
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Übersetzen von Ribosome Affinity Purification (TRAP) zur Untersuchung der Arabidopsis thaliana Wurzelentwicklung in einer zelltypspezifischen Skala
In diesem Artikel
Zusammenfassung
Die Übersetzung der Ribosomenaffinitätsreinigung (TRAP) bietet die Möglichkeit, Entwicklungsprogramme mit minimaler Verarbeitung von Organen und Geweben zu sezieren. Das Protokoll liefert hochwertige RNA aus Zellen, die mit einem grünen fluoreszierenden Protein (GFP)-markierten ribosomalen Untereinheit begezielt sind. Nachgeschaltete Analysewerkzeuge wie qRT-PCR oder RNA-seq zeigen Gewebe- und zelltypspezifische Expressionsprofile auf.
Zusammenfassung
In diesem Artikel geben wir praktische Anweisungen, um translatome Daten von verschiedenen Arabidopsis thaliana Wurzelzelltypen über die übersetzende Ribosome-Affinitätsreinigung (TRAP)-Methode und die konsekutive optimierte Bibliotheksvorbereitung mit niedrigem Input zu erhalten.
Als Ausgangsmaterial verwenden wir Anlagenlinien, die gfP-markiertes ribosomales Protein RPL18 zelltypspezifisch durch den Einsatz geeigneter Promotoren ausdrücken. Vor der Immunreinigung und RNA-Extraktion wird das Gewebe schnappgefroren, was die Gewebeintegrität bewahrt und gleichzeitig die Durchführung von Zeitreihenstudien mit hoher zeitlicher Auflösung ermöglicht. Insbesondere bleiben Zellwandstrukturen intakt, was einen großen Nachteil in alternativen Verfahren wie fluoreszenzaktivierten Zellsortier-basierten Ansätzen darstellt, die auf Gewebeprotoplasting angewiesen sind, um verschiedene Zellpopulationen zu isolieren. Darüber hinaus ist keine Gewebefixierung notwendig wie bei lasercapture mikrodissektionbasierten Techniken, die es ermöglichen, hochwertige RNA zu erhalten.
Die Probenahme aus Subpopulationen von Zellen und die Isolierung der polysomenassoziierten RNA schränke die RNA-Ausbeute jedoch stark ein. Daher ist es notwendig, ausreichend empfindliche Methoden zur Bibliotheksvorbereitung für eine erfolgreiche Datenerfassung durch RNA-seq anzuwenden.
TRAP bietet ein ideales Werkzeug für die Pflanzenforschung, da viele Entwicklungsprozesse zellwandbezogene und mechanische Signalwege beinhalten. Der Einsatz von Promotoren zur Gezielten Anwendung auf bestimmte Zellpopulationen überbrückt die Kluft zwischen Organ- und Einzelzellebene, die wiederum unter geringer Auflösung oder sehr hohen Kosten leiden. Hier wenden wir TRAP zur Untersuchung der Zell-Zell-Kommunikation bei der lateralen Wurzelbildung an.
Einleitung
Angetrieben durch die zunehmende Anwendung von Sequenzierungstechniken der nächsten Generation könnte die räumliche Auflösung in der Entwicklungsbiologie erweitert werden. Zeitgenössische Studien zielen darauf ab, Gewebe bis hin zu spezialisierten Zelltypen, wenn nicht einzellig,1,2,3,4zu sezieren. Zu diesem Zweck wurde in den letzten fünfzig Jahren eine Vielzahl verschiedener Methoden entwickelt (siehe Abbildung 1A)5,6,7,8,9,10,11,12,13,14,15.
Viele Werkzeuge in der Pflanzenwissenschaft waren Anpassungen von Techniken, die in der Tierforschung Pionierarbeit geleistet haben. Dies gilt nicht für die Methode, die wir hier im Detail einführen. Im Jahr 2005, ausgestattet mit einem starken Hintergrund in der Protein-Übersetzung, machte sich das Bailey-Serres Lab daran, ribosomale Proteine für die anschließende Affinitätsreinigung zu entwickeln16. So konnten sie eine zeitaufwändige und arbeitsintensive Polysomenprofilierung vermeiden, die auf einer Ultrazentrifugation mit einem Saccharosegradienten basiert und seit den 1960er Jahren17,,18zur Beurteilung der Übersetzen von Ribosomen verwendet wurde. Das Verfahren wird seitdem als translationale Ribosome-Affinitätsreinigung (TRAP)16bezeichnet. Nach erfolgreichen translatome Studien in Pflanzen, Heiman et al. angepasst TRAP für Tiere19 und andere erweitert seine Anwendung auf Hefe20, Drosophila21, Xenopus22 und Zebrafisch23,24.
Obwohl die genetische Veränderung des Modellsystems eine Voraussetzung für TRAP ist, das seine Anwendung auf Arten beschränkt, die für die genetische Transformation geeignet sind, kann man diesen Einwand gleichzeitig gegen Zielteilmengen von Zellen nutzen, die von besonderem Interesse sind und ansonsten extrem schwierig sind, sich vom intakten Gewebe/Organ25 zu isolieren (z. B. stark verzweigte dendritische Zellen in einem Maushirn oder Pilzhyphen im infizierten Pflanzengewebe). In Pflanzen werden alle Zellen über Zellwände an Ort und Stelle gehalten, die die Grundlage des hydrostatischen Skeletts26bilden. Um eine Pflanzenzelle aus dieser Matrix zu befreien, haben Wissenschaftler die Zelle entweder durch Lasercapture Microdissection (LCM)27 physisch aus ihrem umgebenden Gewebe geschnitten oder die enzymatische Verdauung der Zellwände durchgeführt28. Unter den letztgenannten Zellen, sogenannten Protoplasten, ist die Interessenpopulation fluoreszierend gekennzeichnet und kann durch fluoreszenzaktivierte Zellsortierung (FACS)7abgetrennt werden. LCM erfordert in der Regel, dass eine Probe fixiert und in Wachs eingebettet wird, was letztlich die Qualität ihrer RNA29verschlechtert. FACS-basierte Methoden liefern hochwertige RNA, aber der Protoplasting-Prozess selbst führt zu Unterschieden in der Genexpression30 und Gewebe mit modifizierten und dicken sekundären Zellwänden sind notorisch schwer zu behandeln. Darüber hinaus wird davon ausgegangen, dass viele Entwicklungsprozesse in Anlagen auf mechanisch übertragenen Signalen beruhen und daher die Integrität der Zellwand von größter Bedeutung ist31. Zwei Methoden, die eine Abkürzung verwenden, um die Zellisolation zu umgehen, indem sie auf der Ebene von Nukleii arbeiten, sind fluoreszenzaktivierte nukleare Sortierung (FANS) und Isolierung von Kernen, die in bestimmten Zelltypen (INTACT) markiert sind. Wie in TRAP verwenden sie zelltypspezifische Promotoren, um Kerne zu markieren, die anschließend durch Sortierung angereichert oder heruntergezogen werden, bzw.8,15. Eine große Herausforderung für all diese Ansätze besteht darin, ausreichend RNA-Material aus Teilmengen von Zellen in einem Gewebe zu erhalten. Da TRAP nur einen Bruchteil der zellulären RNAs erfasst, stellt die Probensammlung einen erheblichen Engpass dar. Daher sind besonders sensible Bibliotheksvorbereitungsprotokolle erforderlich, um qualitativ hochwertige Daten aus geringen Eingabemengen zu erzeugen.
Seit seiner Gründung wurde TRAP entweder in Kombination mit DNA-Mikroarrays oder, da die Sequenzierungskosten in den letzten Jahren deutlich gesunken sind, RNA-seq10,32,33verwendet. Eine Vielzahl von Forschungsfragen wurde bereits erläutert, wie in Sablok et al.34begutachtet. Wir sind überzeugt, dass in den kommenden Jahren weitere Berichte folgen werden, da die Technik sehr vielseitig ist, wenn verschiedene Promotoren kombiniert werden, um bestimmte Zelltypen anzusprechen. Schließlich, Dies wird auch in einer induzierbaren Weise getan werden, und kann mit Sondierung der Pflanze Reaktion auf viele biotische und abiotische Stressfaktoren kombiniert werden. Darüber hinaus, wo stabile transgene Linien nicht verfügbar sind, wurden haarige Wurzelexpressionssysteme auch erfolgreich verwendet, um TRAP in Tomaten und medicago35,36durchzuführen.

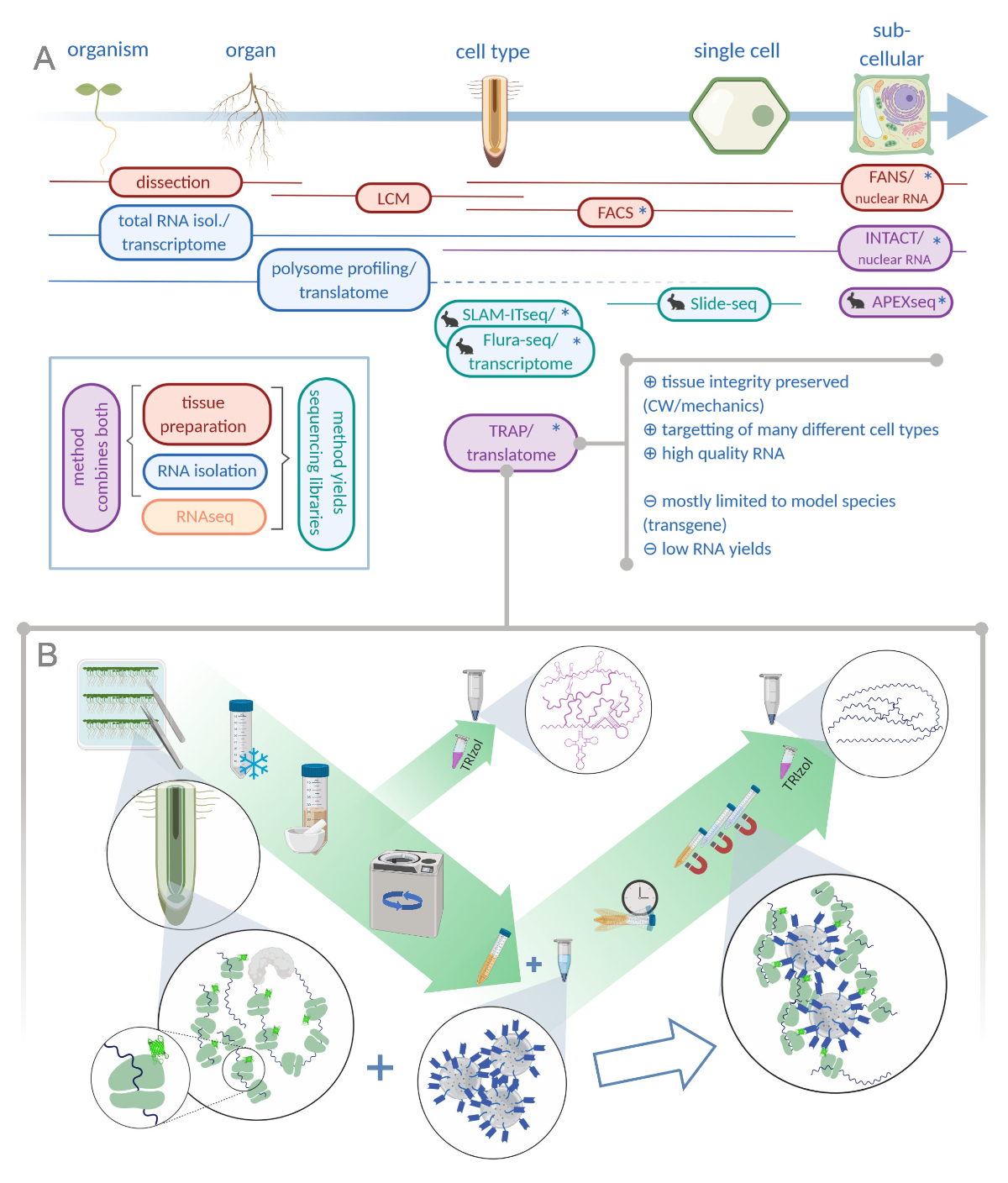
Abbildung 1: Übersetzen der Ribosomenaffinitätsreinigung (TRAP) ergänzt das Analyseportfolio "omics". A. Eine Erhöhung der analytischen Präzision bis hin zur einzelligen oder sogar subzellulären Auflösung kann durch eine Vielzahl von Methoden oder Kombinationen davon erreicht werden. Die Regelung gibt einen Überblick über die derzeit verfügbaren Werkzeuge im Pflanzen- und Tierbereich. Die Gewebesammlung bei zellulärer Auflösung kann durch Protokolle wie LCM oder FACS erreicht werden, die dann an Standard-Transkriptom oder Polysomenprofiling/Translatom-Analyse gekoppelt werden. TRAP und INTACT integrieren sowohl die Gewebeabscheidung als auch die RNA-Isolierung, da sie auf Epitop-Tagging basieren. INTACT-Proben stellen jedoch nur Zellkerne dar und stellen daher einen besonderen Fall der Transkriptomanalyse dar. Ein kleines Kaninchensymbol markiert neu entwickelte Methoden im Tierbereich: Während SLAM-ITseq und Flura-seq auf metabolische Targetting von entstehenden RNAs mit modifizierten Uracil-Basen in Zellen setzen, die das freizügige Enzym exemittieren, verwendet Slide-seq einen beschichteten Glasschlitten mit DNA-Barcodes, die Positionsinformationen im Zellbereich liefern. In APEX-seq wird ein Näherungsetikettierungsansatz verfolgt, um RNAs in bestimmten subzellulären Kompartimenten zu testen. Insbesondere erfordert eine höhere Auflösung oft die Erzeugung von transgenem Material (Sternchen) und diese Methoden werden daher überwiegend für Modellarten verwendet. TRAP eignet sich besonders für pflanzenwissenschaftliche Studien mit Zellwand (CW) oder mechanischer Signalisierung sowie Zellarten, die schwer aus ihrer CW-Matrix freigesetzt werden können. B. Detaillierte Nasslaborschritte des TRAP-Verfahrens: Sämlinge, die GFP-markiertes ribosomales Protein in verschiedenen Zelltypen (z.B. Wurzelendodermis) exzessieren, werden sieben Tage lang auf Petrischalen angebaut und Wurzelmaterial durch Snap Freezing geerntet. Aus dem homogenisierten Rohextrakt wird eine vollständige RNA-Kontrollprobe entnommen, bevor die Trümmer mittels Zentrifugation granuliert werden. Magnetische Anti-GFP-Perlen werden dem gelöschten Extrakt hinzugefügt, um Immunpräzipitation durchzuführen. Nach Inkubation und drei Waschschritten wird die polysome-assoziierte RNA (TRAP/Polysome RNA) direkt über die Phenol-Chlor-Form-Extraktion erhalten. LCM: Lasercapture Mikrodissektion, FACS/FANS: fluoreszenzaktivierte Zell-/Kernsortierung, APEX-seq: Methode basierend auf der entwickelten Ascorbatperoxidase, INTACT: Isolierung von Kernen, die in bestimmten Zelltypen getaggt sind, SLAM-ITseq: Thiol(SH)-verknüpfte Alkylierung zur metabolischen Sequenzierung von RNA im Gewebe, Flura-seq: fluorouracil-markierte RNA-Sequenzierung (Created with Biorender.com)
{kind=link}
Das Ziel dieses Artikels besteht darin, eine detaillierte Beschreibung der TRAP-Methode bereitzustellen, kritische Schritte hervorzuheben und Anleitungen für eine mögliche Bibliotheksvorbereitungsmethode bereitzustellen.
Ein generisches TRAP-Experiment besteht im Wesentlichen aus den folgenden Schritten (siehe auch Abbildung 1B):(1) Vorbereitung von Pflanzenmaterial einschließlich Klonen von Ribosomen-Tagging-Konstrukt, transgener Linienproduktion und -auswahl, Anbau und Füllstoffe von Saatgut, Sterilisation und Beschichtung sowie Stressanwendung/-behandlung (optional) und Gewebeernte; (2) Immunreinigung einschließlich Gewebehomogenisierung und Reinigung des Rohextrakts, Perlenwäsche und Immunreinigung sowie Waschschritte; (3) RNA-Extraktion und Qualitätsbewertung; und (4) Bibliotheksvorbereitung.
Die Arabidopsis-Wurzel ist seit ihrer Einführung als Modellpflanze ein Modellsystem zur Erforschung der Pflanzenentwicklung37,38. Hier wird die Anwendung von TRAP im Kontext der Pflanzen-Lateralwurzelentwicklung präsentiert. Bei Pflanzen beruht der Aufbau des gesamten Wurzelsystems auf der Durchführung dieses Programms und ist daher sehr wichtig für das Überleben des Organismus39. Bei Arabidopsisstammen laterale Wurzeln aus Pericycle-Gewebe, das sich neben Xylemgefäßen befindet und daher als Xylempol-Pericycle (XPP; siehe Abbildung 2C)40bezeichnet wird. Einige XPP-Zellen, die sich tief in der Wurzel befinden, erhalten eine Gründerzellidentität und beginnen sich nach einem lokalen hormonellen Auslöser durch Schwellung und Spaltung antiklinal41zu vermehren. Aufgrund des Vorhandenseins einer starren Zellwandmatrix übt dieser Prozess jedoch mechanische Belastung der umgebenden Gewebe aus. Insbesondere ist die darüber liegende Endodermis betroffen, da sie sich im Weg der seitlichen Wurzelwachstumsachse42,43,44befindet. Tatsächlich muss das sich neu formende Primordium durch die darüber liegende Endodermis-Zelle wachsen (Abbildung 2C2), während Kortex- und Epidermiszellen gerade beiseite geschoben werden, damit das Primordium schließlich45,46entsteht. Jüngste Arbeiten in unserem Labor haben gezeigt, dass die Endodermis aktiv dazu beiträgt, die Proliferation im Pericycle zu bewältigen. Gezielte Blockierung der endodermalen hormonellen Signalisierung reicht aus, um auch die erste Division in den XPP-Zellen47zu hemmen. Daher stellt die Pericycle-Endodermis-Kommunikation einen sehr frühen Checkpoint für die laterale Wurzelentwicklung in Arabidopsisdar. Es ist jedoch nicht bekannt, wie dieser Übersprechen durchgeführt wird. Um dieses Geheimnis zu lüften, haben wir den TRAP-seq-Ansatz gewählt, um XPP- und Endodermale Zellen anzuvisieren. Um uns für Zellen im lateralen Wurzelprogramm anzureichern, imitierten wir den hormonellen Auslöser, indem wir exogen ein Auxin-Analog (1-Naphthalenesessigsäure, NAA)48auftragen, das gleichzeitig erlaubte, die Anfangsphase der lateralen Wurzelbildung zeitlich aufzulösen.
Access restricted. Please log in or start a trial to view this content.
Protokoll
1. Klonen von Transgen, transgenen Linienproduktion und -auswahl
- Klonen Sie den Promotor der Wahl im entsprechenden Eintragsvektor. Verwenden Sie eine rekombinationsbasierte Klonmethode (Materialtabelle) und kombinieren Sie die Promotoren in pDONRP4-P1r. Klonen RPL18 (mit Affinitäts-Tag oder fluoreszierendem Protein der Wahl) unter Verwendung des rekombinationsbasierten Klonens in pDONRP1-P249.
- Kombinieren Sie den Eintragsvektor, der RPL18 enthält, mit dem promoterhaltigen Eintragsvektor in einer Zwei-Fragment-Rekombinationsreaktion in den entsprechenden Zielvektor mit der FAST-roten Selektionskassette50, um die direkte Auswahl transgener Samen zu erleichtern.
- Überprüfen Sie den rekombinierten Vektor durch Sequenzierung und wandeln Sie ihn in geeignete, kompetente Agrobakterien um. Blütendip Arabidopsis Pflanzen und nach 3-4 Wochen Ernte und wählen Sie T1 Samen51.
- Verwenden Sie die Mikroskopie, um gut ausdrückende Linien zu identifizieren und Ausdrucksmuster entsprechend der gemeldeten Promotoraktivität in mehreren unabhängigen Linien zu überprüfen. Wählen Sie Linien aus, die ein repräsentatives Ausdrucksmuster mit einer einzelnen T-DNA-Einfügung anzeigen. Dies könnte helfen, Daseinssalzieren zu minimieren und wird vorteilhaft für genetische Kreuze sein.
- Wählen Sie T3-Nachkommen, die für das Marker-Gen homozygot sind.
2. Vermehrung und Sterilisation
- Zelltypspezifische TRAP isoliert RNA aus einer begrenzten Anzahl von Zielzellen pro Wurzel. Um das benötigte Ausgangsmaterial zu erzeugen, propagieren Sie homozygote Linien. Verwenden Sie zu diesem Zweck Standardwachstumsbedingungen mit besonderem Fokus auf Pilzwachstumskontrolle.
HINWEIS: Wenn einzelne Einfügelinien nicht erhalten werden können, wachsen Chargen in großen Populationen über wenige Generationen, um T-DNA-induzierte transgenerationale Silencing zu vermeiden. - Sterilisieren Sie große Mengen Arabidopsis Samen mit einer Runde Chlorgas und einer Runde von 70% EtOH.
- Die Samen gleichmäßig auf 12 cm x 12 cm quadratische Petrischalen (weniger als 0,3 ml Samen/Platte) verteilen und in einen Trockenbau oder einen anderen geeigneten Behälter stapeln. Vermeiden Sie Klumpen- oder Haufenbildung, da die Samen für das Gas zugänglich sein müssen. Gassterilisation über Nacht mit Bleich- und HCl-Volumen wie berichtet52: 100 ml Bleichmittel (13%) mit 6 ml Conc. HCl in einem 60 L Trockener. Defumigate für mindestens 1 h, bevor die Samen in einem sterilen Behälter gesammelt.
VORSICHT: 37% HCl ist sehr korrosiv und erfordert eine sorgfältige Handhabung. Chlorgas ist giftig, verwenden Sie eine Rauchhaube. - Nehmen Sie 0,1 ml trockene, gassterilisierte Samen pro Platte und mischen Sie sie mit Sterilisationslösung (70% EtOH, 0,01% Tween) bei Raumtemperatur. 20 min inkubieren, EtOH dekanieren und 3-4 Mal mit sterilemH2O waschen.
- Die eingeweichten Samen in 50 ml-Röhrchen geben und mit sterilem 0,1% Agar verdünnen, um 1 ml imbibed Samenschlämme pro Platte (0,1 ml Samen/1 ml Gülle) zu erhalten.
HINWEIS: Aufgrund von Transgenintegrationsereignissen können Anlagenlinien anfällig für verschiedene Sterilisationstechniken sein; insbesondere die EtOH-Inkubationszeit erwies sich als kritisch. In unseren Händen waren die doppelten Sterilisationsschritte notwendig, um Pilzkontaminationen während der Experimente zu vermeiden. Dies ist besonders wichtig, wenn die Durchführung von Zeitreihen als Kontamination eines einzelnen Zeitpunkts das gesamte Experiment behindert. Es kann durchaus sein, dass eine doppelte Sterilisation nicht immer erforderlich ist, abhängig von den lokalen Wachstumsbedingungen.
- Die Samen gleichmäßig auf 12 cm x 12 cm quadratische Petrischalen (weniger als 0,3 ml Samen/Platte) verteilen und in einen Trockenbau oder einen anderen geeigneten Behälter stapeln. Vermeiden Sie Klumpen- oder Haufenbildung, da die Samen für das Gas zugänglich sein müssen. Gassterilisation über Nacht mit Bleich- und HCl-Volumen wie berichtet52: 100 ml Bleichmittel (13%) mit 6 ml Conc. HCl in einem 60 L Trockener. Defumigate für mindestens 1 h, bevor die Samen in einem sterilen Behälter gesammelt.
3. Plating
- Bereiten Sie diese Schritte im Voraus vor. Gießen Sie 1/2 MS-Platten (pH 5.8) mit 1% Agar in den für das Experiment benötigten Mengen (20-30 pro Probe/Zeitpunkt). Schneiden Sie 1 ml Pipettenspitzen, um den Spitzendurchmesser mit einer Rasierklinge auf ca. 3-4 mm zu vergrößern. Autoclave die Tipps. Erstellen Sie einen Schablonenhalter für die Beschichtung von drei Reihen von Samen pro Platte mit quadratischen Petri schale Deckel. Bereiten Sie eine laminare Fließhaube vor, um eine sterile Arbeitsumgebung zu schaffen, und beschriften Sie die zu verarbeitenden Platten.
HINWEIS: Wenn viele Platten gleichzeitig verarbeitet werden, können farbige Etiketten die Beschriftung beschleunigen. - Leere Agarplatten in den Schablonenhalter geben und 1 ml imprägnierte Samen gleichmäßig auf drei Reihen verteilen. Legen Sie die verarbeiteten Platten in Stapeln in den laminaren Fluss, bis die Samen trocken sind (d. h. an der Agaroberfläche kleben). Lassen Sie die Platten nicht länger, da der Agar auch austrocknet.
- Sobald die Samen ausreichend trocken sind, schließen Sie die Deckel und versiegeln Sie jede Platte mit Mikroporenband. Die Samen zwei Tage lang bei 4 °C im Dunkeln stifizieren und anschließend in eine Wachstumskammer geben.
4. Gewebebehandlung (optional)
HINWEIS: In diesem Protokoll skizzieren wir die exogene Behandlung von Arabidopsis-Wurzeln mit der synthetischen Auxin-Variante NAA. Je nach der vorliegenden experimentellen Frage muss dieses Teil angepasst werden oder kann ganz weggelassen werden.
- Bereiten Sie Streifen aus Tissuepapier von 1,5 - 2 cm Höhe und 10 cm Länge vor. Längere Inkubationszeiten erfordern, dass das Gewebe vor der Anwendung autoklaviert wird.
- Entfernen Sie das Mikroporenband von allen Platten, die sich der Hormonbehandlung unterziehen müssen. 1 ml 10 mM NAA (in DMSO gelöst) in 1 L flüssiger, autoklavierter 1/2 MS Lösung (pH 5,8) verdünnen und das Gewebepapier in der Lösung (10 m NAA) einweichen.
- Verwenden Sie eine Pinzette, um einen Streifen Tissuepapier auf jede Wurzelreihe aufzutragen. Verwenden Sie vorsichtig Finger, um Luftblasen zu entfernen. Entleeren Sie überschüssige Flüssigkeit aus der Platte, schließen Sie den Deckel und beschriften Sie die Platte mit der Zeit. Für längere Inkubationszeiten die Platten wieder in die Wachstumskammer legen.
5. Ernte
- Abrufen von Platten für jede biologische Replikation/Zeitpunkt/Behandlung. Sammeln Sie flüssigen Stickstoff in einem sauberen Dewar-Gefäß und Etikettenröhrchen (15 oder 50 ml) für die verschiedenen Gewebeproben. Bereiten Sie einen Styroporhalter vor.
VORSICHT: Machen Sie sich mit flüssigen Stickstoffhandhabungsverfahren vertraut (Belüftung, Frostbisse, potenziell explodierende Rohre). - Öffnen Sie die Platte und entfernen Sie das Tissuepapier mit Zangen, wobei Sie darauf achten, die Wurzeln nicht von der Agaroberfläche zu lösen. Mit einer chirurgischen Klinge einmal pro Reihe entlang der Triebwurzel-Kreuzung in einem einzigen, entschlossenen Schlag schneiden. Reinigen Sie die Klingen zwischen den Proben und tauschen Sie häufig, um Schärfe zu gewährleisten.
- Streichen Sie mit einer Pinzette entlang der Wurzeln jeder Reihe, um sie in drei Bündeln zu sammeln. Schnappen Sie sich die Wurzeln und leeren Sie sie in ein 50 ml Rohr mit flüssigem Stickstoff gefüllt, um gefrieren.
HINWEIS: Versuchen Sie nicht, Wurzeln in dichte Strukturen (wie Kugeln) zu montieren, da sie im nächsten Schritt schwer zu schleifen sind. - Fahren Sie mit allen Platten fort, die eine Probe bilden (in der Reihenfolge der Inkubationszeiten) und gießen Sie überschüssigen flüssigen Stickstoff aus. Verwenden Sie den Rohrdeckel, um ein Verschütten der Wurzeln zu verhindern. Schließen Sie dann den Deckel und sammeln Sie alle Rohre im Dewar-Gefäß. Das Wurzelgewebe bei -80 °C lagern.
6. Immunreinigung
HINWEIS: Dieser Schritt zielt darauf ab, hochwertige TRAP/Polysomen-RNA zu erhalten. Befolgen Sie daher strikt die Ratschläge der guten Praxis für die HANDHABUNG von RNA. Führen Sie alle Schritte in diesem Abschnitt in einer sterilen Bank aus und reinigen Sie alle Geräte und Laborgeräte mit einer RNase-Entfernungslösung(Materialtabelle). Tragen Sie Handschuhe und wechseln Sie sie sofort, wenn sie mit Proben, Eis oder anderen Quellen kontaminiert sind, die nicht gereinigt wurden. Da dies ein sehr wichtiger Aspekt ist, ist ein Abschnitt über die Wiederverwendung von Geräten zusammen mit der Abfallentsorgungsberatung enthalten.
- Puffervorbereitung
- Bereiten Sie Lagerlösungen nach Tabelle 1 und Autoklav (A) oder Filtersterilisation () vor. Sofern nicht anders angegeben, ist das Lösungsmittel RNase-freies Wasser.
- Dithiothreitol (DTT), Phenylmethylsulfonylfluorid (PMSF), Cycloheximid (CHX) und Chloramphenicol (CAM) in ihren Table 1 jeweiligen Lösungsmitteln auflösen und bei -20 °C lagern. Alle anderen Bestände können bei Raumtemperatur bleiben.
- Die Vorräte vormischen - mit den Zutaten 1-4 für Waschpuffer (WB) und 1-6 für Polysomenextraktionspuffer (PEB) - um eine zeitaufwändige Puffermischung vor jeder Extraktion zu vermeiden. Fügen Sie also nur Wasser und die gefrorenen Zutaten (7-10) am Tag der Extraktion hinzu. Halten Sie die vorgemischten Bestände und RNase-freies Wasser bei 4 °C.
HINWEIS: Die DTT-Konzentration beträgt 1/5 der gemeldeten Konzentration von Zanetti et al. 2005, da die Nanokörper-Wechselwirkung mit dem GFP empfindlich gegenüber hohen DTT-Konzentrationen ist.
| Zutaten | Lagerkonzentration | Volumen in ml für 50 ml WB* hinzufügen | Volumen in ml für 50 ml PEB hinzufügen* | ||
| 1 | Tris, pH 9 | Eine | 2 M | 5 | 5 |
| 2 | Kcl | Eine | 2 M | 5 | 5 |
| 3 | EGTA | Eine | 0,5 M | 2.5 | 2.5 |
| 4 | MgCl2 | Eine | 1 M | 1.75 | 1.75 |
| 5 | Pte | Eine | 20% (v/v) | 0 | 2.5 |
| 6 | Waschmittelmischung | Eine | 0 | 2.5 | |
| Tween 20 | 20% (v/v) | ||||
| Triton-X 100 | 20% (v/v) | ||||
| Brij-35 | 20% (mit/v) | ||||
| Igepal | 20% (v/v) | ||||
| 7 | Dtt | ₳ | 0,5 M | 0.1 | 0.1 |
| 8 | PMSF | ₳ | 0,1 M (Isopropanol) | 0.5 | 0.5 |
| 9 | Cycloheximid | ₳ | 25 mg/ml (EtOH) | 0.1 | 0.1 |
| 10 | Chloramphenicol | ₳ | 50 mg/ml (EtOH) | 0.05 | 0.05 |
Tabelle 1: Pufferzusammensetzung und Mischberatung. Zutaten mit den angegebenen Stoffkonzentrationen, die in den angegebenen Mengen gemischt werden, ergeben 50 ml WB oder PEB. Tris: Tris-(Hydroxymethyl)-Aminomethan, EGTA: Ethylenglykol-Bis(-Aminoethylether)-N,N,N',N'-Tetra-Essigsäure, PTE: Polyoxyethylen-(10)-Tridecylether, A: Autoklav, *Bis zu 50 ml mit RNase-freiem Wasser füllen.
- Gewebehomogenisierung/Schleifen
- Zentrifuge abkühlen und Homogenisatoren und Zentrifugenrohre auf Eis legen. Thaw Aliquots von DTT, PMSF, CHX und CAM. PeB und WB aus den Lagerlösungen in 50 ml-Rohren nach Tagesanforderungen mischen und auf Eis abkühlen lassen.
HINWEIS: Fügen Sie PMSF erst kurz vor Gebrauch hinzu, da die Halbwertszeit von PMSF in Wasser nur 30 min beträgt. - Bereiten Sie viel flüssigen Stickstoff in einem Dewar-Gefäß vor und holen Sie Gewebeproben aus -80 °C-Lagerung ab. Tragen Sie Baumwollhandschuhe unter den Standard-Laborhandschuhen, um Verbrennungen durch kalte Mörtel zu verhindern. Gießen Sie flüssigen Stickstoff in Mörtel und Stöße, bis sie kalt genug sind, um das Schleifen zu ermöglichen. Es wird empfohlen, ein System zur Unterscheidung von Mörtel zu entwickeln (Etikett oder in einer bestimmten Reihenfolge zu halten).
- Leere Gewebeprobe in einen Mörtel und sorgfältig mahlen, bis das gesamte Material ein weißes Pulver ist. Fügen Sie bei Bedarf flüssigen Stickstoff hinzu, um das Gewebe gefroren zu halten oder um ein besseres Schleifen zu erleichtern.
- Fügen Sie der Probe 5 ml PEB hinzu und mischen Sie es schnell mit dem Pulver, bevor der Puffer einfriert. Während dieses Beispiel auftaut (von Zeit zu Zeit mischen) verarbeiten Sie eine andere Probe.
- Sobald die Mischung übertragen werden kann, die Gülle in einen Glashomogenisator entleeren und auf Eis halten. Mit zusätzlichen 2 ml PEB den Mörtel und den Stößel abspülen und in die Probe im Homogenisator geben.
HINWEIS: Vermeiden Sie eine vollständig flüssige Probe, da dies den RNA-Abbau ermöglicht. - Die Gülle manuell mahlen, bis der Extrakt homogen ist. Wir empfehlen mindestens 4 bis 5 Tauchgänge.
HINWEIS: Es kann einige zusätzliche Wartezeiten erfordern, damit die Gülle weiter auftauen kann. Der Umgang mit Homogenisatoren erfordert einige Sorgfalt. Wenden Sie keine brachiale Gewalt an und achten Sie auf Saugkräfte. Wenn dies nicht berücksichtigt wird, führt dies zu Verschüttung, Kontamination oder Zerstörung des Homogenisators. - Gießen Sie den Rohwurzelextrakt in ein 50 ml Zentrifugenrohr (auf Eis bleiben).
HINWEIS: In der Regel können mehrere Proben vor der Übertragung gemahlen werden. Paralleles Handling des Schleifens, Übertragens und Homogenisierens ist erforderlich. Versuchen Sie, schnell zu arbeiten, aber nicht überstürzen; ruhig bleiben. Homogenisierte Proben immer auf Eis halten.
- Zentrifuge abkühlen und Homogenisatoren und Zentrifugenrohre auf Eis legen. Thaw Aliquots von DTT, PMSF, CHX und CAM. PeB und WB aus den Lagerlösungen in 50 ml-Rohren nach Tagesanforderungen mischen und auf Eis abkühlen lassen.
- Gesamte RNA-Probensammlung
- Übertragen Sie 200 L Aliquots jeder Rohprobe in ein sauberes Mikrozentrifugenrohr (vorher auf Eis beschriftet und gekühlt).
- Fahren Sie mit der RNA-Extraktion fort, wie sie für TRAP-Proben in den Punkten 7.1 und 7.2 beschrieben ist. Führen Sie diese Schritte aus, während proben in der Zentrifuge gelichtet wird.
- Führen Sie eine DNase-Behandlung mit der resuspendierten Gesamt-RNA durch, um DNA-Kontamination zu beseitigen und die Reaktion mit einem kommerziellen Kit (Table of Materials )zu bereinigen.
HINWEIS: Die Gesamt-RNA-Extraktionen führen in der Regel zu hohen Konzentrationen, und Proben müssen erheblich verdünnt werden. Wir empfehlen, die Konzentration nach Verdünnung durch das empfindliche Qubit-Protokoll zu messen.
- Clearing des Rohextrakts
- Nehmen Sie den Eiskübel mit Proben von 6.2.7 und zentrifugieren Sie ihn 15 min bei 16.000 x g und 4 °C.
HINWEIS: Um die Zentrifuge auszugleichen, paaren Sie Proben entsprechend. Falls dies nicht vollständig möglich ist, passen Sie eine Probe durch Hinzufügen von PEB an. - Gießen Sie den Überstand in ein frisches Zentrifugenrohr (vorher auf Eis gekühlt) und wiederholen Sie die Zentrifugation (15 min bei 16.000 x g und 4 °C). Diese Übertragung kann schnell neben der Zentrifuge durchgeführt werden.
- Während der Rohextrakt abräumt, initiieren Sie das Waschen von GFP-Perlen für Schritt 6.6.
HINWEIS: Bewahren Sie diesen Eiskübel zum Schaukeln auf dem Shaker auf, aber legen Sie ihn nicht wieder in die sterile Bank, da er kontaminiert sein könnte.
- Nehmen Sie den Eiskübel mit Proben von 6.2.7 und zentrifugieren Sie ihn 15 min bei 16.000 x g und 4 °C.
- Perlenwäsche
- Aliquot magnetische GFP-Perlen (#samples x 60 l, Materialtabelle) in ein 1,5 ml-Rohr. Auf den magnetischen Ständer stellen. Sobald die Perlen gesammelt haben, entfernen Sie den Überstand.
- Fügen Sie 1 ml kalte WB, die Perlen wieder aussetzen und sammeln Sie sie wieder. Entsorgen Sie den Waschpuffer und wiederholen Sie ihn noch einmal mit 1 ml WB.
- Schließlich, die Perlen in WB auf das ursprüngliche Volumen in Schritt 6.5.1 verwendet wieder aussetzen.
- Immunreinigung (IP)
- Unmittelbar nach der Zentrifugation den gereinigten Überstand in beschriftete 15 ml-Rohre gießen und 60 l gewaschene Perlen pro Probe hinzufügen.
- Legen Sie alle Proben horizontal in den Eiskübel und legen Sie sie auf einen Shaker. Lassen Sie die Mischung für 2 h brüten, um die GFP-markierten Polysomen an die Perlen zu binden.
- Sammeln Sie die Perlen auf dem magnetischen Ständer für 15 ml Rohre (auf Eis) und fügen Sie PMSF zu den verbleibenden PEB. Entsorgen Sie den Überstand. Gießen Sie ca. 5 ml PEB auf die Perlen und suspendieren Sie sie durch Kippen. Schütteln Sie die Proben für 15 min im gleichen Setup wie in Abschnitt 6.6.2.
- Wiederholen Sie die Wässer mit WB auf insgesamt 3 Wässerungen (1 x PEB, 2 x WB). Fügen Sie vor jedem Pufferaustausch PMSF hinzu.
- Sammeln Sie die Perlen in 1 ml WB und übertragen Sie sie in ein 1,5 ml Rohr. Schließlich sammeln Sie die Perlen noch einmal auf dem magnetischen Ständer und entfernen Sie alle Flüssigkeit. Schließen Sie die Röhre und halten Sie auf Eis, bis alle Proben verarbeitet werden.
- Transportieren Sie die Proben zu einer Dunstabzugshaube für die RNA-Extraktion.
- Abfallentsorgung und Instandsetzung von Laborbedarf.
- Bei guter Laborpraxis (siehe Abschnitt 2.2.1) ergibt das Sterilisationsverfahren eine wässrige NaCl-Lösung. Lassen Sie das Chlorgas sowie die Reste HCl und Bleichmittel in der Dunstabzugshaube abziehen.
- PEB- und WB-Entsorgung: Wenn CHX bei hohem pH-Wert zersetzt, alle Flüssigkeiten sammeln und zu pH>9 bringen. Entsorgen Sie die flüssigen Abfälle in den halogenierten chemischen Abfällen. Alle Feststoffe (Gewebe, serologische Pipetten, Handschuhe usw.) sollten als chemische Abfälle entsorgt werden.
- Sammeln Sie phenolhaltige Flüssigkeiten getrennt sowie phenolverseuchtes Material (Spitzen, Schläuche und Handschuhe).
- Handwäsche Mörtel, Stößel und Homogenisatoren (Schwämme und Bürste) mit Seife und gründlich abspülen. Anschließend das Material bei >220 °C über Nacht backen. Entweder vor der Behandlung in Zinnfolie wickeln oder in einen hitzebeständigen, abgedeckten Behälter geben.
- Bürsten sierende Zentrifugenrohre mit Reinigungsmittel und anschließend Diethylpyrocarbonat (DEPC)-Behandlung in der Dunstabzugshaube. Zu diesem Zweck flüssiges DEPC zu entionisiertem Wasser (1 ml DEPC bis 1 Lh2O) hinzufügen und über Schütteln mischen. Legen Sie die Zentrifugenrohre auf eine autoklavierbare Schale, die verschüttetes DEPC-Wasser fängt. Gießen Sie die Suspension in die Rohre und lassen Sie für 3 h oder über Nacht. DEPC zersetzt sich im nachfolgenden Autoklavierprozess.
VORSICHT: DEPC ist hochgiftig.
7. RNA-Extraktion und QC
- RNA-Extraktion
- Die Tischzentrifuge auf 4 °C abkühlen.
- Fügen Sie 1 ml Säure-Guanidinium-Phenol-basiertes Reagenz (Materialtabelle) zu jeder Probe hinzu, invertieren Sie die Perlen oder die gesamte RNA-Schlämme und brüten sie für 5 min auf Eis. Wirbel nicht!
- Fügen Sie 200 l Chloroform hinzu und brüten Sie 3 min auf Eis. Dann wirbeln die Proben gründlich aus.
- Zur Unterstützung der Phasentrennung zentrifugieren Sie zentrifugieren Sie bei max. Geschwindigkeit für 10-15 min, 4 °C.
- Etikettieren Sie jeweils 1,5 ml Retentionsarme Rohre (Materialtabelle) und aliquot 650 l Isopropanol.
- Nehmen Sie die obere wässrige Phase (ca. 650 l) vorsichtig ein und übertragen Sie sie mit Isopropanol auf die vorbereiteten Rohre. Vermeiden Sie es, die rosa organische Phase zu berühren.
- ÜBER Nacht bei -20 °C die RNA ausfäliert.
HINWEIS: Es wird empfohlen, die Proben in Isopropanol bei -20 °C oder -80 °C zu lagern und bei Bedarf nur in Wasser zu löslich. Wässrige RNA verschlechtert sich sogar bei -80 °C, wenn sie wochenlang/monatelang gelagert wird.
- RNA-Niederschlag
- Die Tischzentrifuge auf 4 °C abkühlen.
- Frisches 80% EtOH mit RNase-freiem Wasser vorbereiten und bei -20 °C abkühlen (5 min bei -80 °C helfen, den Prozess zu beschleunigen).
- Zentrifugieren Sie die Proben mit maximaler Geschwindigkeit (ca. 13.000 x g) für 30 min und entsorgen Sie den Überstand. Das Pellet wird nicht sichtbar sein, so sorgfältig Pipette, als ob es dort war. 1 ml kalte 80% EtOH hinzufügen und das Rohr ein oder zwei Mal invertieren.
- Zentrifugieren Sie wieder für 30 min bei maximaler Geschwindigkeit und wiederholen Sie die Wäsche auf insgesamt zwei Wäschen.
- Drehen Sie 2 min nach unten und entfernen Sie alle Reste EtOH mit einer 10-L-Spitze. Lassen Sie das Pellet 3-5 min (nicht mehr) bei Raumtemperatur trocknen und in 20 l RNase-freiem Wasser wieder aufhängen.
- Halten Sie die Proben auf Eis und führen Sie die Qualitätskontrolle so schnell wie möglich durch. Bewahren Sie die Proben bei -80 °C auf. Vermeiden Sie Gefrier-Tau-Zyklen.
- Qualitätskontrolle mit speziellen Geräten (Materialtabelle) gemäß den Empfehlungen des Herstellers.
8. Bibliotheksvorbereitung
- cDNA-Synthese und -Verstärkung mit dem SMARTer v4 Ultra Low Input RNA Kit
- Berechnen Sie die Verdünnung jeder Probe mit 1,5 ng TRAP-RNA oder Gesamt-RNA in einem Volumen von 4,75 l. Führen Sie alle Reaktionen in PCR-Röhren durch und verdünnen Sie Proben mit frischen Aliquots rNase-freiem Wasser.
- Führen Sie alle Schritte gemäß den Empfehlungen des Herstellers mit 1/2 der Reaktionsvolumina aus. Verstärken Sie die cDNA mit 12-13 PCR-Zyklen.
- Bereinigen Sie die PCR, indem Sie 0,5 l 10x Lysepuffer und 25 'L SPRI-Perlen (Materialtabelle)hinzufügen. Wenn viele Proben verarbeitet werden Lyse Puffer und Perlen können vorgemischt werden. Stellen Sie sicher, dass die Perlen vor dem Pipetieren gleichmäßig verteilt sind.
- Fahren Sie mit dem Protokoll in vollen Reaktionsvolumina (17 L Elutionspuffer) fort. Lassen Sie die Perlen nicht mehr als 3 Minuten trocknen. Überdrocknete Proben können möglicherweise durch längere Inkubationszeiten gerettet werden.
- Messen Sie die Probenkonzentrationen mit dem Qubit HS DNA Kit.
HINWEIS: Das SMARTer v4 Kit kann bis zu 200 pg Eingang tolerieren. Wir haben Bibliotheken in Fällen erhalten, in denen Qubit-Werte nicht ermittelt werden konnten (unter 250 pg, Erkennungsgrenze) mit einer 16-Zyklus-PCR. Das begrenzte Eingabematerial kann jedoch auch zu weniger komplexen Bibliotheken führen.
- Fragmentierung und Adapterligation PCR mit dem Nextera XT DNA Library Preparation Kit
- Verdünnen Sie die cDNA mit RNase-freiem Wasser, um eine Konzentration von 200 pg/l und eine Pipette 1,25 l in einem PCR-Rohr zu erhalten.
- Führen Sie alle Schritte nach Hersteller mit 1/4 der Reaktionsvolumina. Verstärken Sie die cDNA mit 12 PCR-Zyklen und kompatiblen Adaptern für die Samples, die zu einem Sequenzierungspool gehören. Mit den Index Kits A und D von Illumina können bis zu 384 Samples multiplexiert werden.
- Für die PCR-Bereinigung fügen Sie 12,5 L Resuspensionspuffer und 22,5 l SPRI-Perlen (0,9-faches Verhältnis) hinzu. Elute die Probe mit 22 L Elutionspuffer.
HINWEIS: QC und Pooling wurde von der Sequenzierungsfirma (Tabelle der Materialien) durchgeführt und daher war keine perlenbasierte Normalisierung erforderlich. Die enzymatische Fragmentierungsreaktion (Tagmentation) ist sehr empfindlich gegenüber Materialinputs, da jedes Enzym nur einmal schneidet. Überschreiten Sie daher nicht die Konzentrationsempfehlung.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Zur Qualitätsbewertung sollte das oben genannte Verfahren in mehreren Zwischenschritten untersucht werden: Expressionsmustervalidierung in Planta, Qualitätskontrolle der isolierten polysomalen RNA sowie der Endbibliotheken. qRT-PCR mit bekannten Markergenen kann darüber hinaus durchgeführt werden, um die Reaktion auf den Behandlungszustand zu bestätigen oder die experimentellen Bedingungen zu optimieren.
Konfokale A...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Überprüfung des RPL18-Lokalisierungsmusters
Entscheidend, um eine Fehlinterpretation von Daten aus jedem TRAP-Experiment zu vermeiden, ist das richtige Ausdrucksmuster der markierten ribosomalen Untereinheit. Daher ermöglicht die Aufnahme von GFP als Epitop-Tag in RPL18 sehr elegant die Überprüfung des gewünschten Expressionsmusters und nacheinander den Pulldown der Polysomenfraktion aus demselben Gewebe. Invasivere Ansätze zur Sicherstellung der richtigen Promotormuster folgen jiao und Mayerow...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Wir danken Jean-Claude Walser vom Genetic Diversity Center Zürich für die entscheidende fachkundige Beratung in der frühen Phase dieses Projekts. Die Arbeit im Vermeer-Labor wurde durch ein SNF-Professurstipendium (PP00P3_157524) und ein R'EQUIP-Ausrüstungsstipendium (316030_164086) des Schweizerischen Nationalfonds (SNF) unterstützt, das jeMV verliehen wurde.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| Sterilization | |||
| bleach, 13% | Sigma | 71696 | |
| beaker | VWR | 214-1172/74/75 | |
| desiccator with porcelaine plate (DURAN) | Sigma/Merck | Z317454-1EA/Z317594-1EA | |
| EtOH, p.a. | Honeywell | 02860-1L | |
| HCl, 37% | Roth | 4625.1 | |
| Tween 20 | Sigma | P9416 | |
| Plate growth + harvesting | |||
| MS salts, basal salt mixture, incl. MES buffer | Duchefa | M0254 | |
| agar plant for cell culture | Applichem/Panreac | A2111.1000 | |
| DMSO | Sigma | D4540 | |
| forcepts | Rubis Switzerland | 5-SA model | |
| KOH | Fluka | 60370 | |
| micropore/surgical tape | 3M | 1530-0 | |
| NAA | Duchefa | N0903 | |
| petri dishes 120x120 mm | Greiner bio-one | 688102 | |
| scalpel | VWR/Swann-Morton | 233-5454 | |
| tissues, neutral, two-layered | any supplier of your choice | ||
| Immunoprecipitation | |||
| GFP-beads: gtma-100 GFP-Trap_MA | Chromotek | e.g. gtma-100 | |
| Brij-35 | Sigma | P1254-500G | |
| centrifuge tubes (in accordance with centrifuge) | Beckman Coulter | 357001 | |
| Chloramphenicol | Applichem | C0378-25G | |
| cotton gloves | VWR | 113-7355 | |
| Cycloheximide, HPLC grade | Sigma | 01810-1G | |
| DEPC | VWR | E174 | might have long delivery times |
| DTT | Fluka | 43815 | |
| EGTA | Sigma | 3054.3 | |
| homogenizers DUALL 23 | KONTES GLASS CO (via VWR) | SCERSP885450-0023 (set) | SCERSP885451-0023 pestle only - SCERSP885452-0023 cylinder only; long delivery times |
| Igepal CA-360 | Sigma | I3021-100ml | |
| KCl | Sigma | 60130 | |
| MgCl2 hexahydrat | Roth | 2189.2 | |
| mortar and pestle | VWR | 470148-960 & 470019-978 | |
| PMSF | Roche | 10 837 091 001 | |
| Polyoxyethylene-(10)-tridecylether/PTE | Sigma | P2393-500G | |
| RNase-free water | Roth | T143.3 | |
| RNAZap | Thermo Fisher | AM9780/AM9782 | for cleaning surfaces |
| Tris, >99.3% | Roth | AE15.3 | |
| Triton X-100 | Fluka | T8787-250ml | |
| Tween 20 | Sigma | P9416-100ml | |
| RNA extraction | |||
| 2-Propanol, p.a. | Sigma | 33539-1L-GL-R | |
| Chloroform, HPLC grade | Scharlau | CL02181000 | |
| EtOH, p.a. | Honeywell | 02860-1L | |
| low-retention microcentrifuge tubes, 1.5 ml | Eppendorf/Sigma | Z666548-250EA | LoBind |
| RNase-free DNase set | Qiagen | 79254 | |
| RNeasy MiniElute Cleanup Kit | Qiagen | 74204 | |
| TRIzol reagent | ThermoFisher/Ambion | 15596018 | |
| Library preparation | |||
| 15/50 mL Tube Magnetic Separator | Abraxis | PN 472250 | |
| AMPure beads | Beckman Coulter | A63881 | |
| Index Kit A | Illumina | FC-131-2001 | |
| Index Kit D | Illumina | FC-131-2004 | |
| neodymium magnets | Amazon/other | 6 x 1.5 mm range: N42 (NdFeB) | |
| Nextera XT kit | Illumina | FC-131-1024/1096 | https://emea.support.illumina.com/ |
| PCR strips | ThermoScientific | AB-0266 | |
| SMARTer v4 kit | Takara Bioscience | 634892 | https://www.takarabio.com/ |
| Bioanalyzer | Agilent | 2100 Bioanalyzer Instrument | specialized equipment for RNA/DNA quality control |
| Tapestation | Agilent | 4200 Tapestation Instrument | specialized equipment for RNA/DNA quality control |
| Fragment Analyzer | Agilent | 5400 Fragment Analyzer System | specialized equipment for RNA/DNA quality control (high throughput) |
| LabChip | PerkinElmer | LabChip GX Touch Nucleic Acid Analyzer | specialized equipment for RNA/DNA quality control (high throughput) |
| Qubit 4 Fluorometer | ThermoFisher | Q33239 | specialized equipment for RNA/DNA concentration determination |
| qRT-PCR | |||
| GATA23 | Microsynth | fwd: AGTGAGAATGAA AGAAGAGAAGGG; rev: GTGGCTGCGAAT AATATGAATACC | |
| GH3.3 | Microsynth | fwd: CAAACCAATCCT CCAAATGAC; rev: ACTTATCCGCAA CCCGACT | |
| LBD29 | Microsynth | fwd: TCTCCAACAACA GGTTGTGAAT; rev: AAGGAGCCTTAG TAGTGTCTCCA | |
| UBC21 | Microsynth | fwd: TGCGACTCAGGG AATCTTCT; rev: TCATCCTTTCTT AGGCATAGCG | |
| SsoAdvanced Universal SYBR Green | Bio-Rad | #172-5270 | |
| iScript Adv cDNA Kit | Bio-Rad | #172-5038 | |
| miscellaneous | |||
| Falcon tubes 15 ml, Cellstar | Greiner bio-one | 188261 | |
| Falcon tubes 50 ml, Cellstar | Greiner bio-one | 210261 | |
| filter tips 1 ml | Axygen | TF-1000-R-S | |
| filter tips 10 µl | Axygen | TF-10-R-S | |
| filter tips 100 µl | Axygen | TF-100-R-S | |
| filter tips 20 µl | Axygen | TF-20-R-S | |
| filter tips 200 µl | Axygen | TF-200-R-S | |
| microcentrifuge tubes 1.5 ml | SARSTEDT | 72.690.001 | |
| Propidium iodide | Sigma | P4170-100MG | |
| sequencing company | Novogene | en.novogene.com |
Referenzen
- Van Verk, M. C., Hickman, R., Corné, M. J., Pieterse, M., Van Wees, S. C. RNA-Seq: Revelation Of The Messengers. Trends In Plant Science. 18 (4), 175-179 (2013).
- Libault, M., Pingault, L., Zogli, P., Schiefelbein, J. Plant Systems Biology At The Single-Cell Level. Trends In Plant Science. 22 (11), 949-960 (2017).
- Mustroph, A., et al. Profiling Translatomes Of Discrete Cell Populations Resolves Altered Cellular Priorities During Hypoxia In Arabidopsis. Proceedings Of The National Academy Of Sciences Of The United States Of America. 106 (44), 18843-18848 (2009).
- Karve, R., Iyer-Pascuzzi, A. S. Digging Deeper: High-Resolution Genome-Scale Data Yields New Insights Into Root Biology. Current Opinion In Plant Biology. 24, 24-30 (2015).
- Warner, J. R., Knopf, P. M., Rich, A. A Multiple Ribosomal Structure In Protein Synthesis. Proceedings of The National Academy of Sciences of The United States of America. 49 (1), 122-129 (1963).
- Gautam, V., Sarkar, A. K. Laser Assisted Microdissection, An Efficient Technique To Understand Tissue Specific Gene Expression Patterns And Functional Genomics In Plants. Molecular Biotechnology. 57 (4), 299-308 (2015).
- Bargmann, B. O. R., Birnbaum, K. D. Fluorescence Activated Cell Sorting Of Plant Protoplasts. Journal of Visualized Experiments. (36), e1673(2010).
- Deal, R. B., Henikoff, S. The Intact Method For Cell Type-Specific Gene Expression And Chromatin Profiling In Arabidopsis Thaliana. Nature Protocols. 6 (1), 56-68 (2011).
- Dougherty, J. D. The Expanding Toolkit Of Translating Ribosome Affinity Purification. The Journal of Neuroscience: The Official Journal Of The Society For Neuroscience. 37 (50), 12079-12087 (2017).
- Mustroph, A., Juntawong, P., Bailey-Serres, J. Isolation Of Plant Polysomal mRNA By Differential Centrifugation And Ribosome Immunopurification Methods. Methods in Molecular Biology. 553, 109-126 (2009).
- Matsushima, W., et al. SLAM-ITseq: Sequencing Cell Type-Specific Transcriptomes Without Cell Sorting. Development. 145 (13), (2018).
- Basnet, H., et al. Flura-Seq Identifies Organ-Specific Metabolic Adaptations During Early Metastatic Colonization. Elife. 8, (2019).
- Rodriques, S. G., et al. Slide-Seq: A Scalable Technology For Measuring Genome-Wide Expression At High Spatial Resolution. Science. 363 (6434), 1463-1467 (2019).
- Fazal, F. M., et al. Atlas Of Subcellular RNA Localization Revealed By Apex-Seq. Cell. 178 (2), 473-490 (2019).
- Slane, D., Bayer, M. Cell Type-Specific Gene Expression Profiling Using Fluorescence-Activated Nuclear Sorting. Plant Gene Regulatory Networks: Methods And Protocols. Kaufmann, K., Mueller-Roeber, B. , Springer. New York, NY. 27-35 (2017).
- Zanetti, M. E., Chang, I. F., Gong, F., Galbraith, D. W., Bailey-Serres, J. Immunopurification Of Polyribosomal Complexes Of Arabidopsis For Global Analysis Of Gene Expression. Plant Physiology. 138 (2), 624-635 (2005).
- King, H. A., Gerber, A. P. Translatome Profiling: Methods For Genome-Scale Analysis Of mRNA Translation. Briefings In Functional Genomics. 15 (1), 22-31 (2016).
- Mašek, T., Valášek, L., Pospíšek, M. Polysome Analysis And RNA Purification From Sucrose Gradients. RNA: Methods And Protocols. Nielsen, H. , Humana Press. Totowa, NJ. 293-309 (2011).
- Heiman, M., et al. A Translational Profiling Approach For The Molecular Characterization Of Cns Cell Types. Cell. 135 (4), 738-748 (2008).
- Halbeisen, R. E., Scherrer, T., Gerber, A. P. Affinity Purification Of Ribosomes To Access The Translatome. Methods. 48 (3), 306-310 (2009).
- Thomas, A., et al. A Versatile Method For Cell-Specific Profiling Of Translated mRNAs In Drosophila. Plos One. 7 (7), e40276(2012).
- Watson, F. L., et al. Cell Type-Specific Translational Profiling In The Xenopus Laevis Retina. Developmental Dynamics. 241 (12), 1960-1972 (2012).
- Lam, P. Y., Harvie, E. A., Huttenlocher, A. Heat Shock Modulates Neutrophil Motility In Zebrafish. Plos One. 8 (12), e84436(2013).
- Fang, Y., et al. Translational Profiling Of Cardiomyocytes Identifies An Early Jak1/Stat3 Injury Response Required For Zebrafish Heart Regeneration. Proceedings Of The National Academy Of Sciences Of The United States Of America. 110 (33), 13416-13421 (2013).
- Mustroph, A., Zanetti, M. E., Girke, T., Bailey-Serres, J. Isolation And Analysis Of mRNAs From Specific Cell Types Of Plants By Ribosome Immunopurification. Methods In Molecular Biology. 959, 277-302 (2013).
- Monshausen, G. B., Gilroy, S. Feeling Green: Mechanosensing In Plants. Trends In Cell Biology. 19 (5), 228-235 (2009).
- Day, R. C., Grossniklaus, U., Macknight, R. C. Be More Specific! Laser-Assisted Microdissection Of Plant Cells. Trends In Plant Science. 10 (8), 397-406 (2005).
- Sheen, J. Signal Transduction In Maize And Arabidopsis Mesophyll Protoplasts. Plant Physiology. 127 (4), 1466-1475 (2001).
- Datta, S., et al. Laser Capture Microdissection: Big Data From Small Samples. Histology And Histopathology. 30 (11), 1255-1269 (2015).
- Birnbaum, K., et al. A Gene Expression Map Of The Arabidopsis Root. Science. 302 (5652), 1956(2003).
- Hamant, O., Haswell, E. S. Life Behind The Wall: Sensing Mechanical Cues In Plants. BMC Biology. 15 (1), 1354(2017).
- Vragović, K., et al. Translatome Analyses Capture Of Opposing Tissue-Specific Brassinosteroid Signals Orchestrating Root Meristem Differentiation. Proceedings of The National Academy of Sciences of the United States of America. 112 (3), 923-928 (2015).
- Wang, Y., Jiao, Y. Translating Ribosome Affinity Purification (Trap) For Cell-Specific Translation Profiling In Developing Flowers. Methods In Molecular Biology. 1110, 323-328 (2014).
- Sablok, G., Powell, J. J., Kazan, K. Emerging Roles And Landscape Of Translating mRNAs In Plants. Frontiers in Plant Science. 8, 1443(2017).
- Ron, M., et al. Hairy Root Transformation Using Agrobacterium Rhizogenes As A Tool For Exploring Cell Type-Specific Gene Expression And Function Using Tomato As A Model. Plant Physiology. 166 (2), 455-469 (2014).
- Reynoso, M. A., et al. Evolutionary Flexibility In Flooding Response Circuitry In Angiosperms. Science. 365 (6459), 1291-1295 (2019).
- Dolan, L., et al. Cellular Organisation Of The Arabidopsis Thaliana Root. Development. 119 (1), 71(1993).
- Ristova, D., Barbez, E. Root Development. , Springer. New York, NY. (2018).
- Shekhar, V., Stӧckle, D., Thellmann, M., Vermeer, J. E. M. The Role Of Plant Root Systems In Evolutionary Adaptation. Current Topics in Developmental Biology. 131, 55-80 (2019).
- Malamy, J. E., Benfey, P. N. Down And Out In Arabidopsis: The Formation Of Lateral Roots. Trends in Plant Science. 2 (10), 390-396 (1997).
- de Smet, I., et al. Bimodular Auxin Response Controls Organogenesis In Arabidopsis. Proceedings of the National Academy of Sciences of The United States of America. 107 (6), 2705-2710 (2010).
- Péret, B., et al. Arabidopsis Lateral Root Development: An Emerging Story. Trends In Plant Science. 14 (7), 399-408 (2009).
- Vilches-Barro, A., Maizel, A. Talking Through Walls: Mechanisms Of Lateral Root Emergence In Arabidopsis Thaliana. Current Opinion in Plant Biology. 23, 31-38 (2015).
- Porco, S., et al. Lateral Root Emergence In Arabidopsis Is Dependent On Transcription Factor Lbd29 Regulation Of Auxin Influx Carrier Lax3. Development. 143 (18), 3340-3349 (2016).
- Stoeckle, D., Thellmann, M., Vermeer, J. E. Breakout-Lateral Root Emergence In Arabidopsis Thaliana. Current Opinion in Plant Biology. 41, 67-72 (2018).
- Banda, J., et al. Lateral Root Formation In Arabidopsis: A Well-Ordered Lrexit. Trends in Plant Science. 24 (9), 826-839 (2019).
- Vermeer, J. E. M., et al. A Spatial Accommodation By Neighboring Cells Is Required For Organ Initiation In Arabidopsis. Science. 343 (6167), 178-183 (2014).
- Vanneste, S., et al. Cell Cycle Progression In The Pericycle Is Not Sufficient For Solitary Root/Iaa14-Mediated Lateral Root Initiation In Arabidopsis Thaliana. The Plant Cell. 17 (11), 3035-3050 (2005).
- Marques-Bueno, M. M., et al. A Versatile Multisite Gateway-Compatible Promoter And Transgenic Line Collection For Cell Type-Specific Functional Genomics In Arabidopsis. The Plant Journal : For Cell and Molecular Biology. 85 (2), 320-333 (2016).
- Shimada, T. L., Shimada, T., Hara-Nishimura, I. A Rapid And Non-Destructive Screenable Marker, Fast, For Identifying Transformed Seeds Of Arabidopsis Thaliana. The Plant Journal : For Cell and Molecular Biology. 61 (3), 519-528 (2010).
- Clough, S. J., Bent, A. F. Floral Dip: A Simplified Method For Agrobacterium Mediated Transformation Of Arabidopsis Thaliana. The Plant Journal. 16 (6), 735-743 (1998).
- Lindsey, B. E., Rivero, L., Calhoun, C. S., Grotewold, E., Brkljacic, J. Standardized Method For High-Throughput Sterilization Of Arabidopsis Seeds. Journal Of Visualized Experiments. (128), e56587(2017).
- Andersen, T. G., et al. Diffusible Repression Of Cytokinin Signalling Produces Endodermal Symmetry And Passage Cells. Nature. 555, 529-533 (2018).
- Schroeder, A., et al. The Rin: An Rna Integrity Number For Assigning Integrity Values To Rna Measurements. BMC Molecular Biology. 7, 3(2006).
- Vragović, K., Bartom, E., Savaldi-Goldstein, S. Quantitation Of Cell Type-Specific Responses To Brassinosteroid By Deep Sequencing Of Polysome-Associated Polyadenylated RNA. Methods in Molecular Biology. 1564, 81-102 (2017).
- Bertin, B., Renaud, Y., Aradhya, R., Jagla, K., Junion, G. Trap-Rc, Translating Ribosome Affinity Purification From Rare Cell Populations Of Drosophila Embryos. Journal Of Visualized Experiments. (103), e52985(2015).
- Livak, K. J., Schmittgen, T. D. Analysis Of Relative Gene Expression Data Using Real-Time Quantitative PCR And The 2(-Delta Delta C(T)) Method. Methods. 25 (4), 402-408 (2001).
- Jiao, Y., Meyerowitz, E. M. Cell-Type Specific Analysis Of Translating Rnas In Developing Flowers Reveals New Levels Of Control. Molecular Systems Biology. 6, 419(2010).
- Tian, C., et al. A Gene Expression Map Of Shoot Domains Reveals Regulatory Mechanisms. Nature Communications. 10 (1), 141(2019).
- Townsley, B. T., Covington, M. F., Ichihashi, Y., Zumstein, K., Sinha, N. R. Brad-Seq: Breath Adapter Directional Sequencing: A Streamlined, Ultra-Simple And Fast Library Preparation Protocol For Strand Specific mRNA Library Construction. Frontiers in Plant Science. 6, 366(2015).
- Song, Y., et al. A Comparative Analysis Of Library Prep Approaches For Sequencing Low Input Translatome Samples. BMC Genomics. 19 (1), 696(2018).
- Basu, D., Haswell, E. S. Plant Mechanosensitive Ion Channels: An Ocean Of Possibilities. Current Opinion in Plant Biology. 40, 43-48 (2017).
- Brady, S. M., et al. A High-Resolution Root Spatiotemporal Map Reveals Dominant Expression Patterns. Science. 318 (5851), 801(2007).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten