
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Перевод Рибосома сродство очищения (TRAP) для расследования Arabidopsis thaliana Корневая развития в масштабе типа клеток
В этой статье
Резюме
Перевод рибосомной очистки сродства (TRAP) дает возможность вскрыть программы развития с минимальной обработкой органов и тканей. Протокол дает высококачественную РНК из клеток, ориентированных на зеленый флуоресцентный белок (GFP) помечены рибосомной субъединицы. Инструменты анализа вниз по течению, такие как qRT-PCR или RNA-seq, раскрывают профили экспрессии тканей и клеток.
Аннотация
В этой статье мы даем практические инструкции для получения транслатомных данных от различных типов корневых клеток Arabidopsis thaliana с помощью метода очистки сродства рибосомы (TRAP) и последовательной оптимизированной подготовки библиотеки с низким уровнем ввода.
В качестве исходного материала, мы используем растительные линии, которые выражают GFP-тегами рибосомного белка RPL18 в клеточном типе-специфического образом с помощью адекватных промоутеров. Перед иммуноочищением и экстракцией РНК ткань замораживается, что сохраняет целостность тканей и одновременно позволяет проводить исследования временных рядов с высоким временным разрешением. Примечательно, что структуры клеточной стенки остаются нетронутыми, что является основным недостатком в альтернативных процедурах, таких как флуоресценция активированных клеток сортировки на основе подходов, которые полагаются на ткани протоптертодляя изолировать различные популяции клеток. Кроме того, не требуется фиксация тканей, как в лазерных захвата микродиссекции на основе методов, что позволяет высококачественной РНК быть получены.
Однако отбор проб из субпопуляций клеток и только изоляция полисососообразной РНК серьезно ограничивает урожайность РНК. Поэтому необходимо применять достаточно чувствительные методы подготовки библиотекдля для успешного получения данных РНК-сек.
TRAP предлагает идеальный инструмент для исследования растений, так как многие процессы развития включают клеточные стены, связанные с механическими и механическими сигнальными путями. Использование промоутеров для целевых конкретных популяций клеток преодолевает разрыв между органом и одноклеточным уровнем, которые, в свою очередь, страдают от небольшого разрешения или очень высоких затрат. Здесь мы применяем TRAP для изучения клеточной связи в боковом корневом образовании.
Введение
Благодаря все более широкому применению методов секвенирования следующего поколения, пространственное разрешение в биологии развития может быть увеличено. Современные исследования направлены на вскрытие тканей вплоть до специализированных типов клеток, если не одноклеточного уровня1,,2,,3,,4. С этой целью за последние пятьдесят лет было разработано множество различных методов (см. рисунок 1A)5,6,6,7,,88,9,,10,,11,,12,,13,,14,,15.
Многие инструменты в науке о растениях были адаптацией методов, которые были впервые в исследованиях на животных. Это не относится к методу, который мы вводим в деталях здесь. В 2005 году, оснащенный сильным фоном в переводе белка, Лаборатория Бейли-Серрес приступила к разработке рибосомных белков для последующего очищения сродства16. Таким образом, они могли бы избежать трудоемких и трудоемких полисомного профилирования, которое основано на ультрацентрифугации с градиентом сахарозы и было использовано для оценки перевода рибосом с 1960-хгодов 17,18. Метод с тех пор был назван переводной рибосомы очистки сродства (TRAP)16. После успешных исследований транслатома в растениях, Heiman et al. адаптированы TRAP для животных19 и другие расширили свое применение дрожжей20, Drosophila21, Xenopus22 и зебрафиш23,24.
Хотя генетическая модификация модельной системы является необходимым условием для TRAP, которая ограничивает ее применение видов, поддавшихся генетической трансформации, можно одновременно использовать это возражение в целевых подмножествах клеток, которые представляют особый интерес и в противном случае чрезвычайно трудно изолировать от нетронутой ткани / органа25 (например, высоко разветвленные дендритные клетки в мозге мыши или грибковых hyphae инфицированных растительных тканей). В растениях все клетки удерживаются на месте через клеточные стенки, которые составляют основу гидростатического скелета26. Чтобы освободить клетку растений из этой матрицы, ученые либо физически вырезать клетку из окружающей ткани с помощью лазерного захвата микродиссекции (LCM)27 или выполняется ферментативное пищеварение клеточныхстенок 28. Среди последних клеток, так называемых протопластов, популяция, интересующая сяртово,констачированная и может быть отделена с помощью флуоресцентной сортировки клеток (FACS)7. LCM обычно требует, чтобы образец был исправлен и встроен в воск, что в конечном итоге ухудшает качество его РНК29. Методы на основе FACS дают высококачественную РНК, но сам процесс протоптертора вводит различия в экспрессии генов30, а ткани с модифицированными и толстыми вторичными клеточными стенками, как известно, трудно поддаются лечению. Кроме того, многие процессы развития в растениях, как предполагается, полагают, полагаются на механически передаваемые сигналы и, следовательно, целостность клеточной стенки имеет первостепенное значение31. Два метода, которые используют ярлык, чтобы обойти изоляцию клеток, работая на уровне ядер, являются флуоресценция активированной ядерной сортировки (FANS) и изоляции ядер помечены в конкретных типах клеток (INTACT). Как и в TRAP, они используют клеточные тип-специфические промоутеры для того чтобы маркировать ядра, которые затем получают обогащенными через сортировать или вытягивать вниз, соответственно8,,15. Основная проблема для всех этих подходов заключается в том, чтобы получить достаточное количество РНК-материала из подмножества клеток в ткани. Поскольку TRAP захватывает лишь часть клеточных РНК, сбор образцов является значительным узким местом. Поэтому для получения высококачественных данных из низких объемов ввода необходимы особенно чувствительные протоколы подготовки библиотек.
С момента своего создания, TRAP был либо использован в сочетании с ДНК microarrays или, как секвенирование расходы значительно снизились в последние годы, РНК-сек10,32,33. Множество вопросов исследования уже были выяснены, как рассмотрено в Sablok и др.34. Мы убеждены, что больше докладов будет следовать в ближайшие годы, как техника является очень универсальным при объединении различных промоутеров для целевых конкретных типов клеток. В конце концов, это будет сделано даже в индуцируемой образом, и может быть объединена с зондирования реакции растения на многие биотических и абиотических факторов стресса. Кроме того, там, где стабильные трансгенные линии отсутствуют, волосатые системы экспрессии корней также успешно используются для выполнения TRAP в томате и medicago35,36.

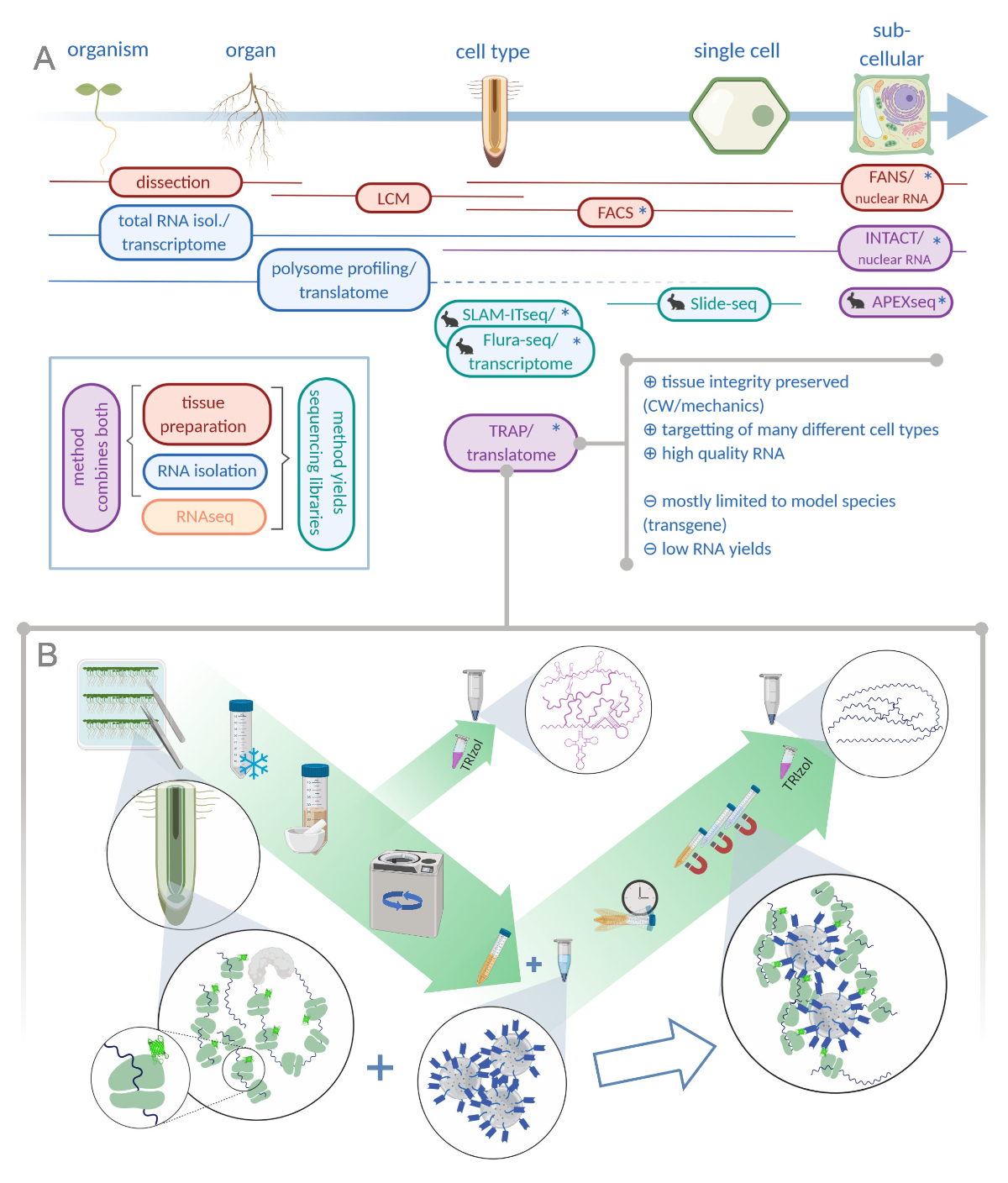
Рисунок 1: Перевод циризующей сродства рибосомы (TRAP) дополняет портфель анализа "омики". О. Повышение уровня аналитической точности, вплоть до одноклеточного или даже субклеточного разрешения может быть достигнуто с помощью множества методов или их комбинаций. Схема дает обзор имеющихся в настоящее время инструментов в области растений и животных. Сбор тканей при клеточном разрешении может быть достигнут с помощью таких протоколов, как LCM или FACS, которые затем соединяются со стандартным транскриптомом или полисомным профилированием/анализом транслатома. TRAP и INTACT интегрируют захват тканей и изоляцию РНК, поскольку они основаны на эпитоп-пометке. Тем не менее, образцы INTACT только ядра клеток и представляет собой, следовательно, особый случай анализа транскриптома. Небольшой значок кролика отмечает недавно разработанные методы в области животных: В то время как SLAM-ITseq и Flura-seq полагаются на метаболическое тарзание зарождающихся РНК с модифицированными базами урацила в клетках, выражающих разрешительный фермент, Slide-seq использует стеклянный слайд с покрытием со штрих-кодами ДНК, которые предоставляют позиционную информацию в клеточном диапазоне. В APEX-seq используется подход к маркировке непосредственной маркировки для пробы РНК в конкретных субклеточных отсеках. Примечательно, что повышенное разрешение часто требует генерации трансгенного материала (звездочек), и эти методы, таким образом, в основном используются для типовых видов. TRAP особенно подходит для исследований растительной науки с участием клеточной стенки (CW) или механической сигнализации, а также клеточных видов, которые трудно выпустить из их матрицы CW. B. Детальные шаги влажной лаборатории процедуры TRAP: Рассада, выражающая GFP-тегами рибосомный белок в различных типах клеток (например, корневые эндодермисы), выращиваются на чашках Петри в течение семи дней, а корневой материал собирают путем замораживания оснастки. Общий образец контроля РНК собирается из гомогенизированного сырого экстракта, прежде чем гранулировать мусор с помощью центрифугации. Магнитные бусы анти-GFP добавляются в очищенный экстракт для выполнения иммунопротеберации. После инкубации и трех этапов стирки, полисомо-ассоциированной РНК (TRAP /полисома РНК) непосредственно получается через экстракцию фенола-хлороформа. LCM: лазерный захват микродиссекции, FACS/ FANS: флуоресценция активированной ячейки / ядерной сортировки, APEX-seq: метод, основанный на инженерии аскорбат перекистой, INTACT: изоляция ядер помечены в конкретных типах клеток, SLAM-ITseq: тиол (SH)-связанных алкилирования для метаболического секвенирования РНК в тканях, Flura-seq: фторурацил помечены РНК секвенирования (Создано с Biorender.com) Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}
Целью данной статьи является предоставление подробного описания метода TRAP, выделение критических шагов и руководство по возможному методу подготовки библиотеки.
Общий эксперимент TRAP будет по существу состоять из следующих шагов (см. также Рисунок 1B):(1) Подготовка растительного материала, включая клонирование рибосом-теговая конструкция, трансгенное производство линии и отбор, выращивание и ссыпая семян, стерилизации и покрытия, а также стрессовое применение/обработка (необязательно) и сбор тканей; (2) иммуноочищение, включая гомогенизацию тканей и очистку сырого экстракта, мытье биса и иммуноочищение, и шаги мытья; (3) добыча РНК и оценка качества; и (4) подготовка библиотеки.
Корень Arabidopsis была модельсистемы для изучения развития растений с момента его введения в качестве модели завода37,38. Здесь применение TRAP демонстрируется в контексте развития бокового корня растений. В растениях, накопление всей корневой системы опирается на выполнение этой программы и поэтому очень важно для выживания организма39. В Arabidopsis, боковые корни происходят из ткани перицикл, который находится рядом с ксилем судов и, следовательно, называется ксилем полюс перицикл (XPP; см. Рисунок 2C)40. Некоторые клетки XPP, которые расположены глубоко внутри корня, приобретают идентичность ячейки основателя и, на местном гормональном триггере, начинают размножаться путем отеков и деления антиклинально41. Однако, из-за наличия жесткой матрицы стенки клетки, этот процесс оказывает механическое давление на окружающие ткани. В частности, поражена надлежащая эндодермис, так как она находится на пути боковой оси роста корня42,,43,,44. Действительно, вновь формирующихся primordium придется расти через надлежащую клетку эндодермис(Рисунок 2C2) в то время как коры и эпидермиз клетки просто отодвинуты в сторону для приморского,наконец,выйти45,46. Недавняя работа в нашей лаборатории показала, что эндодермис активно способствует распространению в перицикле. Целенаправленной блокировки эндодермальной гормональной сигнализации достаточно, чтобы ингибировать даже самое первое деление в клетках XPP47. Таким образом, перицикл эндодермис связи представляет собой очень ранний контрольно-пропускной пункт для бокового развития корней в Arabidopsis. Это, однако, не известно, как этот перекрестный разговор выполняется. Чтобы разгадать эту тайну, мы выбрали trap-seq подход к целевой XPP и эндодермальных клеток. Чтобы обогатить клетки в боковой корневой программе, мы имитировали гормональный триггер, экзогенно применяя аналог auxin (1-нафталенеацетической кислоты, NAA)48, который в то же время позволил временно решить начальную фазу бокового образования корня.
протокол
1. Клонирование трансгенного, трансгенного производства и отбора линий
- Клонпромоутер выбора в соответствующем векторе входа. Используйте метод клонирования на основе рекомбинации(Таблица материалов)и рекомбинируйте промоутеров в pDONRP4-P1r. Clone RPL18 (с тегом сродства или флуоресцентным белком выбора) с помощью рекомбинационного клонирования в pDONRP1-P249.
- Объедините вектор входа, содержащий RPL18 с промоутер-содержащим вектором входа в двух фрагментной реакции рекомбинации в соответствующий вектор назначения с fast-red selection cassette50 для облегчения прямого отбора трансгенных семян.
- Проверьте рекомбинобъединенный вектор путем секвенирования и преобразуйте его в подходящие, компетентные агробактерии. Цветок окунуться Arabidopsis растений и после 3-4 недель урожая и выбрать T1 семена51.
- Используйте микроскопию для выявления хорошо выражающихся линий и проверки моделей выражения в соответствии с сообщениями о деятельности промоутера в нескольких независимых линиях. Выберите строки, показывающие репрезентативную модель выражения с одной вставкой T-DNA. Это может помочь свести к минимуму молчание и будет выгодно для генетических крестов.
- Выберите потомство T3, которое является гомозиготным для гена маркера.
2. Распространение и стерилизация
- Cell type-specific TRAP изолирует РНК от ограниченного числа целевых ячеек на корень. Для генерации необходимого исходного материала распространяйте гомозиготные линии. С этой целью используйте стандартные условия роста с особым акцентом на грибковые меры контроля роста.
ПРИМЕЧАНИЕ: Если одиночные линии вставки не могут быть получены, расти партий в больших популяциях в течение нескольких поколений, чтобы избежать T-ДНК-индуцированной трансгенерации глушить. - Стерилизовать большое количество семян арабидопсиса с одним раундом газа хлора и один раунд 70% EtOH.
- Равномерно распределите семена равномерно на 12 см х 12 см квадратных посуды Петри (менее 0,3 мл семян/плиты) и укладывайте их в обезвреженный или другой подходящий контейнер. Избегайте сгусток или кучи образования, как семена должны быть доступны для газа. Выполните стерилизации газа на ночь с отбеливателем и HCl объемов, как сообщается52: 100 мл отбеливателя (13%) с 6 мл conc. HCl в 60 L desiccator. Defumigate, по крайней мере 1 ч, прежде чем собирать семена в стерильных контейнерах.
ВНИМАНИЕ: 37% HCl очень коррозионный и требует тщательного обращения. Хлор газ является токсичным, использовать дым капот. - Возьмите 0,1 мл сухих, газстерилизованных семян на тарелку и смешайте их с раствором стерилизации (70% EtOH, 0,01% Tween) при комнатной температуре. Инкубировать в течение 20 мин, декантEt EtOH и мыть семена 3-4 раза со стерильным H2O.
- Перенесите пропитанные семена в 50 мл трубок и разбавьте стерильным 0,1% агара, чтобы получить 1 мл впитываемой семян суспензии на тарелку (0,1 мл семян / 1 мл суспензии).
ПРИМЕЧАНИЕ: Из-за трансгенных событий интеграции, растительные линии могут быть восприимчивы к различным методам стерилизации; особенно было установлено, что время инкубации EtOH имеет решающее значение. В наших руках, двойные шаги стерилизации были необходимы, чтобы избежать грибкового загрязнения во время экспериментов. Это особенно важно при выполнении временных рядов, так как загрязнение одной временной точки затрудняет весь эксперимент. Вполне возможно, что двойная стерилизация не всегда необходима, в зависимости от местных условий выращивания.
- Равномерно распределите семена равномерно на 12 см х 12 см квадратных посуды Петри (менее 0,3 мл семян/плиты) и укладывайте их в обезвреженный или другой подходящий контейнер. Избегайте сгусток или кучи образования, как семена должны быть доступны для газа. Выполните стерилизации газа на ночь с отбеливателем и HCl объемов, как сообщается52: 100 мл отбеливателя (13%) с 6 мл conc. HCl в 60 L desiccator. Defumigate, по крайней мере 1 ч, прежде чем собирать семена в стерильных контейнерах.
3. Покрытие
- Подготовьте эти шаги заранее. Налейте 1/2 пластины MS (pH 5.8) с 1% агаром в количествах, необходимых для эксперимента (20-30 на образец/точку времени). Вырезать 1 мл пипетки советы, чтобы увеличить диаметр кончика до около 3-4 мм с лезвием бритвы. Автоклав советы. Создайте держатель шаблона для покрытия трех рядов семян на тарелку с квадратными крышками чашки Петри. Подготовьте ламинарный капот потока, чтобы обеспечить стерильную рабочую среду и пометить пластины для обработки.
ПРИМЕЧАНИЕ: Если многие пластины обрабатываются в то же время, цветные этикетки могут ускорить маркировку. - Поместите пустые агарпластинки в держатель шаблона и равномерно распределите 1 мл впитанных семян на три ряда. Поместите обработанные пластины в стеках в ламинарный поток до тех пор, пока семена не высохнут (т.е. прилипните к поверхности агара). Не оставляйте пластины дольше, как агар высохнет, а также.
- Как только семена достаточно сухие, закройте крышки и запечатать каждую тарелку с микропор лентой. Стратить семена в течение двух дней при 4 градусах По Цельсию в темноте, а затем поместить их в камеру роста.
4. Лечение тканей (по желанию)
ПРИМЕЧАНИЕ: В этом протоколе мы излагаем экзогенное лечение корней арабидопсис с синтетическим вариантом auxin NAA. В зависимости от экспериментального вопроса, эта часть должна быть скорректирована или может быть полностью опущена.
- Приготовьте полоски из бумажной ткани высотой 1,5 - 2 см и 10 см в длину. Расширенные сроки инкубации требуют, чтобы ткань была аутоклавирована перед использованием.
- Удалите микропор ленту со всех пластин, которые должны пройти гормональное лечение. Разбавить 1 мл 10 мМ NAA (растворяется в DMSO) в 1 л жидкости, autoclaved 1'2 MS раствор (pH 5.8) и замочить бумагу в растворе (10 мкм NAA).
- Используйте пинцет, чтобы нанести полоску бумажной ткани на каждый ряд корней. Аккуратно используйте пальцы, чтобы удалить пузырьки воздуха. Пустая лишная жидкость из пластины, закрыть крышку и этикетки пластины со временем. Для продолжительного инкубации поместите пластины обратно в камеру роста.
5. Сбор урожая
- Извлекать пластины для каждой биологической репликации / точки времени / лечения. Сбор жидкого азота в чистом сосуде Dewar и этикетки труб (15 или 50 мл) для различных образцов тканей. Подготовьте держатель пенополистирола.
ВНИМАНИЕ: Ознакомьтесь с процедурами обработки жидкого азота (аэрация, обморожения, потенциально взрывающиеся трубы). - Откройте тарелку и удалите салфетку с помощью щипц, стараясь не отсоединить корни от поверхности агара. С хирургическим лезвием, вырезать один раз в ряд вдоль стрелять корень-соединение в одном, определяется инсульта. Очистите лезвия между образцами и часто обменивайся, чтобы гарантировать резкость.
- С помощью пинцета проведите пальцем по корням каждого ряда, чтобы собрать их в три пучка. Захватите корни и опорожните их в трубку 50 мл, наполненную жидким азотом, чтобы заморозить.
ПРИМЕЧАНИЕ: Не пытайтесь собрать корни в плотные структуры (например, шары), поскольку они трудно измельчить в следующем шаге. - Продолжить со всеми пластинами, которые составляют один образец (в порядке инкубационного времени) и вылить избыток жидкого азота. Используйте крышку трубки, чтобы предотвратить пролитие корней. Затем закройте крышку и соберите все трубки в сосуде Dewar. Храните корневую ткань при -80 градусах Цельсия.
6. Иммуноочищение
ПРИМЕЧАНИЕ: Этот шаг направлен на получение высококачественной TRAP/полисомного РНК. Поэтому строго следуйте рекомендациям по хорошей практике обработки РНК. Выполните все шаги в этом разделе в стерильной скамейке и очистить все оборудование и лабораторное оборудование с RNase удаления решения (Таблица материалов). Носите перчатки и немедленно измените их при загрязнении образца, льда или других источников, которые не были очищены. Поскольку это очень важный аспект, в него включен раздел о повторном использовании оборудования вместе с рекомендациями по удалению отходов.
- Подготовка буфера
- Подготовьте биржевые решения в соответствии с таблицей 1 и автоклавом (A) или фильтром стерилизовать (я). Если иное не указано, растворитель является RNase-свободной воды.
- Растворите и аликвот дитиотритол (DTT), фенилметилсулфолил фторид (PMSF), циклогексимид (CHX) и хлорамфеникол (CAM) в их соответствующих растворителях, как указано в таблице 1, и хранят их при -20 градусов по Цельсию. Все остальные запасы могут оставаться при комнатной температуре.
- Предварительно перемешайте запасы - с ингредиентами 1-4 для буфера для мытья (WB) и 1-6 для полисомного буфера извлечения (PEB) - чтобы избежать многовремени буферсмешиваниа до каждой добычи. Таким образом, только добавить воду и замороженные ингредиенты (7-10) в день извлечения. Держите предварительно смешанные запасы и безRизу воды на 4 градуса Цельсия.
ПРИМЕЧАНИЕ: Концентрация DTT составляет 1/5 от зарегистрированной концентрации от Занетти и др. 2005, так как взаимодействие наивного с GFP чувствительно к высоким концентрациям ДТТ.
| Ингредиенты | Концентрация запасов | Добавить объем в мЛ для 50 мл ВБ | Добавить объем в мЛ для 50 мл ПЭБЗ | ||
| 1 | Трис, рН 9 | A | 2 M | 5 | 5 |
| 2 | Kcl | A | 2 M | 5 | 5 |
| 3 | EGTA | A | 0,5 м | 2.5 | 2.5 |
| 4 | MgCl2 | A | 1 M | 1.75 | 1.75 |
| 5 | Pte | A | 20% (v/v) | 0 | 2.5 |
| 6 | моющее средство смесь | A | 0 | 2.5 | |
| Твина 20 | 20% (v/v) | ||||
| Тритон-X 100 | 20% (v/v) | ||||
| Брий-35 | 20% (w/v) | ||||
| Игепал | 20% (v/v) | ||||
| 7 | Dtt | ₳ | 0,5 м | 0.1 | 0.1 |
| 8 | PMSF | ₳ | 0,1 М (изопропанол) | 0.5 | 0.5 |
| 9 | Циклогексид | ₳ | 25 мг/мл (EtOH) | 0.1 | 0.1 |
| 10 | Хлорамфеникол | ₳ | 50 мг/мл (EtOH) | 0.05 | 0.05 |
Таблица 1: Композиция буфера и советы по смешиванию. Ингредиенты с заданном запасом, смешанные в данных количествах, дают 50 мл ВБ или ПЭБ. Tris: tris-(hydroxymethyl)-аминометан, EGTA: этилен гликоль-бис (З-аминоэтил эфир)-N,N,N,N',N',N'-тетра-ацетической кислоты, PTE: Полиоксиэтилен-(10)-tridecyl эфир, A: автоклав, Заполните до 50 мл с rNase-свободной водой.
- Ткань гомогенизации / шлифовка
- Охладите центрифугу и поместите на лед гомогенизаторы и центрифуги. Оттепель aliquots DTT, PMSF, CHX и CAM. Смешайте PEB и WB из стоковых растворов в трубах мощностью 50 мл в соответствии с требованиями дня (в составе образцов) и охладите на льду.
ПРИМЕЧАНИЕ: Добавить PMSF только незадолго до использования, так как период полураспада PMSF в воде составляет всего 30 минут. - Приготовьте большое количество жидкого азота в сосуде Dewar и извлеките образцы тканей из -80 градусов по Цельсию. Носите хлопчатобумажные перчатки под стандартными лабораторными перчатками, чтобы предотвратить ожоги от холодных минометов. Налейте жидкий азот в ступки и пестики, пока они не остынут достаточно, чтобы позволить шлифовальные. Рекомендуется разработать систему различения минометов (маркировка или хранение в определенном порядке).
- Пустые образцы ткани в ступку и тщательно измельчить, пока весь материал белый порошок. При необходимости добавьте жидкий азот, чтобы сохранить ткань замороженной или облегчить лучшую шлифовку.
- Добавьте 5 мл PEB в образец и быстро смешайте с порошком до того, как буфер замерзнет. В то время как этот образец оттаивает (смешивать время от времени) процесс другой образец.
- Как только смесь может быть передана, опорожните суспензию в стеклянный гомогенизатор и держите на льду. С дополнительным2 мл PEB, промыть раствор и пестик и добавить его в образец в гомогенизатор.
ПРИМЕЧАНИЕ: Избегайте полностью жидкого образца, так как это позволяет деградации РНК. - Измельчить суспензию вручную, пока экстракт не станет однородным. Мы рекомендуем как минимум от 4 до 5 погружений.
ПРИМЕЧАНИЕ: Это может потребовать некоторого дополнительного времени ожидания, чтобы суспензия оттаивать дальше. Обработка гомогенизаторов требует некоторого осмотрительности. Не применяйте грубую силу и остерегайтесь сил всасывания. Если не принимать во внимание, это приведет к разливу, загрязнению или уничтожению гомогенизатора. - Налейте сырой экстракт корня в 50 мл центрифуги трубки (держать на льду).
ПРИМЕЧАНИЕ: Обычно несколько образцов могут быть измельчена перед передачей. Требуется параллельное обращение с шлифовкой, передачей и гомогенизирующим. Старайтесь работать быстро, но не спешите; сохранять спокойствие. Всегда держите однородные образцы на льду.
- Охладите центрифугу и поместите на лед гомогенизаторы и центрифуги. Оттепель aliquots DTT, PMSF, CHX и CAM. Смешайте PEB и WB из стоковых растворов в трубах мощностью 50 мл в соответствии с требованиями дня (в составе образцов) и охладите на льду.
- Общий сбор образцов РНК
- Передача 200 аликотов каждого сырого образца в чистую микроцентрифуговую трубку (помеченную и охлажденный на льду заранее).
- Продолжить экстракцию РНК, как подробно для образцов TRAP в точках 7.1 и 7.2. Делайте эти шаги, пока образцы очищаются в центрифуге.
- Выполните лечение DNase с переподвеской общей РНК для устранения загрязнения ДНК и очистки реакции с помощью коммерческого комплекта (Таблица материалов).
ПРИМЕЧАНИЕ: Общий объем извлечения РНК обычно дает высокие концентрации и образцы должны быть значительно разбавлены. Мы рекомендуем измерять концентрацию после разбавления по чувствительному протоколу Зубита.
- Очистка сырого экстракта
- Возьмите ведро со льдом с образцами от 6.2.7 и центрифуги их в течение 15 мин при 16000 х г и 4 КС.
ПРИМЕЧАНИЕ: Чтобы сбалансировать центрифугу, пара образцов соответственно. В случае, если это не совсем возможно, отрегулируйте один образец, добавив PEB. - Налейте супернатант в свежую центрифугу трубки (охлажденный на льду заранее) и повторите центрифугирование (15 мин при 16000 х г и 4 кв. м). Этот перенос можно быстро выполнить рядом с центрифугой.
- В то время как сырой экстракт очистки, инициировать стирку GFP-бусы для шага 6.6.
ПРИМЕЧАНИЕ: Держите это ведро льда для качания на шейкере, но не место обратно в стерильной скамейке, как это может быть загрязнено.
- Возьмите ведро со льдом с образцами от 6.2.7 и центрифуги их в течение 15 мин при 16000 х г и 4 КС.
- Бисовые стирки
- Аликвот магнитные GFP-бусы (#samples х 60 л, Таблица материалов) в трубку 1,5 мл. Место на магнитной подставке. После того, как шарики собрались, удалить супернатант.
- Добавьте 1 мл холодного ВБ, приостановите бисер и соберите их снова. Отбросьте буфер стирки и повторите еще раз с 1 мл ВБ.
- В конечном счете, повторное присоединение шариков в ВБ до первоначального объема, используемого в шаге 6.5.1.
- Иммуноочищение (IP)
- Сразу же после центрифугации, залить очищенный супернатант в помечены 15 мл труб и добавить 60 л промытых бусин на образец.
- Поместите все образцы горизонтально в ведро со льдом и положите его на шейкер. Пусть смесь инкубировать в течение 2 ч для того, чтобы связать GFP-маркированных полисомы с бисером.
- Соберите бусины на магнитной подставке для 15 мл труб (на льду) и добавить PMSF к остальным PEB. Отбросьте супернатант. Налейте около 5 мл PEB на бисер и resuspend их путем наклона. Встряхните образцы в течение 15 минут в той же установке, что и в разделе 6.6.2.
- Повторите стир с WB в общей сложности 3 стирок (1 х PEB, 2 х WB). Перед каждым буферным обменом добавьте PMSF.
- Соберите шарики в 1 мл ВБ и перенесите их на трубку 1,5 мл. Наконец, соберите бисер еще раз на магнитной подставке и удалите всю жидкость. Закройте трубку и держите на льду до тех пор, пока все образцы не будут обработаны.
- Транспорт образцы дыма капот для извлечения РНК.
- Утилизация отходов и восстановление лабораторных принадлежностей.
- Если выполняется в соответствии с хорошей лабораторной практикой (см. раздел 2.2.1), процедура стерилизации дает aqueous решение NaCl. Оставьте газ хлора, а также остаточный HCl и отбеливатель, чтобы defumigate в дым капот.
- Удаление PEB и WB: По мере того как CHX разлагается при высоком рН, собирайте все жидкости и доведите до рНЗт;9. Утилизировать жидкие отходы в галогенных химических отходах. Все твердые вещества (ткани, серологические пипетки, перчатки и т.д.) должны быть утилизированы в качестве химических отходов.
- Сбор фенолсодержащих жидкостей отдельно, а также фенол загрязненных материалов (советы, трубки и перчатки).
- Растворы для мытья рук, пестики и гомогенизаторы (губки и щетки) с мылом и тщательно промыть. Впоследствии, испечь материал на ночь на йgt;220 градуса по Цельсию. Либо завернуть в фольгу перед обработкой, либо поместить в термостойкую, покрытую контейнер.
- Чистите щеткой центрифуги трубки с моющим средством, а затем диэтилпирокарбонат (DEPC) - лечить в дым капот. С этой целью добавьте жидкий DEPC в деионизированную воду (1 мл DEPC до 1 л H2O) и перемешайте через встряхивание. Поместите центрифуги труб на автоклаванный лоток, который ловит пролитой DEPC-воды. Налейте подвеску в трубки и оставьте на 3 ч или на ночь. DEPC разлагается в последующем процессе автоклавирования.
ВНИМАНИЕ: DEPC является высокотоксичным.
7. Добыча РНК и КК
- Добыча РНК
- Охладите центрифугу столешницы до 4 градусов по Цельсию.
- Добавьте 1 мл кислотно-гуанидинино-фенол-фенола на основе реагента (Таблица материалов) к каждому образцу, инвертировать, чтобы повторно приостановить бисер или общей РНК суспензии и инкубировать в течение 5 минут на льду. Не вихрь!
- Добавьте 200 л хлороформа и инкубировать в течение 3 мин на льду. Затем тщательно вихрь образцов.
- Чтобы помочь фазе разделения, центрифуга на макс. скорость 10-15 мин, 4 градуса по Цельсию.
- Этикетка 1,5 мл с низким удержанием труб (Таблица материалов) и aliquot 650 л изопропанола в каждом.
- Аккуратно возьмите верхнюю ваквистую фазу (около 650 л) и перенесите в подготовленные трубки с изопропанолом. Избегайте прикосновения к розовой органической фазе.
- Осадок РНК ночь при -20 градусах Цельсия.
ПРИМЕЧАНИЕ: Рекомендуется хранить образцы в изопропанола при -20 градусах по Цельсию или -80 градусов по Цельсию и только растворить в воде, когда это необходимо. Aqueous РНК деградирует даже при -80 градусов при хранении в течение нескольких недель/ месяцев.
- Осадки РНК
- Охладите центрифугу столешницы до 4 градусов по Цельсию.
- Приготовьте свежие 80% EtOH с водой без RNase и охладите при -20 градусах По Цельсия (5 мин при -80 градусов по Цельсию помогают ускорить процесс).
- Centrifuge образцы на максимальной скорости (около 13000 х г) в течение 30 минут и отбросить супернатант. Гранулы не будут видны, так тщательно пипетка, как если бы он был там. Добавить 1 мл холодного 80% EtOH и инвертировать трубку один или два раза.
- Центрифуге снова в течение 30 минут на максимальной скорости и повторить мыть в общей сложности два моет.
- Спин вниз в течение 2 мин и удалить все остаточные EtOH с 10 л наконечника. Оставьте гранулы высохнуть в течение 3-5 мин (не более) при комнатной температуре и повторно приостановите в 20 Л L RNase воды.
- Держите образцы на льду и выполнять контроль качества как можно скорее. Приступить к хранению образцов при -80 градусах Цельсия. Избегайте циклов замораживания-оттепели.
- Контроль качества с использованием специального оборудования(Таблица материалов) в соответствии с рекомендациями производителя.
8. Подготовка библиотеки
- синтез кДНК и усиление с SMARTer v4 Ultra Низкий вход РНК Kit
- Рассчитайте разбавление каждого образца, чтобы иметь 1,5 нг TRAP-РНК или общей РНК в объеме 4,75 зЛ. Выполните все реакции в ПЦР-трубках и разбавьте образцы свежими аликотами безрычания без RNase.
- Выполняйте все шаги в соответствии с рекомендациями производителя с объемами реакции. Усиль кДНК 12-13 циклами ПЦР.
- Очистите ПЦР, добавив 0,5 л 10-x буфера лисиса и 25 л бусин SPRI(Таблица материалов). Если многие образцы обработаны лисис буфера и бисер может быть предварительно смешаны. Убедитесь, что бусы равномерно рассредоточены перед пипетой.
- Продолжить с протоколом в полном объеме реакции (17 л буфера elution). Не дайте бисеру высохнуть более 3 минут. Пересушенные образцы потенциально могут быть спасены в течение длительного времени инкубации.
- Измерьте концентрацию образцов с помощью комплекта ДНК Кубит СС.
ПРИМЕЧАНИЕ: SMARTer v4 комплект может терпеть до 200 пг ввода. Мы получили библиотеки в тех случаях, когда значения qubit не могли быть определены (ниже 250 пг, предел обнаружения) с 16 циклом PCR. Однако ограниченный входиновый материал может также давать менее сложные библиотеки.
- Фрагментация и адаптер перевязки PCR с Nextera XT ДНК библиотека Подготовка Комплект
- Разбавить кДНК с rNase свободной воды для получения концентрации 200 пг / л и пипетка 1,25 л в ПЦР-трубки.
- Выполните все шаги в соответствии с производителем с 1/4 объемов реакции. Увеличьте кДНК 12 циклами ПЦР и совместимыми адаптерами для образцов, принадлежащих к одному пулу секвенирования. С индексом Illumina Наборы A и D до 384 образцов могут быть мультиплексированы.
- Для очистки ПЦР добавить 12,5 л буфера повторного подвески и 22,5 л бусинs SPRI (коэффициент 0,9x). образец с 22 злициевого буфера.
ПРИМЕЧАНИЕ: КК и объединение было выполнено последовательности компании (Таблица материалов), и, таким образом, не бис основе нормализации не было необходимости. Реакция ферментативной фрагментации (тегменция) очень чувствительна к вхожгу материала, так как каждый фермент режет только один раз. Поэтому не превышать рекомендацию о концентрации.
Результаты
Для оценки качества вышеупомянутая процедура должна быть исследована на нескольких промежуточных этапах: проверка шаблона выражения в плантах, контроль качества изолированной полисомальной РНК, а также окончательных библиотек. qRT-PCR с использованием известных генов...
Обсуждение
Проверка шаблона локализации RPL18
Решающее значение для того, чтобы избежать неправильного толкования данных из любого эксперимента TRAP, имеет правильную модель выражения помеченного рибосомального подразделения. Таким образом, включение GFP в качестве эпитопа тега RPL18 очень ?...
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Мы хотели бы поблагодарить центр генетического разнообразия цюрихского центра генетического разнообразия за важные консультации экспертов на ранней стадии этого проекта. Работа в лаборатории Vermeer была поддержана грантом sNF Professorship (PP00P3_157524) и грантом на оборудование R'EQUIP (316030-164086) от Швейцарского национального научного фонда (SNSF), присужденного JEMV.
Материалы
| Name | Company | Catalog Number | Comments |
| Sterilization | |||
| bleach, 13% | Sigma | 71696 | |
| beaker | VWR | 214-1172/74/75 | |
| desiccator with porcelaine plate (DURAN) | Sigma/Merck | Z317454-1EA/Z317594-1EA | |
| EtOH, p.a. | Honeywell | 02860-1L | |
| HCl, 37% | Roth | 4625.1 | |
| Tween 20 | Sigma | P9416 | |
| Plate growth + harvesting | |||
| MS salts, basal salt mixture, incl. MES buffer | Duchefa | M0254 | |
| agar plant for cell culture | Applichem/Panreac | A2111.1000 | |
| DMSO | Sigma | D4540 | |
| forcepts | Rubis Switzerland | 5-SA model | |
| KOH | Fluka | 60370 | |
| micropore/surgical tape | 3M | 1530-0 | |
| NAA | Duchefa | N0903 | |
| petri dishes 120x120 mm | Greiner bio-one | 688102 | |
| scalpel | VWR/Swann-Morton | 233-5454 | |
| tissues, neutral, two-layered | any supplier of your choice | ||
| Immunoprecipitation | |||
| GFP-beads: gtma-100 GFP-Trap_MA | Chromotek | e.g. gtma-100 | |
| Brij-35 | Sigma | P1254-500G | |
| centrifuge tubes (in accordance with centrifuge) | Beckman Coulter | 357001 | |
| Chloramphenicol | Applichem | C0378-25G | |
| cotton gloves | VWR | 113-7355 | |
| Cycloheximide, HPLC grade | Sigma | 01810-1G | |
| DEPC | VWR | E174 | might have long delivery times |
| DTT | Fluka | 43815 | |
| EGTA | Sigma | 3054.3 | |
| homogenizers DUALL 23 | KONTES GLASS CO (via VWR) | SCERSP885450-0023 (set) | SCERSP885451-0023 pestle only - SCERSP885452-0023 cylinder only; long delivery times |
| Igepal CA-360 | Sigma | I3021-100ml | |
| KCl | Sigma | 60130 | |
| MgCl2 hexahydrat | Roth | 2189.2 | |
| mortar and pestle | VWR | 470148-960 & 470019-978 | |
| PMSF | Roche | 10 837 091 001 | |
| Polyoxyethylene-(10)-tridecylether/PTE | Sigma | P2393-500G | |
| RNase-free water | Roth | T143.3 | |
| RNAZap | Thermo Fisher | AM9780/AM9782 | for cleaning surfaces |
| Tris, >99.3% | Roth | AE15.3 | |
| Triton X-100 | Fluka | T8787-250ml | |
| Tween 20 | Sigma | P9416-100ml | |
| RNA extraction | |||
| 2-Propanol, p.a. | Sigma | 33539-1L-GL-R | |
| Chloroform, HPLC grade | Scharlau | CL02181000 | |
| EtOH, p.a. | Honeywell | 02860-1L | |
| low-retention microcentrifuge tubes, 1.5 ml | Eppendorf/Sigma | Z666548-250EA | LoBind |
| RNase-free DNase set | Qiagen | 79254 | |
| RNeasy MiniElute Cleanup Kit | Qiagen | 74204 | |
| TRIzol reagent | ThermoFisher/Ambion | 15596018 | |
| Library preparation | |||
| 15/50 mL Tube Magnetic Separator | Abraxis | PN 472250 | |
| AMPure beads | Beckman Coulter | A63881 | |
| Index Kit A | Illumina | FC-131-2001 | |
| Index Kit D | Illumina | FC-131-2004 | |
| neodymium magnets | Amazon/other | 6 x 1.5 mm range: N42 (NdFeB) | |
| Nextera XT kit | Illumina | FC-131-1024/1096 | https://emea.support.illumina.com/ |
| PCR strips | ThermoScientific | AB-0266 | |
| SMARTer v4 kit | Takara Bioscience | 634892 | https://www.takarabio.com/ |
| Bioanalyzer | Agilent | 2100 Bioanalyzer Instrument | specialized equipment for RNA/DNA quality control |
| Tapestation | Agilent | 4200 Tapestation Instrument | specialized equipment for RNA/DNA quality control |
| Fragment Analyzer | Agilent | 5400 Fragment Analyzer System | specialized equipment for RNA/DNA quality control (high throughput) |
| LabChip | PerkinElmer | LabChip GX Touch Nucleic Acid Analyzer | specialized equipment for RNA/DNA quality control (high throughput) |
| Qubit 4 Fluorometer | ThermoFisher | Q33239 | specialized equipment for RNA/DNA concentration determination |
| qRT-PCR | |||
| GATA23 | Microsynth | fwd: AGTGAGAATGAA AGAAGAGAAGGG; rev: GTGGCTGCGAAT AATATGAATACC | |
| GH3.3 | Microsynth | fwd: CAAACCAATCCT CCAAATGAC; rev: ACTTATCCGCAA CCCGACT | |
| LBD29 | Microsynth | fwd: TCTCCAACAACA GGTTGTGAAT; rev: AAGGAGCCTTAG TAGTGTCTCCA | |
| UBC21 | Microsynth | fwd: TGCGACTCAGGG AATCTTCT; rev: TCATCCTTTCTT AGGCATAGCG | |
| SsoAdvanced Universal SYBR Green | Bio-Rad | #172-5270 | |
| iScript Adv cDNA Kit | Bio-Rad | #172-5038 | |
| miscellaneous | |||
| Falcon tubes 15 ml, Cellstar | Greiner bio-one | 188261 | |
| Falcon tubes 50 ml, Cellstar | Greiner bio-one | 210261 | |
| filter tips 1 ml | Axygen | TF-1000-R-S | |
| filter tips 10 µl | Axygen | TF-10-R-S | |
| filter tips 100 µl | Axygen | TF-100-R-S | |
| filter tips 20 µl | Axygen | TF-20-R-S | |
| filter tips 200 µl | Axygen | TF-200-R-S | |
| microcentrifuge tubes 1.5 ml | SARSTEDT | 72.690.001 | |
| Propidium iodide | Sigma | P4170-100MG | |
| sequencing company | Novogene | en.novogene.com |
Ссылки
- Van Verk, M. C., Hickman, R., Corné, M. J., Pieterse, M., Van Wees, S. C. RNA-Seq: Revelation Of The Messengers. Trends In Plant Science. 18 (4), 175-179 (2013).
- Libault, M., Pingault, L., Zogli, P., Schiefelbein, J. Plant Systems Biology At The Single-Cell Level. Trends In Plant Science. 22 (11), 949-960 (2017).
- Mustroph, A., et al. Profiling Translatomes Of Discrete Cell Populations Resolves Altered Cellular Priorities During Hypoxia In Arabidopsis. Proceedings Of The National Academy Of Sciences Of The United States Of America. 106 (44), 18843-18848 (2009).
- Karve, R., Iyer-Pascuzzi, A. S. Digging Deeper: High-Resolution Genome-Scale Data Yields New Insights Into Root Biology. Current Opinion In Plant Biology. 24, 24-30 (2015).
- Warner, J. R., Knopf, P. M., Rich, A. A Multiple Ribosomal Structure In Protein Synthesis. Proceedings of The National Academy of Sciences of The United States of America. 49 (1), 122-129 (1963).
- Gautam, V., Sarkar, A. K. Laser Assisted Microdissection, An Efficient Technique To Understand Tissue Specific Gene Expression Patterns And Functional Genomics In Plants. Molecular Biotechnology. 57 (4), 299-308 (2015).
- Bargmann, B. O. R., Birnbaum, K. D. Fluorescence Activated Cell Sorting Of Plant Protoplasts. Journal of Visualized Experiments. (36), e1673 (2010).
- Deal, R. B., Henikoff, S. The Intact Method For Cell Type-Specific Gene Expression And Chromatin Profiling In Arabidopsis Thaliana. Nature Protocols. 6 (1), 56-68 (2011).
- Dougherty, J. D. The Expanding Toolkit Of Translating Ribosome Affinity Purification. The Journal of Neuroscience: The Official Journal Of The Society For Neuroscience. 37 (50), 12079-12087 (2017).
- Mustroph, A., Juntawong, P., Bailey-Serres, J. Isolation Of Plant Polysomal mRNA By Differential Centrifugation And Ribosome Immunopurification Methods. Methods in Molecular Biology. 553, 109-126 (2009).
- Matsushima, W., et al. SLAM-ITseq: Sequencing Cell Type-Specific Transcriptomes Without Cell Sorting. Development. 145 (13), (2018).
- Basnet, H., et al. Flura-Seq Identifies Organ-Specific Metabolic Adaptations During Early Metastatic Colonization. Elife. 8, (2019).
- Rodriques, S. G., et al. Slide-Seq: A Scalable Technology For Measuring Genome-Wide Expression At High Spatial Resolution. Science. 363 (6434), 1463-1467 (2019).
- Fazal, F. M., et al. Atlas Of Subcellular RNA Localization Revealed By Apex-Seq. Cell. 178 (2), 473-490 (2019).
- Slane, D., Bayer, M., Kaufmann, K., Mueller-Roeber, B. Cell Type-Specific Gene Expression Profiling Using Fluorescence-Activated Nuclear Sorting. Plant Gene Regulatory Networks: Methods And Protocols. , 27-35 (2017).
- Zanetti, M. E., Chang, I. F., Gong, F., Galbraith, D. W., Bailey-Serres, J. Immunopurification Of Polyribosomal Complexes Of Arabidopsis For Global Analysis Of Gene Expression. Plant Physiology. 138 (2), 624-635 (2005).
- King, H. A., Gerber, A. P. Translatome Profiling: Methods For Genome-Scale Analysis Of mRNA Translation. Briefings In Functional Genomics. 15 (1), 22-31 (2016).
- Mašek, T., Valášek, L., Pospíšek, M., Nielsen, H. Polysome Analysis And RNA Purification From Sucrose Gradients. RNA: Methods And Protocols. , 293-309 (2011).
- Heiman, M., et al. A Translational Profiling Approach For The Molecular Characterization Of Cns Cell Types. Cell. 135 (4), 738-748 (2008).
- Halbeisen, R. E., Scherrer, T., Gerber, A. P. Affinity Purification Of Ribosomes To Access The Translatome. Methods. 48 (3), 306-310 (2009).
- Thomas, A., et al. A Versatile Method For Cell-Specific Profiling Of Translated mRNAs In Drosophila. Plos One. 7 (7), e40276 (2012).
- Watson, F. L., et al. Cell Type-Specific Translational Profiling In The Xenopus Laevis Retina. Developmental Dynamics. 241 (12), 1960-1972 (2012).
- Lam, P. Y., Harvie, E. A., Huttenlocher, A. Heat Shock Modulates Neutrophil Motility In Zebrafish. Plos One. 8 (12), e84436 (2013).
- Fang, Y., et al. Translational Profiling Of Cardiomyocytes Identifies An Early Jak1/Stat3 Injury Response Required For Zebrafish Heart Regeneration. Proceedings Of The National Academy Of Sciences Of The United States Of America. 110 (33), 13416-13421 (2013).
- Mustroph, A., Zanetti, M. E., Girke, T., Bailey-Serres, J. Isolation And Analysis Of mRNAs From Specific Cell Types Of Plants By Ribosome Immunopurification. Methods In Molecular Biology. 959, 277-302 (2013).
- Monshausen, G. B., Gilroy, S. Feeling Green: Mechanosensing In Plants. Trends In Cell Biology. 19 (5), 228-235 (2009).
- Day, R. C., Grossniklaus, U., Macknight, R. C. Be More Specific! Laser-Assisted Microdissection Of Plant Cells. Trends In Plant Science. 10 (8), 397-406 (2005).
- Sheen, J. Signal Transduction In Maize And Arabidopsis Mesophyll Protoplasts. Plant Physiology. 127 (4), 1466-1475 (2001).
- Datta, S., et al. Laser Capture Microdissection: Big Data From Small Samples. Histology And Histopathology. 30 (11), 1255-1269 (2015).
- Birnbaum, K., et al. A Gene Expression Map Of The Arabidopsis Root. Science. 302 (5652), 1956 (2003).
- Hamant, O., Haswell, E. S. Life Behind The Wall: Sensing Mechanical Cues In Plants. BMC Biology. 15 (1), 1354 (2017).
- Vragović, K., et al. Translatome Analyses Capture Of Opposing Tissue-Specific Brassinosteroid Signals Orchestrating Root Meristem Differentiation. Proceedings of The National Academy of Sciences of the United States of America. 112 (3), 923-928 (2015).
- Wang, Y., Jiao, Y. Translating Ribosome Affinity Purification (Trap) For Cell-Specific Translation Profiling In Developing Flowers. Methods In Molecular Biology. 1110, 323-328 (2014).
- Sablok, G., Powell, J. J., Kazan, K. Emerging Roles And Landscape Of Translating mRNAs In Plants. Frontiers in Plant Science. 8, 1443 (2017).
- Ron, M., et al. Hairy Root Transformation Using Agrobacterium Rhizogenes As A Tool For Exploring Cell Type-Specific Gene Expression And Function Using Tomato As A Model. Plant Physiology. 166 (2), 455-469 (2014).
- Reynoso, M. A., et al. Evolutionary Flexibility In Flooding Response Circuitry In Angiosperms. Science. 365 (6459), 1291-1295 (2019).
- Dolan, L., et al. Cellular Organisation Of The Arabidopsis Thaliana Root. Development. 119 (1), 71 (1993).
- Ristova, D., Barbez, E. . Root Development. , (2018).
- Shekhar, V., Stӧckle, D., Thellmann, M., Vermeer, J. E. M. The Role Of Plant Root Systems In Evolutionary Adaptation. Current Topics in Developmental Biology. 131, 55-80 (2019).
- Malamy, J. E., Benfey, P. N. Down And Out In Arabidopsis: The Formation Of Lateral Roots. Trends in Plant Science. 2 (10), 390-396 (1997).
- de Smet, I., et al. Bimodular Auxin Response Controls Organogenesis In Arabidopsis. Proceedings of the National Academy of Sciences of The United States of America. 107 (6), 2705-2710 (2010).
- Péret, B., et al. Arabidopsis Lateral Root Development: An Emerging Story. Trends In Plant Science. 14 (7), 399-408 (2009).
- Vilches-Barro, A., Maizel, A. Talking Through Walls: Mechanisms Of Lateral Root Emergence In Arabidopsis Thaliana. Current Opinion in Plant Biology. 23, 31-38 (2015).
- Porco, S., et al. Lateral Root Emergence In Arabidopsis Is Dependent On Transcription Factor Lbd29 Regulation Of Auxin Influx Carrier Lax3. Development. 143 (18), 3340-3349 (2016).
- Stoeckle, D., Thellmann, M., Vermeer, J. E. Breakout-Lateral Root Emergence In Arabidopsis Thaliana. Current Opinion in Plant Biology. 41, 67-72 (2018).
- Banda, J., et al. Lateral Root Formation In Arabidopsis: A Well-Ordered Lrexit. Trends in Plant Science. 24 (9), 826-839 (2019).
- Vermeer, J. E. M., et al. A Spatial Accommodation By Neighboring Cells Is Required For Organ Initiation In Arabidopsis. Science. 343 (6167), 178-183 (2014).
- Vanneste, S., et al. Cell Cycle Progression In The Pericycle Is Not Sufficient For Solitary Root/Iaa14-Mediated Lateral Root Initiation In Arabidopsis Thaliana. The Plant Cell. 17 (11), 3035-3050 (2005).
- Marques-Bueno, M. M., et al. A Versatile Multisite Gateway-Compatible Promoter And Transgenic Line Collection For Cell Type-Specific Functional Genomics In Arabidopsis. The Plant Journal : For Cell and Molecular Biology. 85 (2), 320-333 (2016).
- Shimada, T. L., Shimada, T., Hara-Nishimura, I. A Rapid And Non-Destructive Screenable Marker, Fast, For Identifying Transformed Seeds Of Arabidopsis Thaliana. The Plant Journal : For Cell and Molecular Biology. 61 (3), 519-528 (2010).
- Clough, S. J., Bent, A. F. Floral Dip: A Simplified Method For Agrobacterium Mediated Transformation Of Arabidopsis Thaliana. The Plant Journal. 16 (6), 735-743 (1998).
- Lindsey, B. E., Rivero, L., Calhoun, C. S., Grotewold, E., Brkljacic, J. Standardized Method For High-Throughput Sterilization Of Arabidopsis Seeds. Journal Of Visualized Experiments. (128), e56587 (2017).
- Andersen, T. G., et al. Diffusible Repression Of Cytokinin Signalling Produces Endodermal Symmetry And Passage Cells. Nature. 555, 529-533 (2018).
- Schroeder, A., et al. The Rin: An Rna Integrity Number For Assigning Integrity Values To Rna Measurements. BMC Molecular Biology. 7, 3 (2006).
- Vragović, K., Bartom, E., Savaldi-Goldstein, S. Quantitation Of Cell Type-Specific Responses To Brassinosteroid By Deep Sequencing Of Polysome-Associated Polyadenylated RNA. Methods in Molecular Biology. 1564, 81-102 (2017).
- Bertin, B., Renaud, Y., Aradhya, R., Jagla, K., Junion, G. Trap-Rc, Translating Ribosome Affinity Purification From Rare Cell Populations Of Drosophila Embryos. Journal Of Visualized Experiments. (103), e52985 (2015).
- Livak, K. J., Schmittgen, T. D. Analysis Of Relative Gene Expression Data Using Real-Time Quantitative PCR And The 2(-Delta Delta C(T)) Method. Methods. 25 (4), 402-408 (2001).
- Jiao, Y., Meyerowitz, E. M. Cell-Type Specific Analysis Of Translating Rnas In Developing Flowers Reveals New Levels Of Control. Molecular Systems Biology. 6, 419 (2010).
- Tian, C., et al. A Gene Expression Map Of Shoot Domains Reveals Regulatory Mechanisms. Nature Communications. 10 (1), 141 (2019).
- Townsley, B. T., Covington, M. F., Ichihashi, Y., Zumstein, K., Sinha, N. R. Brad-Seq: Breath Adapter Directional Sequencing: A Streamlined, Ultra-Simple And Fast Library Preparation Protocol For Strand Specific mRNA Library Construction. Frontiers in Plant Science. 6, 366 (2015).
- Song, Y., et al. A Comparative Analysis Of Library Prep Approaches For Sequencing Low Input Translatome Samples. BMC Genomics. 19 (1), 696 (2018).
- Basu, D., Haswell, E. S. Plant Mechanosensitive Ion Channels: An Ocean Of Possibilities. Current Opinion in Plant Biology. 40, 43-48 (2017).
- Brady, S. M., et al. A High-Resolution Root Spatiotemporal Map Reveals Dominant Expression Patterns. Science. 318 (5851), 801 (2007).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены