essere aumentata. Studi contemporanei mirano a sezionare i tessuti verso il basso per tipi di cellule specializzate, se non il livello a cella singola1,2,3,4.4 A tal fine, negli ultimi cinquant'anni è stata ideata una pletora di metodi diversi (vedi Figura 1A)5,6,7,8,9,10,11,12,13,14,15.
Molti strumenti nella scienza delle piante sono stati adattamenti di tecniche che sono state introdotte nella ricerca animale. Questo non è il caso per il metodo che stiamo introducendo in dettaglio qui. Nel 2005, dotato di un forte background nella traduzione delle proteine, il Bailey-Serres Lab ha deciso di progettare proteine ribosomiche per la successiva purificazione dell'affinità16. Così, potrebbero evitare la profilazione polisografica lunga e laboriosa, che si basa sull'ultracentrifugazione con un gradiente di saccarosio ed è stato utilizzato per valutare i ribosomi traslitteranti dal 196017,18. Da allora il metodo è stato indicato come trap (Affinity purificazione)16. Dopo studi translatomi di successo in piante, Heiman et al. adattato TRAP per gli animali19 e altri esteso la sua applicazione al lievito20, Drosophila21, Xenopus22 e pesce zebra23,24.
Anche se la modificazione genetica del sistema modello è un prerequisito per il TRAP, che limita la sua applicazione alle specie suscettibili di trasformazione genetica, si può allo stesso tempo sfruttare questa obiezione per indirizzare sottoinsiemi di cellule che sono di particolare interesse e altrimenti estremamente difficili da isolare dal tessuto intatto / organo25 (ad esempio, cellule dendritiche altamente ramificate in un cervello di topo o iphae fungini intessuto vegetale infetto). Nelle piante, tutte le cellule sono tenute in posizione attraverso pareti cellulari che costituiscono la base dello scheletro idrostatico26. Per liberare una cellula vegetale da questa matrice, gli scienziati hanno fisicamente tagliato la cellula dal tessuto circostante attraverso la microdissezione di cattura laser (LCM)27 o eseguito la digestione enzimatica delle pareti cellulari28. Tra queste ultime cellule, le cosiddette protoplasts, la popolazione di interesse è fluorescente etichettata e può essere separata tramite lo smistamento cellulare attivato dalla fluorescenza (FACS)7. LCM di solito richiede un campione da fissare e incorporare nella cera, che alla fine deteriora la qualità del suo RNA29. I metodi basati su FACS producono RNA di alta qualità, ma il processo di protoplasting stesso introduce differenze nell'espressione genica30 e i tessuti con pareti cellulari secondarie modificate e spesse sono notoriamente difficili da trattare. Inoltre, si presume che molti processi di sviluppo negli impianti si basino su segnali trasmessi meccanicamente e quindi l'integrità della parete cellulare è di fondamentale importanza31. Due metodi, che utilizzano una scorciatoia per aggirare l'isolamento cellulare operando a livello di nucleii, sono lo smistamento nucleare attivato dalla fluorescenza (FANS) e l'isolamento dei nuclei taggati in specifici tipi di cellule (INTACT). Come nel TRAP, usano promotori specifici del tipo di cellula per contrassegnare i nuclei, che successivamente si arricchiscono tramite ordinamento o discesa, rispettivamente8,15. Una grande sfida per tutti questi approcci è ottenere sufficiente materiale di RNA da sottoinsiemi di cellule in un tessuto. Poiché TRAP cattura solo una frazione degli RNA cellulari, la raccolta dei campioni è un notevole collo di bottiglia. Pertanto, sono necessari protocolli di preparazione della libreria particolarmente sensibili per produrre dati di alta qualità da quantità di input basse.
Dalla sua istituzione, TRAP è stato utilizzato in combinazione con microarray di DNA o, come i costi di sequenziamento è sceso in modo significativo negli ultimi anni, RNA-seq10,32,33. Una moltitudine di domande di ricerca è già stato chiarito come esaminato in Sablok et al.34. Siamo convinti che nei prossimi anni seguiranno ulteriori rapporti, poiché la tecnica è molto versatile quando si combinano diversi promotori per indirizzare tipi di cellule specifici. Alla fine, questo sarà fatto anche in modo inducibile, e può essere combinato con sondare la reazione della pianta a molti fattori di stress biotici e abiotici. Inoltre, laddove non sono disponibili linee transgeniche stabili, sono stati utilizzati con successo anche sistemi di espressione delle radici pelose per eseguire trap in pomodoro e medicago35,36.

Figura 1: La traduzione della purificazione dell'affinità del ribosomiare (TRAP) integra il portafoglio di analisi "omiche". R. L'aumento dei livelli di precisione analitica, fino alla risoluzione a cella singola o addirittura subcellulare, può essere raggiunto con una pletora di metodi o combinazioni. Il sistema fornisce una panoramica degli strumenti attualmente disponibili nel campo delle piante e degli animali. La raccolta dei tessuti a risoluzione cellulare può essere ottenuta da protocolli come LCM o FACS, che vengono poi accoppiati a trascrittoma standard o all'analisi di profilazione/traslatoma polisomico. TRAP e INTACT integrano sia la cattura dei tessuti che l'isolamento dell'RNA in quanto si basano sull'epitope-tagging. Tuttavia, INTACT campiona solo i nuclei cellulari e costituisce, quindi, un caso speciale di analisi del trascrittoma. Una piccola icona coniglio segna metodi appena sviluppati nel campo animale: Mentre SLAM-ITseq e Flura-seq si basano sul targeting metabolico di RNA nascenti con basi uracili modificate in cellule che esprimono l'enzima permissivo, Slide-seq fa uso di un vetrino di vetro rivestito con codici a barre del DNA che forniscono informazioni posizionali nella gamma cellulare. In APEX-seq è seguito un approccio di etichettatura di prossimità per campionare gli RNA in specifici compartimenti subcellulari. In particolare, una maggiore risoluzione spesso richiede la generazione di materiale transgenico (asterischi) e questi metodi sono quindi utilizzati prevalentemente per le specie modello. Il TRAP è particolarmente adatto per studi di scienze vegetali che coinvolgono la parete cellulare (CW) o la segnalazione meccanica, nonché le specie cellulari che sono difficili da rilasciare dalla loro matrice CW. B. Le fasi dettagliate del laboratorio umido della procedura TRAP: le piantine che esprimono proteine ribosomiche marcate GFP in tipi di cellule distinte (ad esempio l'endodermide delle radici) vengono coltivate sui piatti Petri per sette giorni e il materiale delle radici raccolto dal congelamento a scatto. Un campione totale di controllo dell'RNA viene raccolto dall'estratto grezzo omogeneo prima di pellere i detriti attraverso la centrifugazione. Perline magnetiche anti-GFP vengono aggiunte all'estratto eliminato per eseguire l'immunoprecipitazioni. Dopo l'incubazione e tre fasi di lavaggio, l'RNA associato al poliso (TRAP/RNA polisopo) viene ottenuto direttamente tramite l'estrazione del fenolo-cloroformio. LCM: microdissezione di cattura laser, FACS/FANS: smistamento cellulare/nucleare attivato dalla fluorescenza, APEX-seq: metodo basato su ingegnerizzato ascorbate perossidasi, INTACT: isolamento dei nuclei contrassegnati in specifici tipi di cellule, SLAM-ITseq: thiol(SH)-linked alchilirazione per il sequenziamento metabolico dell'RNA nel tessuto, Flura-seq: fluorouracil-labeled RNA sequencing (Creato con Biorender.com) Fare clic qui per visualizzare una versione più ampia di questa figura.
{kind=link}
L'obiettivo di questo articolo è fornire una descrizione dettagliata del metodo TRAP, evidenziare i passaggi critici e fornire indicazioni per un possibile metodo di preparazione della libreria.
Un esperimento trap generico consisterà essenzialmente nei seguenti passi (vedi anche Figura 1B): (1) Preparazione del materiale vegetale, compresa la clonazione del costrutto di ribosomi, la produzione e la selezione di linee transgeniche, la coltivazione e l'accumulo di semi, la sterilizzazione e la placcatura e l'applicazione/trattamento dello stress (opzionale) e la raccolta dei tessuti; (2) immunopurificazione, compresa l'omogenesi e la bonifica dei tessuti, il lavaggio delle perline e l'immunopurificazione e le fasi di lavaggio; (3) Estrazione dell'RNA e valutazione della qualità; e (4) la preparazione della biblioteca.
La radice dell'Arabidopsis è stata un sistema modello per studiare lo sviluppo delle piante sin dalla sua introduzione come impianto modello37,38. Qui, l'applicazione di TRAP è presentata nel contesto dello sviluppo delle radici laterali dell'impianto. Nelle piante, l'accumulo dell'intero sistema radicale si basa sull'esecuzione di questo programma ed è quindi molto importante per la sopravvivenza dell'organismo39. In Arabidopsis, le radici laterali provengono dal tessuto periciclo che risiede accanto ai vasi xylem e quindi è chiamato xylem polo periciclo (XPP; vedi Figura 2C)40. Alcune cellule XPP, che si trovano in profondità all'interno della radice, acquisiscono un'identità cellulare fondatore e, su un trigger ormonale locale, iniziano a proliferare gonfiore e dividendo anticlinally41. Tuttavia, a causa della presenza di una matrice rigida della parete cellulare, questo processo esercita uno stress meccanico sui tessuti circostanti. In particolare, l'endodermide sovrastante è interessata, in quanto è nel modo dell'asse di crescita della radice laterale42,43,44. Infatti, il primordio appena formato dovrà crescere attraverso la cellula endodermide sovrastante (Figura 2C2) mentre le cellule corteccia ed epidermide sono solo spinte da parte per il primordio per emergere finalmente45,46. Recenti lavori nel nostro laboratorio hanno dimostrato che l'endodermide sta contribuendo attivamente ad accogliere la proliferazione nel periciclo. Il blocco mirato della segnalazione ormonale endodermica è sufficiente per inibire anche la prima divisione nelle cellule XPP47. Pertanto, la comunicazione pericycle-endodermis costituisce un checkpoint molto precoce per lo sviluppo della radice laterale nell'Arabidopsis. Tuttavia, non è noto come questo crosstalk viene eseguito. Per svelare questo mistero, abbiamo scelto l'approccio TRAP-seq per colpire XPP e cellule endodermiche. Per arricchire per le cellule nel programma della radice laterale, abbiamo imitato il trigger ormonale applicando esogenamente un analogo auxina (1-naftaleneacetic acid, NAA)48, che allo stesso tempo ha permesso di risolvere temporaneamente la fase iniziale di formazione della radice laterale.
Access restricted. Please log in or start a trial to view this content.
Protocollo
1. Clonazione di transgenici, produzione e selezione di linee transgeniche
- Clonare il promotore di scelta nel vettore di immissione appropriato. Utilizzare un metodo di clonazione basato sulla ricombinazione (Table of Materials) e ricombinare i promotori in pDONRP4-P1r. Clone RPL18 (con tag di affinità o proteina fluorescente prescelta) utilizzando la clonazione basata sulla ricombinazione in pDONRP1-P249.
- Combinare il vettore di ingresso contenente RPL18 con il vettore di immissione che lo prompica contiene in una reazione di ricombinazione di due frammenti nel vettore di destinazione appropriato con cassetta di selezione FAST-rosso50 per facilitare la selezione diretta di semi transgenici.
- Verificare il vettore ricombinato sequenziandolo e trasformarlo in agrobatteri adatti e competenti. Fiore immersione piante Arabidopsis e dopo 3-4 settimane di raccolta e selezionare i semi di T151.
- Utilizzare la microscopia per identificare linee ben espressive e verificare i modelli di espressione in base all'attività del promotore segnalato in più righe indipendenti. Selezionare le linee che mostrano un modello di espressione rappresentativo con un singolo inserimento T-DNA. Questo potrebbe contribuire a ridurre al minimo il silenziamento e sarà vantaggioso per le croci genetiche.
- Selezionare la prole T3 che è omozigous per il gene marcatore.
2. Propagazione e sterilizzazione
- Trap specifico del tipo di cellula isola l'RNA da un numero limitato di cellule bersaglio per radice. Per generare il materiale iniziale necessario, propagare linee omozive. A tal fine, utilizzare condizioni di crescita standard con particolare attenzione al controllo della crescita fungina.
NOTA: se non è possibile ottenere singole linee di inserimento, coltivare lotti in grandi popolazioni nel corso di poche generazioni per evitare il silenziamento transgenerazionale indotto da T-DNA. - Sterilizzare grandi quantità di semi di Arabidopsis con un giro di gas cloro e un round del 70% EtOH.
- Stendere i semi uniformemente su piatti Petri quadrati 12 cm x 12 cm (meno di 0,3 mL di semi/piastra) e impilarli in un desiccatore o in un altro contenitore adatto. Evitare la formazione di grumi o cumuli in quanto i semi devono essere accessibili al gas. Eseguire la sterilizzazione del gas durante la notte con i volumi di candeggina e HCl come riportato52: 100 mL di candeggina (13%) con 6 mL di conc. HCl in un desiccatore 60 L. Defumigate per almeno 1 h prima di raccogliere i semi in un contenitore sterile.
AVVISO: il 37% di HCl è altamente corrosivo e richiede un'attenta maneggevolezza. Il gas cloro è tossico, usa una cappa di fumi. - Prendere 0,1 mL di semi secchi sterilizzati a gas per piastra e mescolarli con soluzione di sterilizzazione (70% EtOH, 0,01% Tween) a temperatura ambiente. Incubare per 20 min, decantare EtOH e lavare i semi 3-4 volte con sterile H2O.
- Trasferire i semi imbevuti in tubi da 50 mL e diluire con sterile 0,1% agar per ottenere 1 mL di liquami di semi imbitoni per piastra (0,1 mL di semi/1 mL di liquami).
NOTA: a causa di eventi di integrazione transgenica, le linee di impianto possono essere soggette a diverse tecniche di sterilizzazione; soprattutto il tempo di incubazione dell'EtOH è risultato essere critico. Nelle nostre mani, le doppie fasi di sterilizzazione sono state necessarie per evitare la contaminazione fungina durante gli esperimenti. Ciò è particolarmente importante quando si eseguono serie temporali come contaminazione di un singolo punto temporale ostacola l'intero esperimento. Potrebbe darsi che la doppia sterilizzazione non sia sempre necessaria, a seconda delle condizioni di crescita locali.
- Stendere i semi uniformemente su piatti Petri quadrati 12 cm x 12 cm (meno di 0,3 mL di semi/piastra) e impilarli in un desiccatore o in un altro contenitore adatto. Evitare la formazione di grumi o cumuli in quanto i semi devono essere accessibili al gas. Eseguire la sterilizzazione del gas durante la notte con i volumi di candeggina e HCl come riportato52: 100 mL di candeggina (13%) con 6 mL di conc. HCl in un desiccatore 60 L. Defumigate per almeno 1 h prima di raccogliere i semi in un contenitore sterile.
3. Placcatura
- Preparare questi passaggi in anticipo. Versare 1/2 piastre MS (pH 5,8) con l'1% di agar nelle quantità necessarie per l'esperimento (20-30 per campione/punto temporale). Tagliare 1 mL di punte pipette per ingrandire il diametro della punta a ca. 3-4 mm con una lama di rasoio. Autoclave le punte. Creare un supporto modello per placcare tre file di semi per piatto con coperchi quadrati Petri piatto. Preparare una cappa di flusso laminare per fornire un ambiente di lavoro sterile ed etichettare le piastre da lavorare.
NOTA: se più piastre vengono lavorate contemporaneamente, le etichette colorate possono accelerare l'etichettatura. - Inserire le piastre di agar vuote nel portamodelli e distribuire 1 mL di semi imbibed uniformemente su tre file. Posizionare le piastre lavorate in pile nel flusso laminare fino a quando i semi sono asciutti (cioè, attenersi alla superficie dell'agar). Non lasciare i piatti più a lungo come l'agar si asciugherà pure.
- Una volta che i semi sono sufficientemente asciutti, chiudere i coperchi e sigillare ogni piastra con nastro adesivo. Stratificate i semi per due giorni a 4 gradi centigradi al buio e poi metteteli in una camera di crescita.
4. Trattamento dei tessuti (opzionale)
NOTA: In questo protocollo, illustreremo il trattamento esogeno delle radici dell'Arabidopsis con la variante di auxina sintetica NAA. A seconda della questione sperimentale in questione, questa parte deve essere regolata o può essere omessa completamente.
- Preparare strisce di carta velina di 1,5 - 2 cm di altezza e 10 cm di lunghezza. I tempi di incubazione prolungati richiedono che il tessuto venga autoclaved prima dell'uso.
- Rimuovere il nastro microporo da tutte le piastre che devono sottoporsi al trattamento ormonale. Diluire 1 mL di 10 mM NAA (sciolto in DMSO) in 1 L di liquido, autoclaved 1/2 soluzione MS (pH 5,8) e immergere la carta velina nella soluzione (10 m NAA).
- Utilizzare una pinzetta per applicare una striscia di carta velina su ogni fila di radici. Utilizzare delicatamente le dita per rimuovere le bolle d'aria. Svuotare il liquido in eccesso dalla piastra, chiudere il coperchio ed etichettare la piastra con il tempo. Per lunghi tempi di incubazione, riporre le piastre nella camera di crescita.
5. Raccolta
- Recuperare le piastre per ogni replica biologica/punto temporale/trattamento. Raccogliere l'azoto liquido in un recipiente Dewar pulito ed etichettare i tubi (15 o 50 mL) per i diversi campioni di tessuto. Preparare un supporto di polistirolo.
AVVISO: Acquisire familiarità con le procedure di trattamento dell'azoto liquido (aerazione, congelamento, tubi che esplodono potenzialmente). - Aprire la piastra e rimuovere la carta velina con pinze, facendo attenzione a non staccare le radici dalla superficie dell'agar. Con una lama chirurgica, tagliare una volta per riga lungo il tiro-radice-junction in un singolo colpo determinato. Pulire le lame tra i campioni e scambiare frequentemente per garantire la nitidezza.
- Con una pinzetta, scorri lungo le radici di ogni fila per raccoglierle in tre fasci. Afferra le radici e svuotale in un tubo da 50 mL pieno di azoto liquido per congelare a scatto.
NOTA: Non cercare di assemblare le radici in strutture dense (come le palle) in quanto sono difficili da macinare nel passo successivo. - Procedere con tutte le piastre che costituiscono un campione (nell'ordine dei tempi di incubazione) e versare l'azoto liquido in eccesso. Utilizzare il coperchio del tubo per evitare che le radici si riversino. Quindi chiudere il coperchio e raccogliere tutti i tubi nel vascello Dewar. Conservare il tessuto radicale a -80 gradi centigradi.
6. Immunopurificazione
NOTA: Questo passaggio ha lo scopo di ottenere RNA TRAP/polisome di alta qualità. Pertanto, seguire rigorosamente i consigli di buona pratica per la manipolazione dell'RNA. Eseguire tutti i passaggi in questa sezione in un banco sterile e pulire tutte le attrezzature e i labware con una soluzione di rimozione RNase (Tabella dei materiali). Indossare i guanti e cambiarli immediatamente quando contaminati da campione, ghiaccio o altre fonti che non sono state pulite. Poiché si tratta di un aspetto molto cruciale, è inclusa una sezione sul riutilizzo delle attrezzature e la consulenza allo smaltimento dei rifiuti.
- Preparazione del buffer
- Preparare le soluzioni di stock in base alla tabella 1 e all'autoclave (A) o alla sterilizzazione del filtro. Se non diversamente specificato, il solvente è acqua priva di RNase.
- Sciogliereitol e dithiothreitol (DTT), fluoruro di fenilmethylsulfonyl (PMSF), cicloseximide (CHX) e cloramfenicolo (CAM) nei rispettivi solventi, come indicato nella Tabella 1 e conservarli a -20 gradi centigradi. Tutti gli altri stock possono rimanere a temperatura ambiente.
- Pre-mixare le scorte - con ingredienti 1-4 per il buffer di lavaggio (WB) e 1-6 per il buffer di estrazione polisomistico (PEB) - per evitare la miscelazione del buffer che richiede tempo prima di ogni estrazione. Quindi, aggiungere solo acqua e gli ingredienti congelati (7-10) il giorno dell'estrazione. Mantenere le scorte pre-miscelate e l'acqua senza RNase a 4 gradi centigradi.
NOTA: la concentrazione di DTT è di 1/5 della concentrazione segnalata da .anetti et al.
| Ingredienti | Concentrazione di scorte | Aggiungere il volume in mL per 50 mL di WB | Aggiungere volume in mL per 50 mL di PEB | ||
| 1 | Tris, pH 9 | Un | 2 M | 5 | 5 |
| 2 | Kcl | Un | 2 M | 5 | 5 |
| 3 | EGTA | Un | 0,5 M | 2.5 | 2.5 |
| 4 | MgCl2 | Un | 1 M | 1.75 | 1.75 |
| 5 | Pte | Un | 20% (v/v) | 0 | 2.5 |
| 6 | miscela detergente | Un | 0 | 2.5 | |
| Tween 20 | 20% (v/v) | ||||
| Triton-X 100 | 20% (v/v) | ||||
| Brij-35 | 20% (w/v) | ||||
| Igeope | 20% (v/v) | ||||
| 7 | Dtt | ₳ | 0,5 M | 0.1 | 0.1 |
| 8 | Funzione PMSF | ₳ | 0,1 M (isopropanolo) | 0.5 | 0.5 |
| 9 | Ciclolosmide | ₳ | 25 mg/mL (EtOH) | 0.1 | 0.1 |
| 10 | Cloramfenicolo | ₳ | 50 mg/mL (EtOH) | 0.05 | 0.05 |
Tabella 1: Composizione del buffer e consigli di miscelazione. Gli ingredienti con le concentrazioni di stock date mescolate nelle quantità date producono 50 mL di WB o PEB. Tris: tris-(idxymethyl)-aminomethane, EGTA: etilene glicol-bis(Etere di z-aminoetile)-N,N',N',N'-tetra-acetica acid, PTE: Polyoxyethylene-(10)-tridecyl ether, A: autoclave, - filter-sterilize; - Riempire fino a 50 mL con acqua senza RNase.
- omogeneizzazione/grinding dei tessuti
- Raffreddare la centrifuga e posizionare omogeneizzanti e tubi di centrifuga sul ghiaccio. Aliquote di scongelo, PMSF, CHX e CAM. Mescolare PEB e WB dalle soluzioni di magazzino in tubi da 50 mL in base alle esigenze del giorno (n. di campioni) e raffreddare sul ghiaccio.
NOTA: Aggiungere il PMSF solo poco prima dell'uso, poiché l'emivita del PMSF in acqua è di soli 30 min. - Preparare un sacco di azoto liquido in un recipiente Dewar e recuperare campioni di tessuto da -80 . Indossare guanti di cotone sotto i guanti da laboratorio standard per prevenire le ustioni da mortai freddi. Versare l'azoto liquido in mortai e pestelli fino a quando non sono abbastanza freddi da consentire la macinazione. Si raccomanda di ideare un sistema per distinguere i mortai (etichettare o mantenere in un certo ordine).
- Svuotare il campione di tessuto in un mortaio e macinare con attenzione fino a quando tutto il materiale è una polvere bianca. Se necessario, aggiungere azoto liquido per mantenere il tessuto congelato o per facilitare una migliore macinazione.
- Aggiungere 5 mL di PEB al campione e mescolare rapidamente con la polvere prima che il tampone si congeli. Mentre questo campione si scongela (mix di volta in volta) elaborare un altro campione.
- Non appena la miscela può essere trasferita, svuotare il liquame in un omogeneizzatore di vetro e tenere sul ghiaccio. Con altri 2 mL di PEB, sciacquare il mortaio e il pestello e aggiungerlo al campione nell'omogeneizzatore.
NOTA: Evitare un campione completamente liquido in quanto ciò consente la degradazione dell'RNA. - Macinare manualmente il liquame fino a quando l'estratto è omogeneo. Si consiglia un minimo di 4 a 5 tuffi.
NOTA: Potrebbe essere necessario un ulteriore tempo di attesa per consentire al liquame di scongelarsi ulteriormente. La manipolazione degli omogeneizzatori richiede una certa diligenza. Non applicare la forza bruta e attenzione alle forze di aspirazione. Se non preso in considerazione, questo porterà a fuoriuscite, contaminazioni o distruzione dell'omogeneizzatore. - Versare l'estratto di radice grezza in un tubo di centrifuga di 50 mL (tenere sul ghiaccio).
NOTA: Di solito diversi campioni possono essere macinati prima del trasferimento. È necessaria la movimentazione parallela di macinazione, trasferimento e omogeneizzazione. Cercate di lavorare in fretta, ma non abbiate fretta; stare calmi. Tenere sempre campioni omogeneizzati sul ghiaccio.
- Raffreddare la centrifuga e posizionare omogeneizzanti e tubi di centrifuga sul ghiaccio. Aliquote di scongelo, PMSF, CHX e CAM. Mescolare PEB e WB dalle soluzioni di magazzino in tubi da 50 mL in base alle esigenze del giorno (n. di campioni) e raffreddare sul ghiaccio.
- Raccolta totale di campioni di RNA
- Trasferire 200 aliquote di ogni campione grezzo in un tubo di microcentrifuga pulito (etichettato e raffreddato in anticipo sul ghiaccio).
- Procedere con l'estrazione dell'RNA come dettagliato per i campioni TRAP in punti 7.1 e 7.2. Eseguire questi passaggi durante la compensazione dei campioni nella centrifuga.
- Eseguire un trattamento DNase con l'RNA totale risospeso per eliminare la contaminazione del DNA e ripulire la reazione utilizzando un kit commerciale (Tabella dei materiali).
NOTA: le estrazioni totali di RNA di solito producono alte concentrazioni e i campioni devono essere diluiti considerevolmente. Si consiglia di misurare la concentrazione dopo la diluizione dal protocollo Qubit sensibile.
- Cancellazione dell'estratto grezzo
- Prendere il secchio di ghiaccio con campioni da 6.2.7 e centrifugarli per 15 min a 16.000 x g e 4 gradi centigradi.
NOTA: per bilanciare la centrifuga, accoppiare i campioni di conseguenza. Nel caso in cui ciò non sia del tutto possibile, regolare un campione aggiungendo la PEB. - Versare il supernatante in un tubo di centrifuga fresco (raffreddato in precedenza sul ghiaccio) e ripetere la centrifugazione (15 min a 16.000 x g e 4 gradi centigradi). Questo trasferimento può essere eseguito rapidamente accanto alla centrifuga.
- Mentre l'estratto grezzo è di compensazione, avviare il lavaggio di GFP-perline per il passaggio 6.6.
NOTA: Tenere questo secchio di ghiaccio per dondolare sullo shaker ma non riporre nella panca sterile in quanto potrebbe essere contaminato.
- Prendere il secchio di ghiaccio con campioni da 6.2.7 e centrifugarli per 15 min a 16.000 x g e 4 gradi centigradi.
- Lavaggio del tallone
- Aliquote GFP magnetiche (#samples x 60 l, tabella dei materiali) in un tubo da 1,5 mL. Posizionare sul supporto magnetico. Una volta che le perline hanno raccolto, rimuovere il supernatore.
- Aggiungere 1 mL di WB freddo, risospendere le perline e raccoglierle di nuovo. Eliminare il buffer di lavaggio e ripetere ancora una volta con 1 mL di WB.
- In definitiva, risospendere le perline in WB al volume iniziale utilizzato nel passaggio 6.5.1.
- Immunopurificazione (IP)
- Subito dopo la centrifuga, versare il soltabile eliminato in tubi etichettati da 15 mL e aggiungere 60 l di perline lavate per campione.
- Mettere tutti i campioni orizzontalmente nel secchio di ghiaccio e metterlo su uno shaker. Lasciare incubare la miscela per 2 h per legare i polisos etichettati GFP alle perline.
- Raccogliere le perline sul supporto magnetico per tubi da 15 mL (sul ghiaccio) e aggiungere IL PMSF al restante PEB. Scartare il super-attardato. Versare circa 5 mL di PEB alle perline e rinviderle inclinandole. Agitare i campioni per 15 min nella stessa configurazione della sezione 6.6.2.
- Ripetere i fumi con WB per un totale di 3 lavamenti (1 x PEB, 2 x WB). Prima di ogni scambio di buffer, aggiungere PMSF.
- Raccogliere le perline in 1 mL di WB e trasferirle in un tubo da 1,5 mL. Infine, raccogliere le perline ancora una volta sul supporto magnetico e rimuovere tutto il liquido. Chiudere il tubo e tenere il ghiaccio fino a quando tutti i campioni sono elaborati.
- Trasportare i campioni in una cappa di fumi per l'estrazione dell'RNA.
- Smaltimento dei rifiuti e ricondizionamento delle forniture di laboratorio.
- Se eseguita secondo la buona pratica di laboratorio (vedere la sezione 2.2.1), la procedura di sterilizzazione produce una soluzione NaCl acquosa. Lasciare il gas cloro, così come residua HCl e candeggina, per defumigare nel cappuccio del fume.
- PEB e wb smaltimento: mentre CHX si decompone ad alta pH, raccogli tutti i liquidi e porta al pH>9. Smaltire i rifiuti liquidi nei rifiuti chimici alogenicati. Tutti i solidi (tessuti, pipette sierologiche, guanti, ecc.) devono essere smaltiti come rifiuti chimici.
- Raccogliere i liquidi contenenti fenoli separatamente, così come il materiale contaminato da fenoli (punte, tubi e guanti).
- Mortai lavati a mano, pestelli e omogeneizzatori (spugnole e pennello) con sapone e risciacquare accuratamente. Successivamente, cuocere il materiale a >220 gradi centigradi durante la notte. Avvolgere in lamina di stagnola prima del trattamento o mettere in un contenitore coperto a prova di calore.
- Spazzolare i tubi di centrifugare puliti con detergente e poi diethylpyrocarbonate (DEPC)-trattamento nella cappa fuma. A tal fine, aggiungere DEPC liquido all'acqua deionizzata (1 mL di DEPC a 1 L di H2O) e mescolare tramite agitazione. Posizionare i tubi di centrifuga su un vassoio autoclavable che cattura l'acqua DEPC versata. Versare la sospensione nei tubi e lasciare per 3 ore o per una notte. DEPC si decompone nel successivo processo di autoclaving.
AMMONIenza: IL DEPC è altamente tossico.
7. Estrazione di RNA e QC
- Estrazione dell'RNA
- Raffreddare la centrifuga del tavolo a 4 gradi centigradi.
- Aggiungere 1 mL di reagente a base di acido-guanidinium-fenolo (Tabella dei materiali) ad ogni campione, invertire per risospendere le perline o liquame totale dell'RNA e incubare per 5 min sul ghiaccio. Non vortice!
- Aggiungere 200 l di cloroformio e incubare per 3 min sul ghiaccio. Quindi vortice accuratamente i campioni.
- Per facilitare la separazione di fase, centrifugare al massimo. velocità per 10-15 min, 4 gradi centigradi.
- Etichettatubi a bassa ritenzione da 1,5 ml (Tabella dei materiali) e 650 - L di isopropanolo in ciascuno.
- Prendere con cura la fase acquosa superiore (ca. 650) e trasferirla nei tubi preparati con isopropanolo. Evitare di toccare la fase organica rosa.
- Precipitare l'RNA durante la notte a -20 gradi centigradi.
NOTA: Si raccomanda di conservare i campioni in isopropanolo a -20 o -80 gradi centigradi e solubilizzare solo in acqua quando necessario. L'RNA acquoso si degrada anche a -80 gradi centigradi se conservato per settimane/mesi.
- Precipitazioni nell'RNA
- Raffreddare la centrifuga del tavolo a 4 gradi centigradi.
- Preparare l'80% fresco di EtOH con acqua senza RNase e raffreddare a -20 gradi centigradi (5 min a -80 gradi aiutano ad accelerare il processo).
- Centrifugare i campioni alla massima velocità (ca. 13.000 x g) per 30 min e scartare il sovrentrato. Il pellet non sarà visibile, così attentamente pipetta come se fosse lì. Aggiungere 1 mL di freddo 80% EtOH e invertire il tubo una o due volte.
- Centrifuga di nuovo per 30 min alla massima velocità e ripetere il lavaggio per un totale di due lavaggi.
- Ruotare verso il basso per 2 min e rimuovere tutti i residui EtOH con una punta di 10 .L. Lasciare asciugare il pellet per 3-5 min (non di più) a temperatura ambiente e risospendere in acqua senza RNase da 20.L.
- Conservare i campioni sul ghiaccio ed eseguire il controllo di qualità il più presto possibile. Procedere con l'immagazzinare i campioni a -80 gradi centigradi. Evitare cicli di congelamento-scongelamento.
- Controllo di qualità utilizzando attrezzature dedicate (Tabella dei materiali) secondo le raccomandazioni del produttore.
8. Preparazione della biblioteca
- sintesi e amplificazione del cDNA con il kit RNA SMARTer v4 Ultra Low Input
- Calcolare la diluizione di ogni campione per avere 1,5 ng di RNA TRAP o RNA totale in un volume di 4,75 . Eseguire tutte le reazioni in tubi PCR e diluire campioni con fresco di acqua senza RNase.
- Eseguire tutti i passaggi secondo le raccomandazioni del produttore con 1/2 dei volumi di reazione. Amplifica il cDNA con 12-13 cicli PCR.
- Pulire la PCR aggiungendo 0,5 l of 10x lysis buffer e 25 L di perline SPRI (Tabella dei materiali). Se molti campioni sono trasformati lisi buffer e perline possono essere pre-miscelati. Assicurarsi che le perline siano disperse uniformemente prima di pipettare.
- Procedere con il protocollo in volumi di reazione completi (17 - L di buffer di eluizione). Non lasciare asciugare le perline per più di 3 minuti. I campioni sovraessiccati possono potenzialmente essere salvati da tempi di incubazione prolungati.
- Misurare le concentrazioni del campione con il kit DNA Qubit HS.
NOTA: il kit SMARTer v4 può tollerare fino a 200 pg ingresso. Abbiamo ottenuto librerie nei casi in cui non è stato possibile determinare i valori Qubit (al di sotto di 250 pg, limite di rilevamento) con un PCR a 16 cicli. Tuttavia, il materiale di input limitato potrebbe anche produrre librerie meno complesse.
- Frammentazione e legatura dell'adattatore PCR con il Kit di preparazione della libreria di DNA Nextera XT
- Diluire il cDNA con acqua priva di RNase per ottenere una concentrazione di 200 pg/l e pipetta 1,25 - L in un tubo PCR.
- Eseguire tutti i passaggi secondo il produttore con 1/4 dei volumi di reazione. Amplifica il cDNA con 12 cicli PCR e adattatori compatibili per i campioni che appartengono a un pool di sequenziamento. Con I kit di indice di Illumina A e D è possibile utilizzare fino a 384 campioni.
- Per la pulizia della PCR aggiungere 12,5 litri di buffer di sospensione e 22,5 luna di perline SPRI (rapporto 0,9x). Elutare il campione con 22 l di tampone di eluizione.
NOTA: il QC e il pooling sono stati eseguiti dalla società di sequenziamento (Tabella dei materiali) e pertanto non è stata necessaria alcuna normalizzazione basata su perline. La reazione di frammentazione enzimatica (tagmentation) è molto sensibile all'input del materiale in quanto ogni enzima taglia solo una volta. Pertanto, non superare la raccomandazione di concentrazione.
Access restricted. Please log in or start a trial to view this content.
Risultati
Per la valutazione della qualità, la procedura di cui sopra dovrebbe essere sondata in diversi passaggi intermedi: convalida del modello di espressione nella planta, controllo qualità dell'RNA polisomico isolato e delle librerie finali. QRT-PCR utilizzando geni marcatori noti può, inoltre, essere eseguito per confermare la risposta alla condizione di trattamento o per mettere a punto le condizioni sperimentali.
Analis...
Access restricted. Please log in or start a trial to view this content.
Discussione
Verifica del modello di localizzazione RPL18
Fondamentale per evitare interpretazioni errate dei dati di qualsiasi esperimento TRAP è il modello di espressione corretto della sottounità ribosomica con tag. Pertanto, l'incorporazione di GFP come tag epitope a RPL18 consente molto elegantemente la verifica del modello di espressione desiderato e consecutivamente, la pulldown della frazione polisoma dallo stesso tessuto. Approcci più invasivi per assicurare modelli di promotore adeguati sono seguiti d...
Access restricted. Please log in or start a trial to view this content.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Ringraziamo Jean-Claude Walser del Centro per la diversità genetica di zurighese per la consulenza di esperti cruciali nella prima fase di questo progetto. Il lavoro nel laboratorio di Vermeer è stato sostenuto da una sovvenzione di professori SNF (PP00P3_157524) e da una sovvenzione per attrezzature R'EQUIP (316030_164086) della Fondazione nazionale svizzera per la scienza (SNSF) assegnata a JEMV.
Access restricted. Please log in or start a trial to view this content.
Materiali
| Name | Company | Catalog Number | Comments |
| Sterilization | |||
| bleach, 13% | Sigma | 71696 | |
| beaker | VWR | 214-1172/74/75 | |
| desiccator with porcelaine plate (DURAN) | Sigma/Merck | Z317454-1EA/Z317594-1EA | |
| EtOH, p.a. | Honeywell | 02860-1L | |
| HCl, 37% | Roth | 4625.1 | |
| Tween 20 | Sigma | P9416 | |
| Plate growth + harvesting | |||
| MS salts, basal salt mixture, incl. MES buffer | Duchefa | M0254 | |
| agar plant for cell culture | Applichem/Panreac | A2111.1000 | |
| DMSO | Sigma | D4540 | |
| forcepts | Rubis Switzerland | 5-SA model | |
| KOH | Fluka | 60370 | |
| micropore/surgical tape | 3M | 1530-0 | |
| NAA | Duchefa | N0903 | |
| petri dishes 120x120 mm | Greiner bio-one | 688102 | |
| scalpel | VWR/Swann-Morton | 233-5454 | |
| tissues, neutral, two-layered | any supplier of your choice | ||
| Immunoprecipitation | |||
| GFP-beads: gtma-100 GFP-Trap_MA | Chromotek | e.g. gtma-100 | |
| Brij-35 | Sigma | P1254-500G | |
| centrifuge tubes (in accordance with centrifuge) | Beckman Coulter | 357001 | |
| Chloramphenicol | Applichem | C0378-25G | |
| cotton gloves | VWR | 113-7355 | |
| Cycloheximide, HPLC grade | Sigma | 01810-1G | |
| DEPC | VWR | E174 | might have long delivery times |
| DTT | Fluka | 43815 | |
| EGTA | Sigma | 3054.3 | |
| homogenizers DUALL 23 | KONTES GLASS CO (via VWR) | SCERSP885450-0023 (set) | SCERSP885451-0023 pestle only - SCERSP885452-0023 cylinder only; long delivery times |
| Igepal CA-360 | Sigma | I3021-100ml | |
| KCl | Sigma | 60130 | |
| MgCl2 hexahydrat | Roth | 2189.2 | |
| mortar and pestle | VWR | 470148-960 & 470019-978 | |
| PMSF | Roche | 10 837 091 001 | |
| Polyoxyethylene-(10)-tridecylether/PTE | Sigma | P2393-500G | |
| RNase-free water | Roth | T143.3 | |
| RNAZap | Thermo Fisher | AM9780/AM9782 | for cleaning surfaces |
| Tris, >99.3% | Roth | AE15.3 | |
| Triton X-100 | Fluka | T8787-250ml | |
| Tween 20 | Sigma | P9416-100ml | |
| RNA extraction | |||
| 2-Propanol, p.a. | Sigma | 33539-1L-GL-R | |
| Chloroform, HPLC grade | Scharlau | CL02181000 | |
| EtOH, p.a. | Honeywell | 02860-1L | |
| low-retention microcentrifuge tubes, 1.5 ml | Eppendorf/Sigma | Z666548-250EA | LoBind |
| RNase-free DNase set | Qiagen | 79254 | |
| RNeasy MiniElute Cleanup Kit | Qiagen | 74204 | |
| TRIzol reagent | ThermoFisher/Ambion | 15596018 | |
| Library preparation | |||
| 15/50 mL Tube Magnetic Separator | Abraxis | PN 472250 | |
| AMPure beads | Beckman Coulter | A63881 | |
| Index Kit A | Illumina | FC-131-2001 | |
| Index Kit D | Illumina | FC-131-2004 | |
| neodymium magnets | Amazon/other | 6 x 1.5 mm range: N42 (NdFeB) | |
| Nextera XT kit | Illumina | FC-131-1024/1096 | https://emea.support.illumina.com/ |
| PCR strips | ThermoScientific | AB-0266 | |
| SMARTer v4 kit | Takara Bioscience | 634892 | https://www.takarabio.com/ |
| Bioanalyzer | Agilent | 2100 Bioanalyzer Instrument | specialized equipment for RNA/DNA quality control |
| Tapestation | Agilent | 4200 Tapestation Instrument | specialized equipment for RNA/DNA quality control |
| Fragment Analyzer | Agilent | 5400 Fragment Analyzer System | specialized equipment for RNA/DNA quality control (high throughput) |
| LabChip | PerkinElmer | LabChip GX Touch Nucleic Acid Analyzer | specialized equipment for RNA/DNA quality control (high throughput) |
| Qubit 4 Fluorometer | ThermoFisher | Q33239 | specialized equipment for RNA/DNA concentration determination |
| qRT-PCR | |||
| GATA23 | Microsynth | fwd: AGTGAGAATGAA AGAAGAGAAGGG; rev: GTGGCTGCGAAT AATATGAATACC | |
| GH3.3 | Microsynth | fwd: CAAACCAATCCT CCAAATGAC; rev: ACTTATCCGCAA CCCGACT | |
| LBD29 | Microsynth | fwd: TCTCCAACAACA GGTTGTGAAT; rev: AAGGAGCCTTAG TAGTGTCTCCA | |
| UBC21 | Microsynth | fwd: TGCGACTCAGGG AATCTTCT; rev: TCATCCTTTCTT AGGCATAGCG | |
| SsoAdvanced Universal SYBR Green | Bio-Rad | #172-5270 | |
| iScript Adv cDNA Kit | Bio-Rad | #172-5038 | |
| miscellaneous | |||
| Falcon tubes 15 ml, Cellstar | Greiner bio-one | 188261 | |
| Falcon tubes 50 ml, Cellstar | Greiner bio-one | 210261 | |
| filter tips 1 ml | Axygen | TF-1000-R-S | |
| filter tips 10 µl | Axygen | TF-10-R-S | |
| filter tips 100 µl | Axygen | TF-100-R-S | |
| filter tips 20 µl | Axygen | TF-20-R-S | |
| filter tips 200 µl | Axygen | TF-200-R-S | |
| microcentrifuge tubes 1.5 ml | SARSTEDT | 72.690.001 | |
| Propidium iodide | Sigma | P4170-100MG | |
| sequencing company | Novogene | en.novogene.com |
Riferimenti
- Van Verk, M. C., Hickman, R., Corné, M. J., Pieterse, M., Van Wees, S. C. RNA-Seq: Revelation Of The Messengers. Trends In Plant Science. 18 (4), 175-179 (2013).
- Libault, M., Pingault, L., Zogli, P., Schiefelbein, J. Plant Systems Biology At The Single-Cell Level. Trends In Plant Science. 22 (11), 949-960 (2017).
- Mustroph, A., et al. Profiling Translatomes Of Discrete Cell Populations Resolves Altered Cellular Priorities During Hypoxia In Arabidopsis. Proceedings Of The National Academy Of Sciences Of The United States Of America. 106 (44), 18843-18848 (2009).
- Karve, R., Iyer-Pascuzzi, A. S. Digging Deeper: High-Resolution Genome-Scale Data Yields New Insights Into Root Biology. Current Opinion In Plant Biology. 24, 24-30 (2015).
- Warner, J. R., Knopf, P. M., Rich, A. A Multiple Ribosomal Structure In Protein Synthesis. Proceedings of The National Academy of Sciences of The United States of America. 49 (1), 122-129 (1963).
- Gautam, V., Sarkar, A. K. Laser Assisted Microdissection, An Efficient Technique To Understand Tissue Specific Gene Expression Patterns And Functional Genomics In Plants. Molecular Biotechnology. 57 (4), 299-308 (2015).
- Bargmann, B. O. R., Birnbaum, K. D. Fluorescence Activated Cell Sorting Of Plant Protoplasts. Journal of Visualized Experiments. (36), e1673(2010).
- Deal, R. B., Henikoff, S. The Intact Method For Cell Type-Specific Gene Expression And Chromatin Profiling In Arabidopsis Thaliana. Nature Protocols. 6 (1), 56-68 (2011).
- Dougherty, J. D. The Expanding Toolkit Of Translating Ribosome Affinity Purification. The Journal of Neuroscience: The Official Journal Of The Society For Neuroscience. 37 (50), 12079-12087 (2017).
- Mustroph, A., Juntawong, P., Bailey-Serres, J. Isolation Of Plant Polysomal mRNA By Differential Centrifugation And Ribosome Immunopurification Methods. Methods in Molecular Biology. 553, 109-126 (2009).
- Matsushima, W., et al. SLAM-ITseq: Sequencing Cell Type-Specific Transcriptomes Without Cell Sorting. Development. 145 (13), (2018).
- Basnet, H., et al. Flura-Seq Identifies Organ-Specific Metabolic Adaptations During Early Metastatic Colonization. Elife. 8, (2019).
- Rodriques, S. G., et al. Slide-Seq: A Scalable Technology For Measuring Genome-Wide Expression At High Spatial Resolution. Science. 363 (6434), 1463-1467 (2019).
- Fazal, F. M., et al. Atlas Of Subcellular RNA Localization Revealed By Apex-Seq. Cell. 178 (2), 473-490 (2019).
- Slane, D., Bayer, M. Cell Type-Specific Gene Expression Profiling Using Fluorescence-Activated Nuclear Sorting. Plant Gene Regulatory Networks: Methods And Protocols. Kaufmann, K., Mueller-Roeber, B. , Springer. New York, NY. 27-35 (2017).
- Zanetti, M. E., Chang, I. F., Gong, F., Galbraith, D. W., Bailey-Serres, J. Immunopurification Of Polyribosomal Complexes Of Arabidopsis For Global Analysis Of Gene Expression. Plant Physiology. 138 (2), 624-635 (2005).
- King, H. A., Gerber, A. P. Translatome Profiling: Methods For Genome-Scale Analysis Of mRNA Translation. Briefings In Functional Genomics. 15 (1), 22-31 (2016).
- Mašek, T., Valášek, L., Pospíšek, M. Polysome Analysis And RNA Purification From Sucrose Gradients. RNA: Methods And Protocols. Nielsen, H. , Humana Press. Totowa, NJ. 293-309 (2011).
- Heiman, M., et al. A Translational Profiling Approach For The Molecular Characterization Of Cns Cell Types. Cell. 135 (4), 738-748 (2008).
- Halbeisen, R. E., Scherrer, T., Gerber, A. P. Affinity Purification Of Ribosomes To Access The Translatome. Methods. 48 (3), 306-310 (2009).
- Thomas, A., et al. A Versatile Method For Cell-Specific Profiling Of Translated mRNAs In Drosophila. Plos One. 7 (7), e40276(2012).
- Watson, F. L., et al. Cell Type-Specific Translational Profiling In The Xenopus Laevis Retina. Developmental Dynamics. 241 (12), 1960-1972 (2012).
- Lam, P. Y., Harvie, E. A., Huttenlocher, A. Heat Shock Modulates Neutrophil Motility In Zebrafish. Plos One. 8 (12), e84436(2013).
- Fang, Y., et al. Translational Profiling Of Cardiomyocytes Identifies An Early Jak1/Stat3 Injury Response Required For Zebrafish Heart Regeneration. Proceedings Of The National Academy Of Sciences Of The United States Of America. 110 (33), 13416-13421 (2013).
- Mustroph, A., Zanetti, M. E., Girke, T., Bailey-Serres, J. Isolation And Analysis Of mRNAs From Specific Cell Types Of Plants By Ribosome Immunopurification. Methods In Molecular Biology. 959, 277-302 (2013).
- Monshausen, G. B., Gilroy, S. Feeling Green: Mechanosensing In Plants. Trends In Cell Biology. 19 (5), 228-235 (2009).
- Day, R. C., Grossniklaus, U., Macknight, R. C. Be More Specific! Laser-Assisted Microdissection Of Plant Cells. Trends In Plant Science. 10 (8), 397-406 (2005).
- Sheen, J. Signal Transduction In Maize And Arabidopsis Mesophyll Protoplasts. Plant Physiology. 127 (4), 1466-1475 (2001).
- Datta, S., et al. Laser Capture Microdissection: Big Data From Small Samples. Histology And Histopathology. 30 (11), 1255-1269 (2015).
- Birnbaum, K., et al. A Gene Expression Map Of The Arabidopsis Root. Science. 302 (5652), 1956(2003).
- Hamant, O., Haswell, E. S. Life Behind The Wall: Sensing Mechanical Cues In Plants. BMC Biology. 15 (1), 1354(2017).
- Vragović, K., et al. Translatome Analyses Capture Of Opposing Tissue-Specific Brassinosteroid Signals Orchestrating Root Meristem Differentiation. Proceedings of The National Academy of Sciences of the United States of America. 112 (3), 923-928 (2015).
- Wang, Y., Jiao, Y. Translating Ribosome Affinity Purification (Trap) For Cell-Specific Translation Profiling In Developing Flowers. Methods In Molecular Biology. 1110, 323-328 (2014).
- Sablok, G., Powell, J. J., Kazan, K. Emerging Roles And Landscape Of Translating mRNAs In Plants. Frontiers in Plant Science. 8, 1443(2017).
- Ron, M., et al. Hairy Root Transformation Using Agrobacterium Rhizogenes As A Tool For Exploring Cell Type-Specific Gene Expression And Function Using Tomato As A Model. Plant Physiology. 166 (2), 455-469 (2014).
- Reynoso, M. A., et al. Evolutionary Flexibility In Flooding Response Circuitry In Angiosperms. Science. 365 (6459), 1291-1295 (2019).
- Dolan, L., et al. Cellular Organisation Of The Arabidopsis Thaliana Root. Development. 119 (1), 71(1993).
- Ristova, D., Barbez, E. Root Development. , Springer. New York, NY. (2018).
- Shekhar, V., Stӧckle, D., Thellmann, M., Vermeer, J. E. M. The Role Of Plant Root Systems In Evolutionary Adaptation. Current Topics in Developmental Biology. 131, 55-80 (2019).
- Malamy, J. E., Benfey, P. N. Down And Out In Arabidopsis: The Formation Of Lateral Roots. Trends in Plant Science. 2 (10), 390-396 (1997).
- de Smet, I., et al. Bimodular Auxin Response Controls Organogenesis In Arabidopsis. Proceedings of the National Academy of Sciences of The United States of America. 107 (6), 2705-2710 (2010).
- Péret, B., et al. Arabidopsis Lateral Root Development: An Emerging Story. Trends In Plant Science. 14 (7), 399-408 (2009).
- Vilches-Barro, A., Maizel, A. Talking Through Walls: Mechanisms Of Lateral Root Emergence In Arabidopsis Thaliana. Current Opinion in Plant Biology. 23, 31-38 (2015).
- Porco, S., et al. Lateral Root Emergence In Arabidopsis Is Dependent On Transcription Factor Lbd29 Regulation Of Auxin Influx Carrier Lax3. Development. 143 (18), 3340-3349 (2016).
- Stoeckle, D., Thellmann, M., Vermeer, J. E. Breakout-Lateral Root Emergence In Arabidopsis Thaliana. Current Opinion in Plant Biology. 41, 67-72 (2018).
- Banda, J., et al. Lateral Root Formation In Arabidopsis: A Well-Ordered Lrexit. Trends in Plant Science. 24 (9), 826-839 (2019).
- Vermeer, J. E. M., et al. A Spatial Accommodation By Neighboring Cells Is Required For Organ Initiation In Arabidopsis. Science. 343 (6167), 178-183 (2014).
- Vanneste, S., et al. Cell Cycle Progression In The Pericycle Is Not Sufficient For Solitary Root/Iaa14-Mediated Lateral Root Initiation In Arabidopsis Thaliana. The Plant Cell. 17 (11), 3035-3050 (2005).
- Marques-Bueno, M. M., et al. A Versatile Multisite Gateway-Compatible Promoter And Transgenic Line Collection For Cell Type-Specific Functional Genomics In Arabidopsis. The Plant Journal : For Cell and Molecular Biology. 85 (2), 320-333 (2016).
- Shimada, T. L., Shimada, T., Hara-Nishimura, I. A Rapid And Non-Destructive Screenable Marker, Fast, For Identifying Transformed Seeds Of Arabidopsis Thaliana. The Plant Journal : For Cell and Molecular Biology. 61 (3), 519-528 (2010).
- Clough, S. J., Bent, A. F. Floral Dip: A Simplified Method For Agrobacterium Mediated Transformation Of Arabidopsis Thaliana. The Plant Journal. 16 (6), 735-743 (1998).
- Lindsey, B. E., Rivero, L., Calhoun, C. S., Grotewold, E., Brkljacic, J. Standardized Method For High-Throughput Sterilization Of Arabidopsis Seeds. Journal Of Visualized Experiments. (128), e56587(2017).
- Andersen, T. G., et al. Diffusible Repression Of Cytokinin Signalling Produces Endodermal Symmetry And Passage Cells. Nature. 555, 529-533 (2018).
- Schroeder, A., et al. The Rin: An Rna Integrity Number For Assigning Integrity Values To Rna Measurements. BMC Molecular Biology. 7, 3(2006).
- Vragović, K., Bartom, E., Savaldi-Goldstein, S. Quantitation Of Cell Type-Specific Responses To Brassinosteroid By Deep Sequencing Of Polysome-Associated Polyadenylated RNA. Methods in Molecular Biology. 1564, 81-102 (2017).
- Bertin, B., Renaud, Y., Aradhya, R., Jagla, K., Junion, G. Trap-Rc, Translating Ribosome Affinity Purification From Rare Cell Populations Of Drosophila Embryos. Journal Of Visualized Experiments. (103), e52985(2015).
- Livak, K. J., Schmittgen, T. D. Analysis Of Relative Gene Expression Data Using Real-Time Quantitative PCR And The 2(-Delta Delta C(T)) Method. Methods. 25 (4), 402-408 (2001).
- Jiao, Y., Meyerowitz, E. M. Cell-Type Specific Analysis Of Translating Rnas In Developing Flowers Reveals New Levels Of Control. Molecular Systems Biology. 6, 419(2010).
- Tian, C., et al. A Gene Expression Map Of Shoot Domains Reveals Regulatory Mechanisms. Nature Communications. 10 (1), 141(2019).
- Townsley, B. T., Covington, M. F., Ichihashi, Y., Zumstein, K., Sinha, N. R. Brad-Seq: Breath Adapter Directional Sequencing: A Streamlined, Ultra-Simple And Fast Library Preparation Protocol For Strand Specific mRNA Library Construction. Frontiers in Plant Science. 6, 366(2015).
- Song, Y., et al. A Comparative Analysis Of Library Prep Approaches For Sequencing Low Input Translatome Samples. BMC Genomics. 19 (1), 696(2018).
- Basu, D., Haswell, E. S. Plant Mechanosensitive Ion Channels: An Ocean Of Possibilities. Current Opinion in Plant Biology. 40, 43-48 (2017).
- Brady, S. M., et al. A High-Resolution Root Spatiotemporal Map Reveals Dominant Expression Patterns. Science. 318 (5851), 801(2007).
Access restricted. Please log in or start a trial to view this content.
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati