Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Un cultivo de explantes de cola 3-D para estudiar la segmentación de vertebrados en el pez cebra
En este artículo
Resumen
Aquí, presentamos el protocolo para el cultivo de tejidos 3-D del eje posterior del cuerpo del pez cebra, lo que permite el estudio en vivo de la segmentación de vertebrados. Este modelo de explant proporciona control sobre el alargamiento del eje, alteración de las fuentes de morfógenos y obtención de imágenes vivas a nivel de tejido de resolución subcelular.
Resumen
Los embriones de vertebrados modelan su eje corporal principal como somitas repetitivas, los precursores de las vértebras, los músculos y la piel. Las somitas se segmentan progresivamente desde el mesodermo presomático (PSM) a medida que el extremo de la cola del embrión se alarga posteriormente. Los somitas se forman con periodicidad regular y escala en tamaño. El pez cebra es un organismo modelo popular, ya que es genéticamente manejable y tiene embriones transparentes que permiten obtener imágenes vivas. Sin embargo, durante la somitogénesis, los embriones de peces se envuelven alrededor de una yema grande y redondeada. Esta geometría limita las imágenes vivas de tejido PSM en embriones de pez cebra, particularmente a resoluciones más altas que requieren una distancia de trabajo objetiva cercana. Aquí, presentamos un método aplanado de la cultura del tejido de 3-D para la proyección de imagen viva de los explantes de la cola del pez cebra. Los explantes de cola imitan los embriones intactos mostrando una desaceleración proporcional de la elongación del eje y el acortamiento de las longitudes de somita rostrocaudal. Además, somos capaces de detener la velocidad de elongación del eje a través del cultivo de explantes. Esto, por primera vez, nos permite desenredar la entrada química de los gradientes de señalización de la entrada mecanicista de la elongación axial. En estudios futuros, este método se puede combinar con una configuración microfluídica para permitir perturbaciones farmacéuticas controladas por el tiempo o la detección de la segmentación de vertebrados sin ningún problema de penetración de fármacos.
Introducción
La segmentación metamérica de organismos es ampliamente utilizada en la naturaleza. Las estructuras repetidas son esenciales para la funcionalidad de órganos laterales como vértebras, músculos, nervios, vasos, extremidades u hojas en un plan corporal1. Como resultado de tales restricciones fisiológicas y geométricas de la simetría axial, la mayoría de los filos de Bilateria-tales como anélidos, artrópodos, y cordados-exhiben la segmentación de sus tejidos embrionarios (e.g., ectodermo, mesodermo) antero-posteriorly.
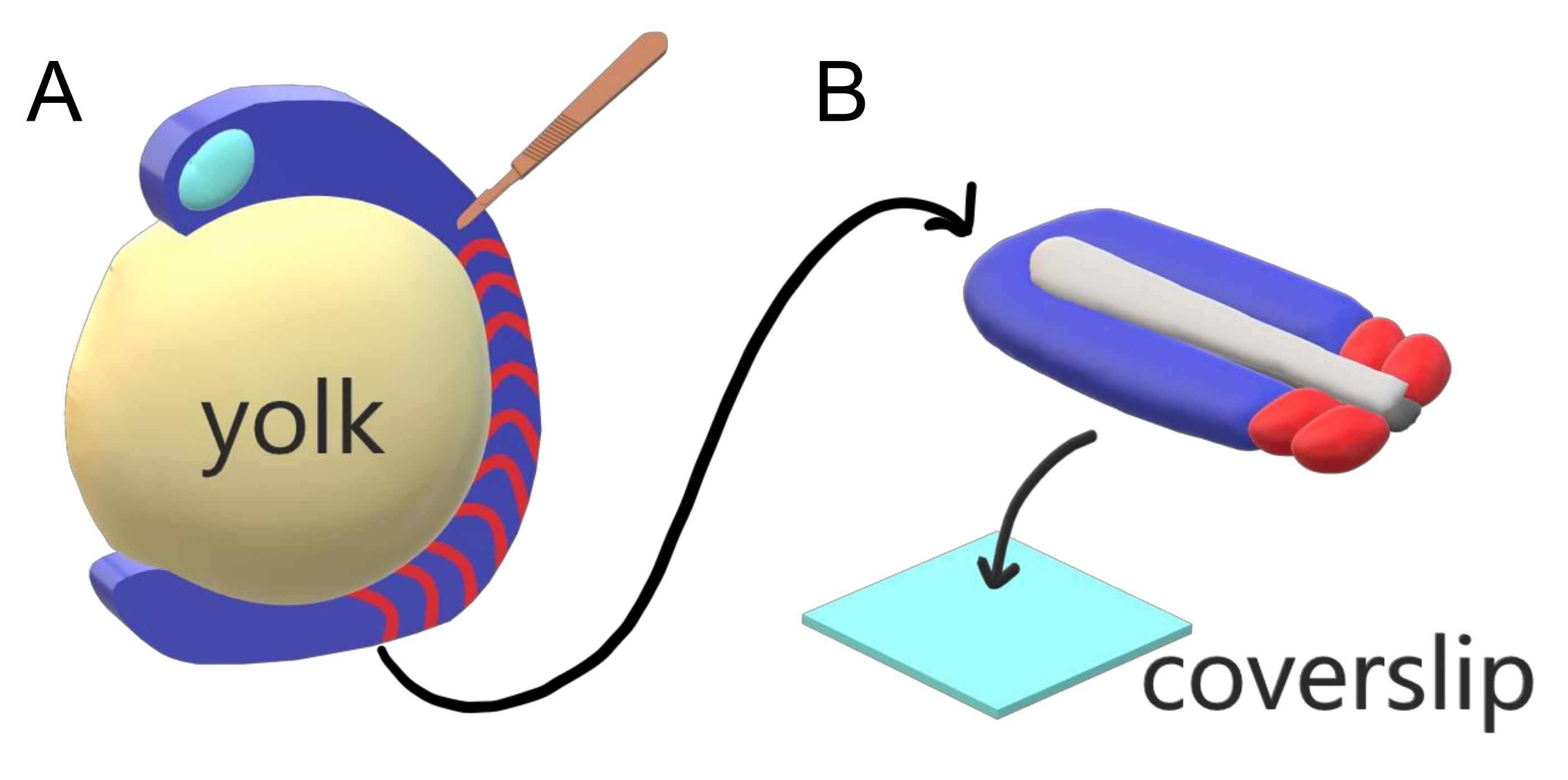
Los embriones de vertebrados segmentan secuencialmente su mesodermo paraxial a lo largo del eje principal del cuerpo en somitas con intervalos específicos de la especie, recuentos y distribuciones de tamaño. A pesar de tal robustez entre embriones individuales dentro de una especie, la segmentación de somita es versátil entre las especies de vertebrados. La segmentación ocurre en un vasto régimen de intervalos de tiempo (de 25 min en el pez cebra a 5 h en humanos), tamaños (de ~20 μm en somitas de cola de pez cebra a ~200 μm en somites de tronco de ratones) y recuentos (de 32 en pez cebra a ~300 en serpientes de maíz)2. Más interesante aún, los embriones de peces pueden desarrollarse en un amplio rango de temperaturas (desde ~ 20.5 ° C hasta 34 ° C para el pez cebra) mientras mantienen sus somitas intactas con distribuciones de tamaño adecuadas al compensar tanto los intervalos de segmentación como las velocidades de elongación axial. Más allá de estas interesantes características, el pez cebra se mantiene como un organismo modelo útil para estudiar la segmentación en vertebrados debido al desarrollo externo, síncrono y transparente de una plenitud de embriones hermanos, así como sus herramientas genéticas accesibles. Negativamente desde una perspectiva de microscopía, los embriones de teleósteo se desarrollan en una yema esférica voluminosa, estirando y redondeando el tejido gastrulante a su alrededor (Figura 1A). En este artículo, presentamos un cultivo de explantes de tejido 3-D aplanado para colas de pez cebra. Este sistema de explantes evita las restricciones esféricas de la masa de yema, lo que permite el acceso a imágenes vivas de alta resolución de embriones de peces para el modelado de somita.

Figura 1:Sistema de explante de cámara deslizante para embriones de pez cebra. (A) Los embriones de pez cebra tienen ventajas para la obtención de imágenes vivas, como la transparencia del tejido embrionario gastrulado (azul), pero el tejido se forma alrededor de una masa de yema esférica voluminosa (amarillo) que impide obtener imágenes casi objetivas y de alta resolución en embriones intactos. Los explantes de cola se pueden diseccionar comenzando con un cuchillo microquirúrgico (marrón) cortado del tejido anterior de somitas (rojo) y continuando en el borde con la yema posterior. (B) Los explantes de cola disecados pueden colocarse en un cubrebocas (azul claro) dorsoventralmente; mantener el tejido neural (gris claro) en la parte superior y la notocorda (gris oscuro) en la parte inferior. Haga clic aquí para ver una versión más amplia de esta figura.
{kind=link}
Protocolo
Este protocolo implica el uso de embriones de vertebrados vivos menores de 1 día después de la fertilización. Todos los experimentos con animales se realizaron bajo las pautas éticas del Cincinnati Children's Hospital Medical Center; los protocolos de animales fueron revisados y aprobados por el Comité Institucional de Cuidado y Uso de Animales (Protocolo # 2017-0048).
1. Recolección de embriones
- Configure pares de peces cebra en tanques de cruce la noche antes del día de recolección de embriones. Para un control preciso de la estadificación del desarrollo embrionario, utilice barreras entre las parejas de apareamiento.
- Levante las barreras antes del tiempo de desove preferido y recoja los huevos en 15 minutos en una placa de Petri de 100 mm.
- Limpie los escombros de la placa de Petri. Si se recogen más de 50 embriones de un solo embrague, divida el embrague en múltiples placas de Petri en consecuencia.
- Incubar embriones en agua del sistema de peces a 28 °C hasta que alcancen el 50% de la etapa epibólica (5 horas después de la fertilización). También se puede utilizar un medio estandarizado de crecimiento embrionario como E3 en lugar del agua del sistema de acuario hasta el paso 3.2.
- Retire los óvulos no fertilizados debajo de un estereoscopio y mueva los embriones a una incubadora de 23,5 °C durante la noche (O/N). Los embriones deben estar en la etapa de 8-10 somites la mañana siguiente al día de recolección.
2. Preparación de la herramienta
- Esterilice la hoja del cuchillo de microcirugía, las puntas de las agujas (utilizadas para la disección del tejido) y la pipeta Pasteur de vidrio remojando etanol al 100% (EtOH) y acristalamiento contra incendios.
- Utilice dos capas de cinta transparente (~ 100-120 μm de espesor) en portaobjetos de microscopio de 25 mm x 75 mm. Corte los pozos cuadrados de ~ 18 mm x 18 mm en el centro de la cinta de cada diapositiva que cubre con un bisturí.
- Limpie las cámaras de diapositivas preparadas con 70% de EtOH. Estos pozos tendrán ~40 μL de medio.
3. Preparación de la muestra
- Decorionato de embriones usando la punta de dos jeringas de aguja bajo un estereoscopio. Transfiera embriones a una placa de Petri separada con agua del sistema de peces para enjuagar.
- Utilizando una pipeta Pasteur de vidrio estéril vidriada al fuego, transfiera embriones en una placa de Petri de 6 cm que contenga un medio de disección (medio de cultivo celular Leibovitz-15 con L-Glutamina sin Fenol Rojo, 0,8 mM CaCl2 y 1× solución antibiótico-antimicótica).
NOTA: Continúe utilizando una pipeta de vidrio esterilizado para todas las transferencias que siguen este paso.- Utilice una placa de Petri de vidrio para el procedimiento de explantación para evitar las virutas de poliestireno durante la disección.
- Ponga 50 μL de medio de crecimiento tisular (medio de disección y 10% de FBS) en la cámara de deslizamiento.
- Estabilice un embrión para la disección debajo del estereoscopio con una punta de aguja en la intersección entre la yema y el tejido cerca del cerebro posterior.
- Manteniendo el tejido embrionario estable con una aguja, utilice el cuchillo microquirúrgico con la hoja sostenida a 45° para cortar el tejido anterior al cerebro posterior y pelar la yema del tejido embrionario comenzando desde el anterior y moviéndose hacia el brote de la cola (Figura 1A).
NOTA: Tenga cuidado de no perder el tejido de la piel mientras limpia la yema. La piel se desprendería fácilmente como un tejido elástico de una sola capa que flanquea alrededor del embrión durante la disección, por lo que es fácil de reconocer. - Una vez que la yema se haya eliminado por completo del cuerpo embrionario, corte el tejido cutáneo que flanquea del tailbud. Manteniendo los últimos 3-4 somites formados intactos, corte el tejido más anterior (explant de eje completo).
- La yema debe desprenderse principalmente intacta de este procedimiento. En caso de una yema rota, gránulos significativos de yema pueden permanecer unidos a la superficie ventral del tejido. Si es así, use una herramienta de pestañas para limpiar suavemente los gránulos de yema restantes.
NOTA: Un desequilibrio de los tejidos de la piel en los lados laterales de un explant no permitiría que el tejido mantenga una dirección de crecimiento recta. El explant en su lugar se doblará hacia el lado de la piel más estirada. Este desequilibrio se puede corregir bajo el estereoscopio rompiendo la capa de la piel con la ayuda del cuchillo microquirúrgico. - Para explantes sin piel, presione una punta de la capa de piel con aguja y pelar el explantador de tejido con el cuchillo microquirúrgico. Estos explantes no alargarán su eje corporal en cultivo.
- Además de los explantes de eje completo, se pueden hacer explantes alternativos en este paso. Por ejemplo, diseccione los somitas ya segmentados usando el cuchillo microquirúrgico (explantes de PSM completos) o diseccione el PSM en su medio anteroposterior (explantes de PSM medio). Consulte la sección 5.1 para una aplicación de dichos explantes alternativos.
- La yema debe desprenderse principalmente intacta de este procedimiento. En caso de una yema rota, gránulos significativos de yema pueden permanecer unidos a la superficie ventral del tejido. Si es así, use una herramienta de pestañas para limpiar suavemente los gránulos de yema restantes.
- Transfiera inmediatamente el explant diseccionado a una cubierta de 22 mm x 22 mm en la que se realizará la imagen.
- Coloque el explant plano sobre el eje dorsoventral, el lado ventral tocando el cubrebocas (Figura 1B). Retire suavemente el exceso de medios alrededor del explant de tejido utilizando una pipeta de punta filtrada de 20 μL.
NOTA: La transferencia retrasada de explantes disecados a la cubierta da lugar a deformaciones del tejido, pues se releva de las restricciones geométricas de la yema.
- Coloque el explant plano sobre el eje dorsoventral, el lado ventral tocando el cubrebocas (Figura 1B). Retire suavemente el exceso de medios alrededor del explant de tejido utilizando una pipeta de punta filtrada de 20 μL.
- Voltee rápida y cuidadosamente la tapa con el explant sobre la cámara de diapositivas llena de medio de crecimiento.
- Para evitar la formación de burbujas, coloque un lado de la cubierta cuadrada en la cámara de cinta y suelte el otro lado suavemente. Tenga cuidado de no mover / deformar el explant en este paso.
- Elimine suavemente el exceso de sangrado de medios de la cámara presionando la cámara deslizante sobre un tejido de laboratorio. El cubrebocas se sentará establemente en la cámara deslizante para obtener imágenes en vivo, debido a la tensión superficial del medio líquido sin ningún tipo de sellado.
- Para el cultivo a largo plazo (>6 horas), utilice una cámara más grande. En tales casos se pueden utilizar cubiertas rectangulares de 22 mm x 50 mm junto con dos carriles paralelos de capas de cinta en diapositivas. Se puede dejar un espacio de ~ 1 mm de ancho entre dos carriles de cinta para facilitar el acceso de aire al medio de crecimiento.
- Repita los pasos 3.3-3.8 para preparar más explantes. Los explantes preparados alargarán su eje de cuerpo A-P con una velocidad promedio de ~ 30 μm / h y segmentarán sus somites con intervalos de ~ 40 min a 25 ° C (Figura 2A, Video 1).
- Para los explantes no alargados, aplique una presión suave a los lados del portaobjetos que sujetan la muestra en el paso 3.8.2 mientras succione el exceso de medios en un tejido de laboratorio. Alternativamente, los explantes se pueden cultivar en cámaras deslizantes de una sola capa de cinta. Además, la activación química de la superficie de la cámara de deslizamiento con colágeno tipo I dará como resultado explantes no alargados(Figura 2B,Video 2).
- Realice el recubrimiento de la cámara con colágeno tipo I de antemano cubriendo completamente las cámaras deslizantes con 15-20 mL de solución de colágeno predilutado a temperatura ambiente durante 1 h. Utilice una campana de flujo laminar para este protocolo para mantener la esterilidad. Enjuague cuidadosamente las cámaras con medio de disección al final.
- Para los embriones mayores de 15 años en etapa somita, monte lateralmente el tejido explantado de cola en lugar de un montaje plano (dorsoventral) (Video 3). Para prevenir las contracciones musculares, incluir solución de tricaina al 0,004% en los medios de cultivo como agente anestésico3.
- Para los explantes no alargados, aplique una presión suave a los lados del portaobjetos que sujetan la muestra en el paso 3.8.2 mientras succione el exceso de medios en un tejido de laboratorio. Alternativamente, los explantes se pueden cultivar en cámaras deslizantes de una sola capa de cinta. Además, la activación química de la superficie de la cámara de deslizamiento con colágeno tipo I dará como resultado explantes no alargados(Figura 2B,Video 2).
4. Adquisición de imágenes en vivo
- Muestras de imagen, ya sea en un ámbito de disección para imágenes de luz transmitidas a campo ancho de tamaños y períodos de segmentación de somite, o con microscopía de iluminación estructurada / confocal / lámina de luz utilizando líneas de peces reporteros transgénicos.
- Equilibre la temperatura de los explantes de tejido con la temperatura ambiente de la proyección de imagen por lo menos 15 minutos.
- Para un control de temperatura más preciso, utilice un sistema de control de temperatura comercial montado en un microscopio invertido.
- Establezca los intervalos del marco de adquisición de la imagen en 2 - 10 min dependiendo del proceso biológico de interés.
NOTA: La segmentación de somita de pez cebra es un proceso rápido, que oscila entre 20 - 55 min para temperaturas viables de 30 °C a 21,5 °C en embriones enteros. Los explantes se alargarán y segmentarán ~ 30% más lento que los embriones enteros.- Preste atención a dejar suficiente retraso entre conjuntos de adquisiciones de canales para evitar la posible fototoxicidad del tejido vivo. No exponga el tejido al haz de excitación durante más de la mitad de la duración de la imagen y baje la intensidad del haz tanto como sea posible.
NOTA: La acumulación de especies reactivas de oxígeno (ROS) es generalmente la principal causa de fototoxicidad en muestras vivas4. El ácido ascórbico como limpiador del ROS se puede complementar al medio de crecimiento en la concentración de 4 milímetros para amortiguar actividad del ROS y para aliviar phototoxicity. Los efectos adversos de la fototoxicidad pueden ser difíciles de notar durante las imágenes en vivo. Los explantes de cola son ventajosos en este aspecto ya que algunos marcadores visuales de fototoxicidad como la detención mitótica, el desarrollo de tejidos impedidos (es decir, la formación de somitas, el alargamiento de la cola) y la desintegración del tejido son más fáciles de notar. Por favor refiérase a la referenciaproporcionada 4 para una discusión detallada.
- Preste atención a dejar suficiente retraso entre conjuntos de adquisiciones de canales para evitar la posible fototoxicidad del tejido vivo. No exponga el tejido al haz de excitación durante más de la mitad de la duración de la imagen y baje la intensidad del haz tanto como sea posible.
- Utilice embriones inyectados de ARN en etapa unicelular para adquirir imágenes 4-D destinadas a ser segmentadas y analizadas a nivel de resolución celular.
- Utilice 300 pg de ARN de membranas transcritas in vitro y plásmidos marcadores de reportero fluorescente nucleares como pCS-membrana-cerruleanFP (plásmido Addgene #53749) o pCS-memb-mCherry (plásmido Addgene #53750) en combinación con pCS2+ H2B-mTagBFP2 (plásmido Addgene #99267) o pCS2+ H2B-TagRFP-T (Plásmido Addgene #99271) en inyecciones. Para una película de muestra con membrana celular y marcadores de núcleos, consulte el video 4.
NOTA: El tamaño celular promedio del tejido PSM es de aproximadamente ~ 5 μm de diámetro, de los cuales los núcleos comprenden ~ 3 - 4 μm. Se debe registrar un tamaño de píxel de ~0,5 μm y una sección z de ~1 μm para una segmentación de celda adecuada.
- Utilice 300 pg de ARN de membranas transcritas in vitro y plásmidos marcadores de reportero fluorescente nucleares como pCS-membrana-cerruleanFP (plásmido Addgene #53749) o pCS-memb-mCherry (plásmido Addgene #53750) en combinación con pCS2+ H2B-mTagBFP2 (plásmido Addgene #99267) o pCS2+ H2B-TagRFP-T (Plásmido Addgene #99271) en inyecciones. Para una película de muestra con membrana celular y marcadores de núcleos, consulte el video 4.
5. Inmunostaining de explantes de cola
NOTA: Los tejidos cultivados después de varios escenarios de disección (alargados, no alargados, brotes de cola disecados, medio PSM, etc.) como explantes de cola montados en planos5 se pueden recuperar de las cámaras de deslizamiento para futuras cuantificaciones inmunosutenidas de proteínas de interés. Aquí, presentamos el protocolo utilizado para la quinasa regulada señal extracelular di-fosforilada (ppERK) tinción de explantes como FGF señalización gradiente readout.
- Después de la formación de somites hasta la etapa deseada, cambie cautelosamente el cubrebocas a la mitad de la esquina de la cámara deslizante sin levantar.
- Con la ayuda de ~ 100 μL de medio de disección suplementario en una pipeta Pasteur de vidrio, recupere los explantes del portaobjetos y transfiéralos a una placa de cultivo celular de 64 pozos.
NOTA: A partir de este paso, todos los reemplazos de soluciones se pueden realizar bajo un alcance de disección con una pipeta de vidrio separada para muestras fijas. Esto asegurará no perder tejidos explantes en pozos o transferirlos en el medio. - Después de transferir todos los explantes, chupe el medio excesivo de los pozos uno por uno y ponga 100 μL de paraformadehído al 4% en PBS (PFA) en cada pozo.
PRECAUCIÓN: PFA es una solución tóxica con efectos cancerígenos. Se debe usar el EPP adecuado durante la manipulación. - Fije los explantes en una placa de 64 pozos a temperatura ambiente durante 1 h en una coctelera.
- Los explantes tisulares son más sensibles a las deformaciones que los embriones enteros. Ajuste la velocidad de la coctelera en consecuencia.
- Lave el fijador con 150 μL de PBS-Tw (0,1% Tween20 en PBS) tres veces. Recoja el primer lavado en un recipiente específico de "Residuos de PFA".
- Deshidrate los explantes en pasos de 4×5 min reemplazando ~ 40 μL de solución cada vez con metanol al 100% (MeOH).
PRECAUCIÓN: MeOH es un producto químico tóxico que es volátil e inflamable. Trabaje en un espacio bien ventilado y use el EPP adecuado para su manejo. - Como último paso de la deshidratación, retire toda la solución de los pozos y reemplácela con 100 μL de MeOH. Incubar a -20 °C durante 15 min.
NOTA: Utilice un contenedor específico de "MeOH Waste" para recoger las soluciones hasta el paso 5.11. - Añadir 50 μL de MeOH y agitar a temperatura ambiente durante 5 min.
- Rehidrate los explantes en pasos de 4×5 min reemplazando ~ 40 μL de solución cada vez con PBS-T (0.1% Triton-X 100 en PBS). Utilice un contenedor específico de "Residuos de MeOH" para recoger las soluciones.
- Como último paso de la rehidratación, retire toda la solución de los pozos y reemplácela con 100 μL de PBS-T.
- Para la permeabilización tisular tratar explantes con 1.5% Triton-X 100 en PBS durante 20 min a temperatura ambiente en la coctelera.
- Lavar las muestras con MAB-D-T (0,1% de detergente Triton-X 100 y 1% de dimetil sulfóxido (DMSO) en 150 mM NaCl 100 mM de tampón de ácido maleico pH 7,5) 3×5 min.
PRECAUCIÓN: DMSO es inflamable y un mutágeno tóxico. Se debe usar el EPP adecuado durante la manipulación. - Incubar explantes en solución de bloqueo sérico de 100 μL/pozo (2% de suero fetal bovino en MAB-D-T) durante 2 horas a temperatura ambiente.
- Reemplace toda la solución de bloqueo con una solución de anticuerpo primario de 50-100 μL/pozo (dilución 1:1000 de anticuerpo monoclonal de ratón contra ppERK en el bloqueo sérico). Incubar muestras O/N (>16 h) a 4 °C en coctelera.
- Lave la solución de anticuerpos primarios con MAB-D-T 5×5 min.
- Incubar muestras en solución de anticuerpos secundarios (Alexa Fluor 597 cabra anti-ratón IgG2b (1:200) y Hoechst 33342 (1:5000) en MAB-D-T) O/N a 4 °C en una coctelera o durante 3 h a temperatura ambiente.
NOTA: A partir de este paso, cubra la placa de 64 pocillos con papel de aluminio para evitar la exposición a la luz de las muestras secundarias tratadas con anticuerpos.
PRECAUCIÓN: Hoechst 33342 es un carcinógeno potencial. Se debe usar el EPP adecuado durante la manipulación. - Lave la solución de anticuerpos secundarios con PBS-Tw 3×5 min.
- Fijar muestras con PFA durante 15 minutos a temperatura ambiente.
- Lave el fijador con PBS-Tw y equilibre las muestras dentro del 60% de glicerol. Monte explantes en portaobjetos de microscopio con esmalte de uñas y glicerol al 60% para la toma de imágenes. Para obtener resultados representativos de inmunotenstenimiento, consulte la Figura 3.
Resultados
Este protocolo permite el cultivo geométrico plano de explantes de cola de pez cebra vivos. El cultivo de tejidos presenta tres ventajas principales sobre embriones enteros: 1) control de la velocidad de elongación del eje, 2) control sobre varias fuentes de señalización (morfógeno) por disección simple, y 3) cerca de objetivos, alta ampliación y altas imágenes en vivo de NA.
Las cámaras deslizantes no tratadas químicamente permiten que el explante de la cola alargue su eje principal...
Discusión
Este artículo presenta un protocolo detallado de una técnica del explant de la cultura del tejido que desarrollamos y utilizamos recientemente5 para los embriones del pez cebra. Nuestra técnica se basa en los métodos de explantación anteriores en el polluelo8 y el pez cebra9,10,11 organismos modelo. Los explantes de cola preparados con este protocolo pueden sobrevivir hasta >1...
Divulgaciones
Los autores no tienen nada que revelar y no declaran ningún conflicto de intereses.
Agradecimientos
Agradecemos a la AECOM Zebrafish Core Facility y Cincinnati Children's Veterinary Services por el mantenimiento de peces, al Cincinnati Children's Imaging Core por la asistencia técnica, a Didar Saparov por la asistencia con la producción de video y a Hannah Seawall por editar el manuscrito. La investigación reportada en esta publicación fue apoyada por el Instituto Nacional de Ciencias Médicas Generales de los Institutos Nacionales de Salud bajo el Número de Premio R35GM140805 a E.M.Ö. El contenido es responsabilidad exclusiva de los autores y no representa necesariamente los puntos de vista oficiales de los Institutos Nacionales de Salud.
Materiales
| Name | Company | Catalog Number | Comments |
| 1 mL Sub-Q Syringe with PrecisionGlide Needle | Becton, Dickinson and Co. | REF 309597 | for dechorionating embryos and manipulations |
| 200 Proof Ethanol, Anhydrous | Decon Labs | 2701 | for immunostaining |
| Antibiotic Antimycotic Solution (100×) | Sigma-Aldrich | A5955 | for tissue dissection media |
| Calcium Chloride Anhydrous, Powder | Sigma-Aldrich | 499609 | for tissue dissection media |
| Dimethylsulfoxide | Sigma-Aldrich | D5879 | for immunostaining |
| Disposable Scalpel, #10 Stainless Steel | Integra-Miltex | MIL4-411 | for preparing tape slide wells |
| Ethyl 3-aminobenzoate methanesulfonate salt (Tricaine) | Sigma-Aldrich | 886-86-2 | (optional) for anesthesizing tissues older than 20 somites stage |
| Fetal Bovine Serum (FBS) | ThermoFisher | A3160601 | additional for tissue culture media |
| Goat anti-Mouse IgG2b, Alexa Fluor 594 | Invitrogen | Cat#A-21145; RRID: AB_2535781 | secondary antibody for immunostaining |
| L-15 Medium with L-Glutamine w/o Phenol Red | GIBCO | 21083-027 | for tissue dissection media |
| Methanol | Sigma-Aldrich | 179337 | for immunostaining |
| Microsurgical Corneal Knife 2.85 mm Angled Tip Double Bevel Blade | Surgical Specialties | 72-2863 | for tissue dissection |
| Mouse monoclonal anti-ppERK | Sigma-Aldrich | Cat#M8159; RRID:AB_477245 | for ppERK immunostaining |
| NucRed Live 647 ReadyProbes Reagent | Invitrogen | R37106 | (optional) for live staining of cell nuclei |
| Paraformaldehyde Powder, 95% | Sigma-Aldrich | 158127 | for fixation of samples for immunostaining |
| Rat Tail Collagen Coating Solution | Sigma-Aldrich | 122-20 | (optional) for chemically activating slide chambers |
| Stage Top Incubator | Tokai Hit | tokai-hit-stxg | (optional) for temperature control during live imaging |
| Transparent Tape 3/4'' | Scotch | S-9782 | for preparing tape slide wells |
| Triton X-100 | Sigma-Aldrich | X100 | for immunostaining |
| Tween 20 | Sigma-Aldrich | P1379 | for immunostaining |
| Zebrafish: Tg(actb2:2xMCP-NLS-EGFP) | Campbell et al., 2015 | ZFIN: ZDB-TGCONSTRCT-150624-4 | transgenic fish with nuclear localized EGFP |
| Zebrafish: Tg(Ola.Actb:Hsa.HRAS-EGFP) | Cooper et al., 2005 | ZFIN: ZDB-TGCONSTRCT-070117-75 | transgenic fish with cell membrane localized EGFP |
Referencias
- Assheton, R. . Growth in length: Embryological Essays. , (1916).
- Gomez, C., et al. Control of segment number in vertebrate embryos. Nature. 454 (7202), 335-339 (2008).
- Westerfield, M. . The Zebrafish Book: a guide for the laboratory use of zebrafish (Danio rerio), 3rd edition. , (1995).
- Icha, J., Weber, M., Waters, J. C., Norden, C. Phototoxicity in live fluorescence microscopy, and how to avoid it. BioEssays. 39 (1700003), (2017).
- Simsek, M. F., Ozbudak, E. M. Spatial fold change of Fgf signaling encodes positional information for segmental determination in zebrafish. Cell Reports. 24 (1), 66-78 (2018).
- Dubrulle, J., Pourquié, O. fgf8 mRNA decay establishes a gradient that couples axial elongation to patterning in the vertebrate embryo. Nature. 427 (6973), 419-422 (2004).
- Diez del Corral, R., et al. Opposing FGF and Retinoid Pathways Control Ventral Neural Pattern, Neuronal Differentiation, and Segmentation during Body Axis Extension. Neuron. 40 (1), 65-79 (2003).
- Stern, H. M., Hauschka, S. D. Neural tube and notochord promote in vitro myogenesis in single somite explants. Developmental Biology. 167 (1), 87-103 (1995).
- Langenberg, T., Brand, M., Cooper, M. S. Imaging brain development and organogenesis in zebrafish using immobilized embryonic explants. Developmental Dynamics. 228 (3), 464-474 (2003).
- Picker, A., Roellig, D., Pourquié, O., Oates, A. C., Brand, M. Tissue micromanipulation in zebrafish embryos. Methods in molecular biology. 546 (11), 153-172 (2009).
- Manning, A. J., Kimelman, D. Tbx16 and Msgn1 are required to establish directional cell migration of zebrafish mesodermal progenitors. Developmental Biology. 406 (2), 172-185 (2015).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental Dynamics. 203 (3), 253-310 (1995).
- Kaufmann, A., Mickoleit, M., Weber, M., Huisken, J. Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 139, 3242-3247 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados