
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Культура эксплантата 3-D хвоста для изучения сегментации позвоночных у рыбок данио
В этой статье
Резюме
Здесь мы представляем протокол для 3-D культуры тканей задней оси тела рыбок данио, позволяющий изучать сегментацию позвоночных в реальном времени. Эта эксплантная модель обеспечивает контроль над удлинением оси, изменением источников морфогенов и субклеточным разрешением на тканевую визуализацию на уровне живую.
Аннотация
Эмбрионы позвоночных моделируют свою основную ось тела как повторяющиеся сомиты, предшественники позвонков, мышц и кожи. Сомит прогрессивно отрезок от пресомитической мезодермы (PSM), поскольку хвостовой конец эмбриона удлиняется к заднему лонгу. Сомиты формируются с регулярной периодичностью и масштабом в размерах. Рыбка данио является популярным модельным организмом, поскольку она генетически податлива и имеет прозрачные эмбрионы, которые позволяют проводить живую визуализацию. Тем не менее, во время сомитогенеза эмбрионы рыбы оборачиваются вокруг большого, округлого желтка. Эта геометрия ограничивает живую визуализацию ткани PSM у эмбрионов рыбок данио, особенно при более высоких разрешениях, которые требуют близкого объективного рабочего расстояния. Здесь мы представляем метод сплющенной 3-D культуры тканей для живой визуализации эксплантов хвоста рыбки данио. Хвостовые экспланты имитируют интактные эмбрионы, демонстрируя пропорциональное замедление удлинения оси и укорочение ростроковых длин сомита. Кроме того, мы можем остановить скорость удлинения оси благодаря культуре эксплантата. Это впервые позволяет нам распутать химический вход сигнальных градиентов от механистического входа осевого удлинения. В будущих исследованиях этот метод может быть объединен с микрофлюидной установкой, чтобы обеспечить контролируемые по времени фармацевтические возмущения или скрининг сегментации позвоночных без каких-либо проблем с проникновением лекарств.
Введение
Метамерная сегментация организмов широко используется в природе. Повторяющиеся структуры необходимы для функциональности боковых органов, таких как позвонки, мышцы, нервы, сосуды, конечности или листья в плане тела1. В результате таких физиологических и геометрических ограничений осевой симметрии большинство типов Bilateria, таких как кольчатые черви, членистоногие и хордовые, демонстрируют сегментацию своих эмбриональных тканей (например, эктодермы, мезодермы) с задней стороны.
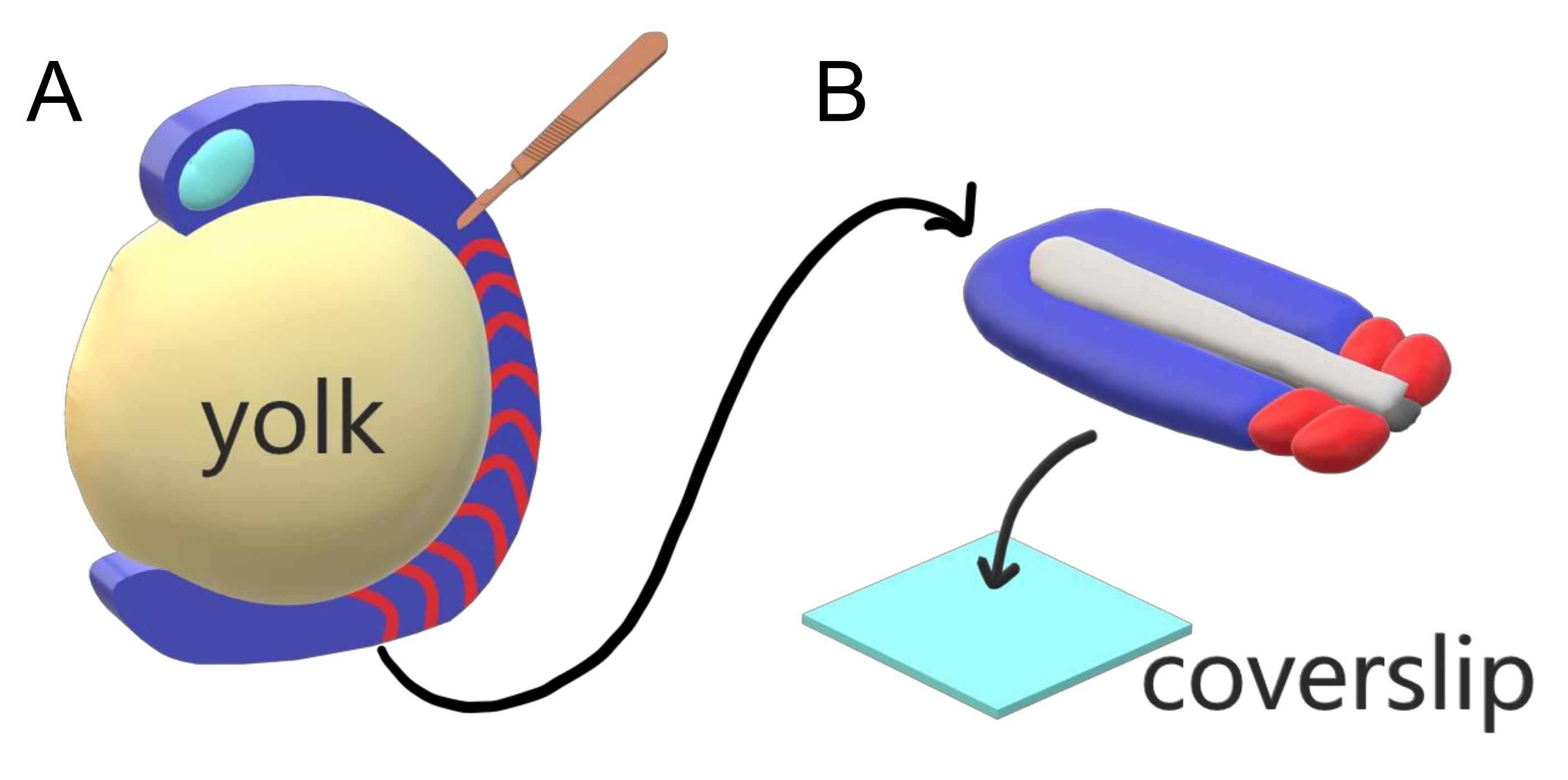
Эмбрионы позвоночных последовательно сегментируют свою параксиальную мезодерму вдоль главной оси тела в сомиты с видоспецифичными интервалами, подсчетами и распределением размеров. Несмотря на такую устойчивость среди отдельных эмбрионов внутри вида, сегментация сомита универсальна между видами позвоночных. Сегментация происходит в обширном режиме временных интервалов (от 25 мин у рыбок данио до 5 ч у человека), размеров (от ~20 мкм в хвостовых сомитах рыбок данио до ~200 мкм у хоботных сомитов мышей) и подсчетов (от 32 у рыбок данио до ~300 у кукурузных змей)2. Что еще более интересно, эмбрионы рыб могут развиваться в широком диапазоне температур (от ~ 20,5 ° C до 34 ° C для рыбок данио), сохраняя при этом свои сомиты нетронутыми с правильным распределением размеров, компенсируя как интервалы сегментации, так и скорости осевого удлинения. Помимо таких интересных особенностей, рыбки данио остаются полезным модельным организмом для изучения сегментации у позвоночных из-за внешнего, синхронного и прозрачного развития обильной популяции эмбрионов братьев и сестер, а также их доступных генетических инструментов. Неблагоприятно с точки зрения микроскопии, телеостные эмбрионы развиваются на громоздком сферическом желтке, растягивая и округляя гаструлирующие ткани вокруг него(рисунок 1А). В этой статье мы представляем сплющенный 3-D тканевый эксплант культуры для хвостов рыбок данио. Эта эксплантная система обходит сферические ограничения желточной массы, позволяя получить доступ к живым изображениям эмбрионов рыб с высоким разрешением для сомит-паттерна.

Рисунок 1:Slide Chamber Explant System for Zebrafish Embryos. (A) Эмбрионы рыбок данио имеют преимущества для живой визуализации, такие как прозрачность гаструлирующей эмбриональной ткани (синий), но ткань образуется вокруг громоздкой сферической желточной массы (желтый), которая предотвращает почти объективную визуализацию с высоким разрешением у неповрежденных эмбрионов. Хвост эксплантов можно рассечь, начиная с микрохирургического ножа (коричневого), вырезанного из ткани с передней части сомитов (красного) и продолжая на границе с желтком с задней стороны. (B) Рассеченные хвостовые экспланты могут быть размещены на покровном (светло-синем) дорсовентральном; сохранение нервной ткани (светло-серого) сверху и нотохорды (темно-серого) внизу. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
протокол
Этот протокол предполагает использование живых эмбрионов позвоночных моложе 1 дня после оплодотворения. Все эксперименты на животных проводились в соответствии с этическими принципами Медицинского центра детской больницы Цинциннати; Протоколы по животным были рассмотрены и одобрены Институциональным комитетом по уходу за животными и их использованию (Протокол No 2017-0048).
1. Сбор эмбрионов
- Установите пары рыбок данио в аквариумах для скрещивания в ночь перед днем сбора эмбрионов. Для точного контроля за постановкой развития эмбриона используйте барьеры между спаривающимися парами.
- Поднимите барьеры до предпочтительного времени нереста и соберите икру в течение 15 минут в чашку Петри 100 мм.
- Очистите мусор от чашки Петри. Если из одной кладки собрано более 50 эмбрионов, разделите кладку на несколько чашек Петри соответственно.
- Инкубировать эмбрионы в воде рыбной системы при 28 °C до тех пор, пока они не достигнут стадии эпиболии 50% (через 5 часов после оплодотворения). Стандартизированная среда роста эмбрионов, такая как E3, также может использоваться вместо воды аквариумной системы до этапа 3.2.
- Удалите неоплодотворенные яйцеклетки под стереоскопом и переместите эмбрионы в инкубатор при 23,5 °C в течение ночи (O/N). Эмбрионы должны быть на стадии 8-10 сомитов утром следующего дня сбора.
2. Подготовка инструмента
- Стерилизуйте лезвие микрохирургии, наконечники игл (используются для рассечения ткани) и стеклянную пипетку Пастера путем замачивания в 100% этаноле (EtOH) и огненной глазури.
- Используйте два слоя прозрачной ленты (толщиной ~100-120 мкм) на слайдах микроскопа 25 мм x 75 мм. Вырежьте квадратные колодцы ~18 мм х 18 мм в центре ленты каждой горки, покрывающей скальпелем.
- Протрите подготовленные скользящие камеры 70% EtOH. Эти скважины будут вмещать ~ 40 мкл среды.
3. Пробоподготовка
- Дехорионать эмбрионы используют кончиком двух игольчатых шприцев под стереоскопом. Перенесите эмбрионы в отдельную чашку Петри с рыбьей системой для промывки.
- Используя огнеупорную стерильную стеклянную пипетку Пастера, переносят эмбрионы в 6 см чашку Петри, содержащую среду для расслоения (питательная среда клеток Лейбовица-15 с L-глютамином без фенола Красного, 0,8 мМ CaCl2 и 1× антибиотико-антимикотический раствор).
ПРИМЕЧАНИЕ: Продолжайте использовать стерилизованную стеклянную пипетку для всех переносов после этого шага.- Используйте стеклянную чашку Петри для процедуры эксплантирования, чтобы избежать полистирольных чипов во время рассечения.
- Поместите 50 мкл питательной среды (среды рассечения и 10% FBS) в слайдовую камеру.
- Стабилизируют эмбрион для рассечения под стереоскопом кончиком иглы на пересечении желточной ткани вблизи задвяза.
- Сохраняя эмбриональную ткань стабильной с помощью иглы, используйте микрохирургический нож с лезвием, удерживаемым под 45°, чтобы разрезать ткань, переднюю к задней части мозга, и отклеить желток от эмбриональной ткани, начиная с передней и двигаясь к хвостовой почке(рисунок 1A).
ПРИМЕЧАНИЕ: Будьте осторожны, чтобы не потерять ткани кожи во время очистки желтка. Кожа будет легко отслаиваться как фланкинговая однослойная эластичная ткань вокруг эмбриона во время рассечения, поэтому ее легко распознать. - Как только желток будет полностью удален из эмбрионального тела, отрежьте фланкирующую кожную ткань от хвостового бутончика. Сохраняя последние образовавшуюся 3-4 сомита неповрежденными, вырезают более переднюю ткань (полноосевой эксплант).
- Желток должен отрываться в основном неповрежденным от этой процедуры. В случае разрыва желтка значительные гранулы желтка могут оставаться прикрепленными к вентральной поверхности ткани. Если это так, используйте инструмент для ресцев, чтобы аккуратно очистить оставшиеся гранулы желтка.
ПРИМЕЧАНИЕ: Дисбаланс тканей кожи на боковых сторонах экспланта не позволит ткани поддерживать прямое направление роста. Эксплант вместо этого будет изгибаться в сторону более растянутой кожи. Этот дисбаланс можно исправить под стереоскопом, разорвав слой кожи с помощью микрохирургического ножа. - Для эксплантов без кожи надавите на кончик слоя кожи иглой и отклейте ткань, эксплантированную микрохирургическим ножом. Эти экспланты не будут удлинять свою ось тела в культуре.
- В дополнение к полноосевым эксплантам, на этом этапе могут быть изготовлены альтернативные экспланты. Например, рассекают уже сегментированные сомиты с помощью микрохирургического ножа (полные экспланты PSM) или рассекают PSM на его половину переднезаднего (полу-PSM explants). Пожалуйста, ознакомьтесь с разделом 5.1 для применения таких альтернативных эксплантов.
- Желток должен отрываться в основном неповрежденным от этой процедуры. В случае разрыва желтка значительные гранулы желтка могут оставаться прикрепленными к вентральной поверхности ткани. Если это так, используйте инструмент для ресцев, чтобы аккуратно очистить оставшиеся гранулы желтка.
- Немедленно перенесите рассеченный эксплант на крышку 22 мм x 22 мм, на которой будет выполняться визуализация.
- Расположите эксплант плоско на дорсовентральной оси, вентральная сторона коснется чехловогослипа (рисунок 1В). Аккуратно удалите лишнюю жиму вокруг тканевой эксплант с помощью пипетки с фильтрованным наконечником 20 мкл.
ПРИМЕЧАНИЕ: Задержка переноса рассеченных эксплантов в покровный лист приводит к деформации ткани, так как она освобождается от геометрических ограничений желтка.
- Расположите эксплант плоско на дорсовентральной оси, вентральная сторона коснется чехловогослипа (рисунок 1В). Аккуратно удалите лишнюю жиму вокруг тканевой эксплант с помощью пипетки с фильтрованным наконечником 20 мкл.
- Быстро и осторожно переверните крышку эксплантом над заполненной камерой слайда со средой роста.
- Чтобы предотвратить образование пузырьков, поместите сторону квадратного покровного слипа на ленточную камеру и осторожно отпустите другую сторону. Позаботьтесь о том, чтобы не переместить /не деформировать эксплант на этом этапе.
- Осторожно удалите лишнюю среду, истекав кровью из камеры, надавливая скользящей камерой на лабораторную ткань. Крышка будет стабильно сидеть на слайд-камере для визуализации в реальном времени из-за поверхностного натяжения жидкой среды без какого-либо уплотнения.
- Для длительного культивирования (>6 часов) используйте камеру большего размера. В таких случаях можно использовать прямоугольные крышки 22 мм x 50 мм вместе с двумя параллельными полосами слоев ленты на слайдах. Между двумя ленточными полосами можно оставить зазор шириной ~1 мм, чтобы облегчить доступ воздуха в питательную среду.
- Повторите шаги 3.3-3.8, чтобы подготовить больше эксплантов. Подготовленные экспланты удлиняют свою ось тела A-P со средней скоростью ~ 30 мкм / ч и сегментируют свои сомиты с интервалами ~ 40 минут при 25 ° C(Рисунок 2A,Видео 1).
- Для неудлиняющихся эксплантов нанесите мягкое давление на стороны слайда, удерживающего образец на этапе 3.8.2, высасывая избыток среды на лабораторную ткань. В качестве альтернативы экспланты могут культивированы в однослойных скользящих камерах. Кроме того, химическая активация поверхности слайдовой камеры коллагеном типа I приведет к неудлиняющимся эксплантам(рисунок 2B,видео 2).
- Заранее выполняют покрытие камеры коллагеном I типа, полностью покрыв скользящие камеры 15-20 мл предварительно разбвеченного раствора коллагена при комнатной температуре в течение 1 ч. Используйте ламинарную вытяжку для этого протокола для поддержания стерильности. Тщательно промойте камеры со средой для рассечения на конце.
- Для эмбрионов старше 15 лет установите ткань хвоста эксплантат сбоку вместо плоского (дорсовентрального) крепления(Видео 3). Для предотвращения мышечных подергиваний в качестве анестетика3включайте 0,004% раствор трикаина в культуральную муляж.
- Для неудлиняющихся эксплантов нанесите мягкое давление на стороны слайда, удерживающего образец на этапе 3.8.2, высасывая избыток среды на лабораторную ткань. В качестве альтернативы экспланты могут культивированы в однослойных скользящих камерах. Кроме того, химическая активация поверхности слайдовой камеры коллагеном типа I приведет к неудлиняющимся эксплантам(рисунок 2B,видео 2).
4. Получение живого изображения
- Образцы изображений либо на рассечении для широкоугольной визуализации светопропуска размеров и периодов сегментации сомита, либо со структурированной микроскопией освещения / конфокального / светового листа с использованием трансгенных репортерных рыбьих линий.
- Уравновешивают температуру тканей эксплантов с помощью визуализации комнатной температуры в течение не менее 15 мин.
- Для более точного контроля температуры используйте коммерческую систему контроля температуры, установленную на инвертированный микроскоп.
- Установите интервалы кадра получения изображения на 2 - 10 минут в зависимости от интересующего биологического процесса.
ПРИМЕЧАНИЕ: Сегментация сомита рыбок данио является быстрым процессом, в диапазоне от 20 до 55 мин при жизнеспособных температурах от 30 ° C до 21,5 ° C у целых эмбрионов. Экспланты будут удлибатеться и сегментироваться ~ на 30% медленнее, чем целые эмбрионы.- Обратите внимание на то, чтобы оставить достаточную задержку между наборами каналов захвата, чтобы избежать возможной фототоксичности для живой ткани. Не подвергайте ткань воздействию пучка возбуждения более половины продолжительности визуализации и максимально снижайте интенсивность пучка.
ПРИМЕЧАНИЕ: Накопление активных форм кислорода (АФК), как правило, является основной причиной фототоксичности в живых образцах4. Аскорбиновая кислота в качестве мусорщика АФК может быть дополнена в питательную среду при концентрации 4 мМ для буферизации активности АФК и облегчения фототоксичности. Побочные эффекты фототоксичности может быть трудно заметить во время живой визуализации. Хвостовые экспланты являются выгодными в этом аспекте, поскольку некоторые визуальные маркеры фототоксичности, такие как митотическая остановка, затрудненное развитие тканей (т. Е. Образование сомитов, удлинение хвоста) и распадающаяся ткань, легче заметить. Пожалуйста, обратитесь к предоставленной ссылке4 для подробного обсуждения.
- Обратите внимание на то, чтобы оставить достаточную задержку между наборами каналов захвата, чтобы избежать возможной фототоксичности для живой ткани. Не подвергайте ткань воздействию пучка возбуждения более половины продолжительности визуализации и максимально снижайте интенсивность пучка.
- Используйте одноклеточную РНК, введенную эмбрионам, для получения 4-D изображений, предназначенных для сегментации и анализа на уровне клеточного разрешения.
- Используйте 300 пг РНК из транскрибированных мембран in vitro и ядерных флуоресцентных репортерных маркерных плазмид, таких как pCS-мембрана-ceruleanFP (Addgene plasmid #53749) или pCS-memb-mCherry (Addgene plasmid #53750) в сочетании с pCS2+ H2B-mTagBFP2 (Addgene plasmid #99267) или pCS2+ H2B-TagRFP-T (Addgene plasmid #99271) в инъекциях. Образец фильма с маркерами клеточной мембраны и ядер см. в видео 4.
ПРИМЕЧАНИЕ: Средний размер клеток ткани PSM составляет около ~5 мкм в диаметре, из которых ядра составляют ~3 - 4 мкм. Размер пикселя ~0,5 мкм и z-сечение ~1 мкм должны быть записаны для правильной сегментации клеток.
- Используйте 300 пг РНК из транскрибированных мембран in vitro и ядерных флуоресцентных репортерных маркерных плазмид, таких как pCS-мембрана-ceruleanFP (Addgene plasmid #53749) или pCS-memb-mCherry (Addgene plasmid #53750) в сочетании с pCS2+ H2B-mTagBFP2 (Addgene plasmid #99267) или pCS2+ H2B-TagRFP-T (Addgene plasmid #99271) в инъекциях. Образец фильма с маркерами клеточной мембраны и ядер см. в видео 4.
5. Иммуноокрашивание хвостовых эксплантов
ПРИМЕЧАНИЕ: Ткани, выращенные после различных сценариев рассечения (удлинение, неудлинение, рассечение хвостовой почки, половина PSM и т. Д.), Как плоского хвостовые экспланты5, могут быть извлечены из слайдовых камер для дальнейшего иммуноокрашивания количественных размеров интересующих белков. Здесь мы представляем протокол, используемый для окрашивания эксплантов дифосфорилированной внеклеточной регулируемой киназой (ppERK) в качестве считывания градиента сигналов FGF.
- После формирования сомитов до нужного этапа осторожно перекладывайте крышку наполовину в угол скользящей камеры без подъема.
- С помощью ~100 мкл дополнительной среды для рассечения в стеклянной пипетке Пастера извлекают экспланты из слайда и переносят в 64-скважинную пластину для культивовки клеток.
ПРИМЕЧАНИЕ: Начиная с этого этапа, все замены раствора могут быть выполнены под рассечением с помощью отдельной стеклянной пипетки для фиксированных образцов. Это позволит не терять эксплантные ткани в колодцах или переносить их между ними. - После переноса всех эксплантов высасывайте избыточную среду из скважин по одному и помещайте в каждую скважину 100 мкл 4% параформальдегида в ПБС (ПФА).
ВНИМАНИЕ: ПФА является токсичным раствором с канцерогенным действием. При обращении следует использовать правильные СИЗ. - Зафиксируйте экспланты в 64-скважинной пластине при комнатной температуре в течение 1 ч на шейкере.
- Тканевые экспланты более чувствительны к деформациям, чем целые эмбрионы. Отрегулируйте скорость шейкерного встряхивания соответствующим образом.
- Вымойте фиксатор 150 мкл PBS-Tw (0,1% Tween20 в PBS) три раза. Соберите первую стирку в специальный контейнер «PFA Waste».
- Обезвоживать экспланты на 4×5 мин, заменяя ~40 мкл раствора каждый раз 100% метанолом (MeOH).
ВНИМАНИЕ: MeOH является токсичным химическим веществом, которое является летучим и легковоспламеняющимся. Работайте в хорошо проветриваемом помещении и используйте надлежащие СИЗ для обработки. - В качестве последней ступени обезвоживания удалите весь раствор из скважин и замените 100 мкл MeOH. Инкубировать при -20 °C в течение 15 мин.
ПРИМЕЧАНИЕ: Используйте специальный контейнер "MeOH Waste" для сбора растворов до шага 5.11. - Добавьте 50 мкл MeOH и встряхните при комнатной температуре в течение 5 мин.
- Эксплантировать экспланты 4×5 мин путем замены ~40 мкл раствора каждый раз PBS-T (0,1% Triton-X 100 в PBS). Используйте специальный контейнер «MeOH Waste» для сбора решений.
- В качестве последней ступени регидратации удалите весь раствор из скважин и замените 100 мкл PBS-T.
- Для пермеабилизации тканей обрабатывают экспланты 1,5% Тритон-Х 100 в ПБС в течение 20 мин при комнатной температуре на шейкере.
- Промывайте образцы MAB-D-T (0,1% моющего средства Triton-X 100 и 1% диметилсульфоксида (DMSO) в 150 мМ NaCl 100 мМ буфера малеиновой кислоты pH 7,5) 3×5 мин.
ВНИМАНИЕ: ДМСО является легковоспламеняющимся и токсичным мутагеном. При обращении следует использовать правильные СИЗ. - Инкубировать экспланты в 100 мкл/хорошо блокирующий раствор сыворотки (2% фетальная бытовая сыворотка в MAB-D-T) в течение 2 часов при комнатной температуре.
- Заменить весь блокирующий раствор раствором первичного антитела 50-100 мкл/хорошо (разведение моноклональных мышиных антител против ppERK в сывороточной блокаде 1:1000). Инкубировать образцы O/N (>16 ч) при 4 °C на шейкере.
- Промыть раствор первичных антител МАБ-D-Т 5×5 мин.
- Инкубировать образцы во вторичном растворе антител (Alexa Fluor 597 козьи против мыши IgG2b (1:200) и Hoechst 33342 (1:5000) в MAB-D-T) O/N при 4 °C на шейкере или в течение 3 ч при комнатной температуре.
ПРИМЕЧАНИЕ: Начиная с этой ступени, покройте 64-скважинную пластину алюминиевой фольгой, чтобы избежать светового воздействия вторичных образцов, обработанных антителами.
ВНИМАНИЕ: Hoechst 33342 является потенциальным канцерогеном. При обращении следует использовать правильные СИЗ. - Промыть раствор вторичных антител PBS-Tw 3×5 мин.
- Зафиксируйте образцы с помощью PFA в течение 15 минут при комнатной температуре.
- Промыть фиксатор PBS-Tw и уравновесить образцы в пределах 60% глицерина. Установите экспланты на слайды микроскопа с лаком для ногтей и 60% глицерином для визуализации. Для репрезентативных результатов иммуноокрашивания, см. Рисунок 3.
Результаты
Этот протокол позволяет культивировать плоскую геометрическую культивирование живых эксплантов хвоста рыбки данио. Культура тканей имеет три основных преимущества перед целыми эмбрионами: 1) контроль скорости удлинения оси, 2) контроль над различными источниками сигнализации (морфог...
Обсуждение
В этой статье представлен подробный протокол метода эксплантатов тканевых культур, который мы разработали и недавно использовали5 для эмбрионов рыбок данио. Наша методика основана на предыдущих методах эксплантата у цыплят8 и рыбок данио9,
Раскрытие информации
Авторам нечего раскрывать и заявлять об отсутствии конфликта интересов.
Благодарности
Мы благодарим AECOM Zebrafish Core Facility и Cincinnati Children's Veterinary Services за содержание рыбы, Cincinnati Children's Imaging Core за техническую помощь, Didar Saparov за помощь в производстве видео и Hannah Seawall за редактирование рукописи. Исследования, представленные в этой публикации, были поддержаны Национальным институтом общих медицинских наук Национальных институтов здравоохранения под номером R35GM140805 для E.M.Ö. Содержание является исключительной ответственностью авторов и не обязательно отражает официальную точку зрения Национальных институтов здравоохранения.
Материалы
| Name | Company | Catalog Number | Comments |
| 1 mL Sub-Q Syringe with PrecisionGlide Needle | Becton, Dickinson and Co. | REF 309597 | for dechorionating embryos and manipulations |
| 200 Proof Ethanol, Anhydrous | Decon Labs | 2701 | for immunostaining |
| Antibiotic Antimycotic Solution (100×) | Sigma-Aldrich | A5955 | for tissue dissection media |
| Calcium Chloride Anhydrous, Powder | Sigma-Aldrich | 499609 | for tissue dissection media |
| Dimethylsulfoxide | Sigma-Aldrich | D5879 | for immunostaining |
| Disposable Scalpel, #10 Stainless Steel | Integra-Miltex | MIL4-411 | for preparing tape slide wells |
| Ethyl 3-aminobenzoate methanesulfonate salt (Tricaine) | Sigma-Aldrich | 886-86-2 | (optional) for anesthesizing tissues older than 20 somites stage |
| Fetal Bovine Serum (FBS) | ThermoFisher | A3160601 | additional for tissue culture media |
| Goat anti-Mouse IgG2b, Alexa Fluor 594 | Invitrogen | Cat#A-21145; RRID: AB_2535781 | secondary antibody for immunostaining |
| L-15 Medium with L-Glutamine w/o Phenol Red | GIBCO | 21083-027 | for tissue dissection media |
| Methanol | Sigma-Aldrich | 179337 | for immunostaining |
| Microsurgical Corneal Knife 2.85 mm Angled Tip Double Bevel Blade | Surgical Specialties | 72-2863 | for tissue dissection |
| Mouse monoclonal anti-ppERK | Sigma-Aldrich | Cat#M8159; RRID:AB_477245 | for ppERK immunostaining |
| NucRed Live 647 ReadyProbes Reagent | Invitrogen | R37106 | (optional) for live staining of cell nuclei |
| Paraformaldehyde Powder, 95% | Sigma-Aldrich | 158127 | for fixation of samples for immunostaining |
| Rat Tail Collagen Coating Solution | Sigma-Aldrich | 122-20 | (optional) for chemically activating slide chambers |
| Stage Top Incubator | Tokai Hit | tokai-hit-stxg | (optional) for temperature control during live imaging |
| Transparent Tape 3/4'' | Scotch | S-9782 | for preparing tape slide wells |
| Triton X-100 | Sigma-Aldrich | X100 | for immunostaining |
| Tween 20 | Sigma-Aldrich | P1379 | for immunostaining |
| Zebrafish: Tg(actb2:2xMCP-NLS-EGFP) | Campbell et al., 2015 | ZFIN: ZDB-TGCONSTRCT-150624-4 | transgenic fish with nuclear localized EGFP |
| Zebrafish: Tg(Ola.Actb:Hsa.HRAS-EGFP) | Cooper et al., 2005 | ZFIN: ZDB-TGCONSTRCT-070117-75 | transgenic fish with cell membrane localized EGFP |
Ссылки
- Assheton, R. . Growth in length: Embryological Essays. , (1916).
- Gomez, C., et al. Control of segment number in vertebrate embryos. Nature. 454 (7202), 335-339 (2008).
- Westerfield, M. . The Zebrafish Book: a guide for the laboratory use of zebrafish (Danio rerio), 3rd edition. , (1995).
- Icha, J., Weber, M., Waters, J. C., Norden, C. Phototoxicity in live fluorescence microscopy, and how to avoid it. BioEssays. 39 (1700003), (2017).
- Simsek, M. F., Ozbudak, E. M. Spatial fold change of Fgf signaling encodes positional information for segmental determination in zebrafish. Cell Reports. 24 (1), 66-78 (2018).
- Dubrulle, J., Pourquié, O. fgf8 mRNA decay establishes a gradient that couples axial elongation to patterning in the vertebrate embryo. Nature. 427 (6973), 419-422 (2004).
- Diez del Corral, R., et al. Opposing FGF and Retinoid Pathways Control Ventral Neural Pattern, Neuronal Differentiation, and Segmentation during Body Axis Extension. Neuron. 40 (1), 65-79 (2003).
- Stern, H. M., Hauschka, S. D. Neural tube and notochord promote in vitro myogenesis in single somite explants. Developmental Biology. 167 (1), 87-103 (1995).
- Langenberg, T., Brand, M., Cooper, M. S. Imaging brain development and organogenesis in zebrafish using immobilized embryonic explants. Developmental Dynamics. 228 (3), 464-474 (2003).
- Picker, A., Roellig, D., Pourquié, O., Oates, A. C., Brand, M. Tissue micromanipulation in zebrafish embryos. Methods in molecular biology. 546 (11), 153-172 (2009).
- Manning, A. J., Kimelman, D. Tbx16 and Msgn1 are required to establish directional cell migration of zebrafish mesodermal progenitors. Developmental Biology. 406 (2), 172-185 (2015).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental Dynamics. 203 (3), 253-310 (1995).
- Kaufmann, A., Mickoleit, M., Weber, M., Huisken, J. Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 139, 3242-3247 (2012).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены