
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Une culture d’explantation de queue 3D pour étudier la segmentation des vertébrés chez le poisson zèbre
Dans cet article
Résumé
Ici, nous présentons le protocole pour la culture tissulaire 3D de l’axe postérieur du corps du poisson zèbre, permettant l’étude vivante de la segmentation des vertébrés. Ce modèle d’explant fournit le contrôle sur l’élongation d’axe, l’altération des sources morphogènes, et l’imagerie vivante au niveau du tissu de résolution subcellulaire.
Résumé
Les embryons de vertébrés façonnent leur axe principal du corps comme des somites répétitives, les précurseurs des vertèbres, des muscles et de la peau. Les somites segmentent progressivement du mésoderme présomitique (PSM) à mesure que la queue de l’embryon s’allonge postérieurement. Les somites se forment avec une périodicité régulière et une échelle de taille. Le poisson zèbre est un organisme modèle populaire car il est génétiquement tractable et possède des embryons transparents qui permettent l’imagerie vivante. Néanmoins, au cours de la somitogenèse, les embryons de poisson sont enroulés autour d’un gros jaune arrondi. Cette géométrie limite l’imagerie en direct des tissus PSM dans les embryons de poissons zèbres, en particulier à des résolutions plus élevées qui nécessitent une distance de travail objective étroite. Ici, nous présentons une méthode de culture tissulaire 3D aplati pour l’imagerie vivante des explants de queue de poisson zèbre. Les explants de queue imitent des embryons intacts en montrant un ralentissement proportionnel de l’allongement de l’axe et un raccourcissement des longueurs de somite rostrocaudales. Nous sommes en outre en mesure de caler la vitesse d’allongement de l’axe grâce à la culture explant. Ceci, pour la première fois, nous permet de démêler l’entrée chimique des gradients de signalisation de l’entrée mécaniste de l’allongement axial. Dans de futures études, cette méthode peut être combinée avec une configuration microfluidique pour permettre des perturbations pharmaceutiques contrôlées dans le temps ou le dépistage de la segmentation des vertébrés sans aucun problème de pénétration des médicaments.
Introduction
La segmentation métamérique des organismes est largement utilisée dans la nature. Les structures répétées sont essentielles pour la fonctionnalité des organes latéraux tels que les vertèbres, les muscles, les nerfs, les vaisseaux, les membres ou les feuilles dans un plan corporel1. En raison de telles contraintes physiologiques et géométriques de la symétrie axiale, la plupart des embranchements de Bilateria- tels que les annélides, les arthropodes et les chordés- présentent une segmentation de leurs tissus embryonnaires (par exemple, ectoderme, mésoderme) antéro-postérieur.
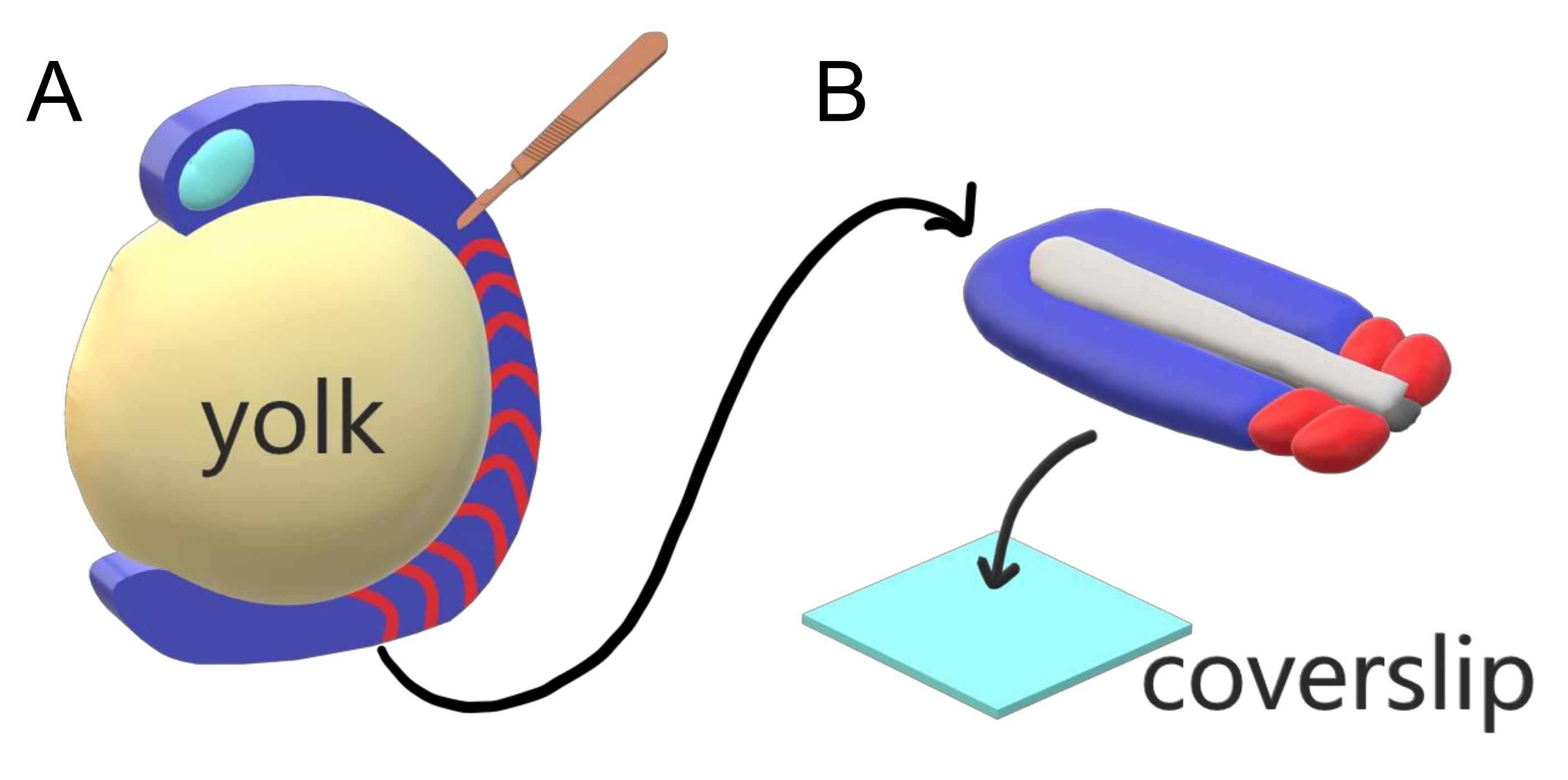
Les embryons de vertébrés segmentent séquentiellement leur mésoderme paraxial le long de l’axe principal du corps en somites avec des intervalles, des comptes et des distributions de taille spécifiques à l’espèce. Malgré une telle robustesse parmi les embryons individuels au sein d’une espèce, la segmentation de la somite est polyvalente entre les espèces de vertébrés. La segmentation se produit dans un vaste régime d’intervalles de temps (de 25 min chez le poisson zèbre à 5 h chez l’homme), de tailles (de ~20 μm dans les somites de queue de poisson zèbre à ~200 μm dans les somites de tronc de souris) et de comptages (de 32 chez les poissons zèbres à ~300 chez les serpents de maïs)2. Plus intéressant encore, les embryons de poissons peuvent se développer dans une large gamme de températures (de ~ 20,5 ° C à 34 ° C pour le poisson zèbre) tout en gardant leurs somites intactes avec des distributions de taille appropriées en compensant à la fois les intervalles de segmentation et les vitesses d’allongement axiales. Au-delà de ces caractéristiques intéressantes, le poisson zèbre reste un organisme modèle utile pour étudier la segmentation chez les vertébrés en raison du développement externe, synchrone et transparent d’une plénitude d’embryons de frères et sœurs ainsi que de leurs outils génétiques accessibles. Du point de vue de la microscopie, les embryons de téléostéos se développent sur un jaune sphérique volumineux, étirant et arrondissant le tissu gazeux qui l’entoure (Figure 1A). En cet article, nous présentons une culture aplatie d’explant de tissu 3D pour des queues de poisson zèbre. Ce système d’explantation contourne les contraintes sphériques de la masse vitellin, permettant l’accès à l’imagerie vivante haute résolution d’embryons de poissons pour le modelage de somite.

Figure 1: Système d’explantation à chambre coulissante pour les embryons de poisson zèbre. (A) Les embryons de poisson zèbre présentent des avantages pour l’imagerie vivante, tels que la transparence du tissu embryonnaire gastrulating (bleu), mais le tissu se forme autour d’une masse de jaune sphérique volumineuse (jaune) qui empêche l’imagerie proche de l’objectif et à haute résolution dans les embryons intacts. Les explants de queue peuvent être disséqués en commençant par un couteau microchirurgical (brun) coupé du tissu antérieur des somites (rouge) et en continuant à la frontière avec le jaune postérieurement. (B) Les explants de queue disséqués peuvent être placés sur une lamelle de couverture (bleu clair) dorsoventralement; garder le tissu neural (gris clair) sur le dessus et notochord (gris foncé) en bas. Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Protocole
Ce protocole implique l’utilisation d’embryons vertébrés vivants plus jeunes que 1 jour après la fécondation. Toutes les expériences sur les animaux ont été effectuées selon les directives éthiques du Cincinnati Children’s Hospital Medical Center; les protocoles relatifs aux animaux ont été examinés et approuvés par le Comité institutionnel de protection et d’utilisation des animaux (Protocole no 2017-0048).
1. Collecte d’embryons
- Installer des paires de poissons zèbres dans des réservoirs traversants la nuit précédant le jour de la collecte d’embryons. Pour un contrôle précis de la stadification du développement de l’embryon, utilisez des barrières entre les paires d’accouplement.
- Soulevez les barrières avant l’heure de frai préférée et recueillez les œufs dans les 15 minutes dans une boîte de Pétri de 100 mm.
- Nettoyer les débris de la boîte de Pétri. Si plus de 50 embryons sont collectés à partir d’une seule couvée, divisez la couvée en plusieurs boîtes de Pétri en conséquence.
- Incuber des embryons dans l’eau du système poissonnier à 28 °C jusqu’à ce qu’ils atteignent 50 % d’épiboly (5 heures après la fécondation). Un milieu de croissance embryonnaire normalisé tel que E3 peut également être utilisé à la place de l’eau du système d’aquarium jusqu’à l’étape 3.2.
- Retirer les œufs non fécondés sous un stéréoscope et déplacer les embryons vers un incubateur à 23,5 °C pendant la nuit (O/N). Les embryons doivent être à un stade de 8 à 10 somites le matin suivant le jour de la collecte.
2. Préparation de l’outil
- Stérilisez la lame du couteau de microchirurgie, les pointes d’aiguille (utilisées pour la dissection du tissu) et la pipette Pasteur en verre en trempant dans de l’éthanol à 100% (EtOH) et du vitrage feu.
- Utilisez deux couches de ruban transparent (~ 100-120 μm d’épaisseur) sur des lames de microscope de 25 mm x 75 mm. Coupez ~ 18 mm x 18 mm puits carrés au centre du ruban adhésif de chaque diapositive recouvert d’un scalpel.
- Essuyez les chambres à lame préparées avec 70% d’EtOH. Ces puits retiendront environ 40 μL de milieu.
3. Préparation de l’échantillon
- Embryons de dechorionate utilisant la pointe de deux seringues d’aiguille sous un stéréoscope. Transférer les embryons dans une boîte de Pétri séparée avec de l’eau du système de poisson à rincer.
- À l’aide d’une pipette Pasteur en verre stérile vitré au feu, transférer des embryons dans une boîte de Petri de 6 cm contenant un milieu de dissection (milieu de culture cellulaire Leibovitz-15 avec de la L-Glutamine sans rouge de phénol, 0,8 mMcaCl2 et 1× solution antibiotique-antimycotique).
REMARQUE : Continuez d’utiliser une pipette en verre stérilisé pour tous les transferts suivant cette étape.- Utilisez une boîte de Petri en verre pour la procédure d’explantation afin d’éviter les copeaux de polystyrène pendant la dissection.
- Mettre 50 μL de milieu de croissance tissulaire (milieu de dissection et 10 % de FBS) dans la chambre à glissière.
- Stabiliser un embryon pour la dissection sous le stéréoscope avec une pointe d’aiguille à l’intersection jaune-tissu près du cerveau postérieur.
- En maintenant le tissu embryonnaire stable à l’arme d’une aiguille, utilisez le couteau microchirurgical dont la lame est maintenue à 45° pour séparer le tissu antérieur au cerveau postérieur et décoller le jaune du tissu embryonnaire à partir de l’antérieur et en se déplaçant vers le bourgeon caudale(figure 1A).
REMARQUE: Veillez à ne pas perdre le tissu cutané lors du nettoyage du jaune. La peau se décollerait facilement comme un tissu élastique flanquant à une seule couche autour de l’embryon pendant la dissection, il est donc facile à reconnaître. - Une fois que le jaune est complètement retiré du corps embryonnaire, coupez le tissu cutané flanquant du tailbud. En gardant les 3-4 derniers somites formés intacts, coupez le tissu antérieur (explant à axe complet).
- Le jaune doit se détacher principalement intact de cette procédure. En cas de rupture du jaune, des granules importants de jaune peuvent rester attachés à la surface ventrale du tissu. Si c’est le cas, utilisez un outil de cils pour nettoyer doucement les granules de jaune restants.
REMARQUE: Un déséquilibre des tissus cutanés sur les côtés latéraux d’une explantation ne permettrait pas au tissu de maintenir une direction de croissance droite. L’explant se plie plutôt vers le côté d’une peau plus étirée. Ce déséquilibre peut être corrigé sous le stéréoscope en rompant la couche cutanée à l’aide d’un couteau microchirurgical. - Pour les explants sans peau, appuyez sur une pointe de la couche de peau avec une aiguille et décollez le tissu explant avec le couteau microchirurgical. Ces explants n’allferont pas leur axe corporel en culture.
- En plus des explants d’axe complet, des explants alternatifs peuvent être fabriqués à cette étape. Par exemple, disséquer les somites déjà segmentés à l’aide du couteau microchirurgical (explants psm complets) ou disséquer le PSM dans son demi-antéropostérieur (explants mi-PSM). Veuillez consulter la section 5.1 pour une application de ces explants de remplacement.
- Le jaune doit se détacher principalement intact de cette procédure. En cas de rupture du jaune, des granules importants de jaune peuvent rester attachés à la surface ventrale du tissu. Si c’est le cas, utilisez un outil de cils pour nettoyer doucement les granules de jaune restants.
- Transférer immédiatement l’explant disséqué sur une lamelle de 22 mm x 22 mm sur laquelle l’imagerie sera effectuée.
- Disposer l’explant à plat sur l’axe dorsoventral, la face ventrale touchant la lamelle(figure 1B). Retirez délicatement l’excès de milieux autour de l’explant de tissu à l’aide d’une pipette à pointe filtrée de 20 μL.
REMARQUE: Le transfert retardé des explants disséqués à la lamelle entraîne des déformations du tissu, car il est soulagé des contraintes géométriques du jaune.
- Disposer l’explant à plat sur l’axe dorsoventral, la face ventrale touchant la lamelle(figure 1B). Retirez délicatement l’excès de milieux autour de l’explant de tissu à l’aide d’une pipette à pointe filtrée de 20 μL.
- Retournez rapidement et soigneusement la lame de couverture avec l’explant sur la chambre à glissière remplie de milieu de croissance.
- Pour éviter la formation de bulles, placez un côté de la lamelle de couverture carrée sur la chambre à ruban adhésif et relâchez doucement l’autre côté. Veillez à ne pas déplacer/déformer l’explant dans cette étape.
- Retirez délicatement l’excès de saignements de milieux hors de la chambre en appuyant sur la chambre à glissière sur un tissu de laboratoire. La lamelle de couverture reposera de manière stable sur la chambre à glissière pour l’imagerie en direct, en raison de la tension superficielle du milieu liquide sans aucune étanchéité.
- Pour la culture à long terme (>6 heures), utilisez une chambre plus grande. Des lamelle rectangulaires de 22 mm x 50 mm ainsi que deux voies parallèles de couches de ruban adhésif sur des glissières peuvent être utilisées dans de tels cas. Un espace d’environ 1 mm de large peut être laissé entre deux voies de ruban pour faciliter l’accès de l’air dans le milieu de croissance.
- Répétez les étapes 3.3-3.8 pour préparer plus d’explants. Les explants préparés allineront leur axe corporel A-P avec une vitesse moyenne d’environ 30 μm/h et segmenteront leurs somites avec des intervalles d’environ 40 minutes à 25 °C(Figure 2A,Vidéo 1).
- Pour les explants non allongés, appliquer une pression douce sur les côtés de la lame qui retient l’échantillon à l’étape 3.8.2 tout en aspirant l’excès de milieu sur un tissu de laboratoire. Alternativement, les explants peuvent être cultivés dans des chambres à glissière à couche de ruban unique. En outre, l’activation chimique de la surface de la chambre à glissière avec du collagène de type I entraînera des explants non allongés(Figure 2B,Vidéo 2).
- Effectuer le revêtement de la chambre avec du collagène de type I à l’avance en couvrant entièrement les chambres à glissière avec 15-20 mL de solution de collagène prédiluée à température ambiante pendant 1 h. Utilisez une hotte à flux laminaire pour ce protocole afin de maintenir la stérilité. Rincer soigneusement les chambres avec un milieu de dissection à la fin.
- Pour les embryons de plus de 15 somite, monter latéralement le tissu explantaire de la queue au lieu d’un support plat (dorsoventral) (Vidéo 3). Pour prévenir les contractions musculaires, inclure une solution de tricaine à 0,004% dans les milieux de culture en tant qu’agent anesthésique3.
- Pour les explants non allongés, appliquer une pression douce sur les côtés de la lame qui retient l’échantillon à l’étape 3.8.2 tout en aspirant l’excès de milieu sur un tissu de laboratoire. Alternativement, les explants peuvent être cultivés dans des chambres à glissière à couche de ruban unique. En outre, l’activation chimique de la surface de la chambre à glissière avec du collagène de type I entraînera des explants non allongés(Figure 2B,Vidéo 2).
4. Acquisition d’images en direct
- Échantillons d’images soit sur une portée de dissection pour l’imagerie de lumière transmise à large champ des tailles et des périodes de segmentation de somite, soit avec éclairage structuré / confocal / microscopie à feuille de lumière utilisant des lignes de poissons rapporteurs transgéniques.
- Équilibrer la température des explants tissulaires avec la température ambiante d’imagerie pendant au moins 15 min.
- Pour un contrôle de température plus précis, utilisez un système commercial de contrôle de la température monté sur un microscope inversé.
- Définissez les intervalles de trame d’acquisition d’image sur 2 à 10 min en fonction du processus biologique d’intérêt.
REMARQUE: La segmentation de la somite du poisson zèbre est un processus rapide, allant de 20 à 55 min pour des températures viables de 30 ° C à 21,5 ° C dans des embryons entiers. Les explants s’allongeront et se segmenteront environ 30% plus lentement que les embryons entiers.- Faites attention à laisser suffisamment de délai entre les séries d’acquisitions de canaux pour éviter une phototoxicité possible des tissus vivants. N’exposez pas le tissu au faisceau d’excitation pendant plus de la moitié de la durée de l’imagerie et abaissez l’intensité du faisceau autant que possible.
NOTA : L’accumulation d’espèces réactives de l’oxygène (ROS) est généralement la principale cause de phototoxicité dans les échantillons vivants4. L’acide ascorbique en tant qu’extracteur de ROS peut être complété au milieu de croissance à une concentration de 4 mM pour amortir l’activité des ROS et soulager la phototoxicité. Les effets indésirables de la phototoxicité peuvent être difficiles à remarquer lors de l’imagerie en direct. Les explants de queue sont avantageux à cet égard puisque certains marqueurs visuels de phototoxicité tels que l’arrêt mitotique, le développement tissulaire entravé (c.-à-d. formation de somites, allongement de la queue) et la désintégration des tissus sont plus faciles à remarquer. Veuillez vous référer à la référence4 fournie pour une discussion détaillée.
- Faites attention à laisser suffisamment de délai entre les séries d’acquisitions de canaux pour éviter une phototoxicité possible des tissus vivants. N’exposez pas le tissu au faisceau d’excitation pendant plus de la moitié de la durée de l’imagerie et abaissez l’intensité du faisceau autant que possible.
- Utilisez des embryons injectés d’ARN au stade cellulaire unique pour acquérir des images 4D destinées à être segmentées et analysées au niveau de résolution cellulaire.
- Utilisez 300 pg d’ARN provenant de membranes transcrites in vitro et de plasmides marqueurs rapporteurs fluorescents nucléaires tels que pCS-membrane-ceruleanFP (plasmide Addgene #53749) ou pCS-memb-mCherry (plasmide Addgene #53750) en combinaison avec pCS2+ H2B-mTagBFP2 (plasmide Addgene #99267) ou pCS2+ H2B-TagRFP-T (Addgene plasmid #99271) dans les injections. Pour un exemple de film avec membrane cellulaire et marqueurs de noyaux s’il vous plaît voir Vidéo 4.
REMARQUE: La taille moyenne des cellules du tissu PSM est d’environ ~ 5 μm de diamètre, dont les noyaux comprennent ~ 3 - 4 μm. Une taille de pixel d’environ 0,5 μm et une section z d’environ 1 μm doivent être enregistrées pour une segmentation cellulaire appropriée.
- Utilisez 300 pg d’ARN provenant de membranes transcrites in vitro et de plasmides marqueurs rapporteurs fluorescents nucléaires tels que pCS-membrane-ceruleanFP (plasmide Addgene #53749) ou pCS-memb-mCherry (plasmide Addgene #53750) en combinaison avec pCS2+ H2B-mTagBFP2 (plasmide Addgene #99267) ou pCS2+ H2B-TagRFP-T (Addgene plasmid #99271) dans les injections. Pour un exemple de film avec membrane cellulaire et marqueurs de noyaux s’il vous plaît voir Vidéo 4.
5. Immunomarquage des explants de la queue
REMARQUE: Les tissus cultivés après divers scénarios de dissection (allongé, non allongé, bourgeon de queue disséqué, demi-PSM, etc.) sous forme d’explants de queue montés à plat5 peuvent être récupérés à partir de chambres à lame pour d’autres quantifications immunostaining des protéines d’intérêt. Ici, nous présentons le protocole utilisé pour la coloration de la kinase régulée par signal extracellulaire di-phosphorylée (ppERK) des explants en tant que lecture du gradient de signalisation FGF.
- Après la formation de somites jusqu’au stade souhaité, déplacez prudemment la lame de couverture à mi-chemin vers le coin de la chambre à glissière sans soulever.
- À l’aide d’environ 100 μL de milieu de dissection supplémentaire dans une pipette Pasteur en verre, récupérez les explants de la lame et transférez-les dans une plaque de culture cellulaire à 64 puits.
REMARQUE: À partir de cette étape, tous les remplacements de solution peuvent être effectués sous une portée de dissection avec une pipette en verre distincte pour les échantillons fixes. Cela garantira de ne pas perdre de tissus explant dans les puits ou de les transférer entre les deux. - Après avoir transféré toutes les explants, aspirez le milieu excessif hors des puits un par un et mettez 100 μL de paraformaldéhyde à 4% dans du PBS (PFA) dans chaque puits.
ATTENTION : Le PFA est une solution toxique aux effets cancérigènes. Un EPI approprié doit être utilisé pendant la manipulation. - Fixer les explants dans une plaque de 64 puits à température ambiante pendant 1 h sur un agitateur.
- Les explants tissulaires sont plus sensibles aux déformations que les embryons entiers. Ajustez la vitesse du shaker en conséquence.
- Laver le fixateur avec 150 μL de PBS-Tw (0,1 % tween20 dans pbs) trois fois. Collectez le premier lavage dans un récipient spécifique « PFA Waste ».
- Déshydrater les explants par étapes de 4×5 min en remplaçant ~40 μL de solution à chaque fois par du méthanol à 100% (MeOH).
ATTENTION : Le MeOH est un produit chimique toxique qui est volatil et inflammable. Travaillez dans un espace bien ventilé et utilisez un EPI approprié pour la manipulation. - Comme dernière étape de déshydratation, retirer toute la solution des puits et la remplacer par 100 μL de MeOH. Incuber à -20 °C pendant 15 min.
REMARQUE: Utilisez un conteneur spécifique « MeOH Waste » pour collecter les solutions jusqu’à l’étape 5.11. - Ajouter 50 μL de MeOH et agiter à température ambiante pendant 5 min.
- Réhydrater les explants par étapes de 4×5 min en remplaçant ~40 μL de solution à chaque fois par du PBS-T (0,1 % de Triton-X 100 dans du PBS). Utilisez un conteneur spécifique « MeOH Waste » pour collecter les solutions.
- Comme dernière étape de la réhydratation, retirer toute la solution des puits et la remplacer par 100 μL de PBS-T.
- Pour la perméabilisation tissulaire, traiter les explants avec 1,5% de Triton-X 100 dans du PBS pendant 20 min à température ambiante sur le shaker.
- Laver les échantillons avec du détergent MAB-D-T (0,1 % de détergent Triton-X 100 et 1 % de diméthylsulfoxyde (DMSO) dans un tampon d’acide maléique 150 mM NaCl 100 mM pH 7,5) 3×5 min.
ATTENTION : Le DMSO est inflammable et mutagène toxique. Un EPI approprié doit être utilisé pendant la manipulation. - Incuber les plantes dans une solution de blocage sérique de 100 μL/puits (2 % de sérum fœtal bovin dans du MAB-D-T) pendant 2 heures à température ambiante.
- Remplacer toute la solution bloquante par une solution d’anticorps primaires de 50 à 100 μL/puits (dilution 1:1000 de l’anticorps monoclonal de souris contre le ppERK dans le bloc sérique). Incuber les échantillons O/N (>16 h) à 4 °C sur agitateur.
- Laver la solution d’anticorps primaires avec MAB-D-T 5×5 min.
- Incuber des échantillons dans une solution d’anticorps secondaires (Alexa Fluor 597 chèvre anti-souris IgG2b (1:200) et Hoechst 33342 (1:5000) dans MAB-D-T) O/N à 4 °C sur un agitateur ou pendant 3 h à température ambiante.
REMARQUE: À partir de cette étape, couvrez la plaque de 64 puits avec du papier d’aluminium pour éviter l’exposition légère d’échantillons d’anticorps secondaires traités.
ATTENTION : Hoechst 33342 est un cancérogène potentiel. Un EPI approprié doit être utilisé pendant la manipulation. - Laver la solution d’anticorps secondaire avec PBS-Tw 3×5 min.
- Fixer les échantillons avec PFA pendant 15 minutes à température ambiante.
- Laver le fixateur avec pbs-tw et équilibrer les échantillons dans 60% de glycérol. Monter des explants sur des lames de microscope avec du vernis à ongles et 60% de glycérol pour l’imagerie. Pour des résultats d’immunomarquage représentatifs, veuillez consulter la figure 3.
Résultats
Ce protocole permet la culture géométrique plate d’explants de queue de poisson zèbre vivants. La culture de tissu présente trois avantages principaux au-dessus des embryons entiers : 1) contrôle de la vitesse d’élongation d’axe, 2) contrôle au-dessus de diverses sources de signalisation (morphogène) par dissection simple, et 3) presque-objectif, grossissement élevé et formation image vivante élevée de NA.
Les chambres à lame chimiquement non traitées permettent à l’expl...
Discussion
Cet article présente un protocole détaillé d’une technique d’explant de culture de tissu que nous avons développée et employée récemment5 pour des embryons de poisson zèbre. Notre technique s’appuie sur les méthodes explant précédentes chez les poussins8 et les poissonszèbres 9,10,11 organismes modèles. Les explants de queue préparés avec ce protocole peuvent s...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer et ne déclarent aucun conflit d’intérêts.
Remerciements
Nous remercions l’AECOM Zebrafish Core Facility et les Cincinnati Children’s Veterinary Services pour l’entretien du poisson, le Cincinnati Children’s Imaging Core pour l’assistance technique, Didar Saparov pour l’aide à la production vidéo et Hannah Seawall pour l’édition du manuscrit. La recherche rapportée dans cette publication a été soutenue par le National Institute of General Medical Sciences des National Institutes of Health sous le numéro de bourse R35GM140805 à E.M.Ö. Le contenu relève de la seule responsabilité des auteurs et ne représente pas nécessairement les opinions officielles des National Institutes of Health.
matériels
| Name | Company | Catalog Number | Comments |
| 1 mL Sub-Q Syringe with PrecisionGlide Needle | Becton, Dickinson and Co. | REF 309597 | for dechorionating embryos and manipulations |
| 200 Proof Ethanol, Anhydrous | Decon Labs | 2701 | for immunostaining |
| Antibiotic Antimycotic Solution (100×) | Sigma-Aldrich | A5955 | for tissue dissection media |
| Calcium Chloride Anhydrous, Powder | Sigma-Aldrich | 499609 | for tissue dissection media |
| Dimethylsulfoxide | Sigma-Aldrich | D5879 | for immunostaining |
| Disposable Scalpel, #10 Stainless Steel | Integra-Miltex | MIL4-411 | for preparing tape slide wells |
| Ethyl 3-aminobenzoate methanesulfonate salt (Tricaine) | Sigma-Aldrich | 886-86-2 | (optional) for anesthesizing tissues older than 20 somites stage |
| Fetal Bovine Serum (FBS) | ThermoFisher | A3160601 | additional for tissue culture media |
| Goat anti-Mouse IgG2b, Alexa Fluor 594 | Invitrogen | Cat#A-21145; RRID: AB_2535781 | secondary antibody for immunostaining |
| L-15 Medium with L-Glutamine w/o Phenol Red | GIBCO | 21083-027 | for tissue dissection media |
| Methanol | Sigma-Aldrich | 179337 | for immunostaining |
| Microsurgical Corneal Knife 2.85 mm Angled Tip Double Bevel Blade | Surgical Specialties | 72-2863 | for tissue dissection |
| Mouse monoclonal anti-ppERK | Sigma-Aldrich | Cat#M8159; RRID:AB_477245 | for ppERK immunostaining |
| NucRed Live 647 ReadyProbes Reagent | Invitrogen | R37106 | (optional) for live staining of cell nuclei |
| Paraformaldehyde Powder, 95% | Sigma-Aldrich | 158127 | for fixation of samples for immunostaining |
| Rat Tail Collagen Coating Solution | Sigma-Aldrich | 122-20 | (optional) for chemically activating slide chambers |
| Stage Top Incubator | Tokai Hit | tokai-hit-stxg | (optional) for temperature control during live imaging |
| Transparent Tape 3/4'' | Scotch | S-9782 | for preparing tape slide wells |
| Triton X-100 | Sigma-Aldrich | X100 | for immunostaining |
| Tween 20 | Sigma-Aldrich | P1379 | for immunostaining |
| Zebrafish: Tg(actb2:2xMCP-NLS-EGFP) | Campbell et al., 2015 | ZFIN: ZDB-TGCONSTRCT-150624-4 | transgenic fish with nuclear localized EGFP |
| Zebrafish: Tg(Ola.Actb:Hsa.HRAS-EGFP) | Cooper et al., 2005 | ZFIN: ZDB-TGCONSTRCT-070117-75 | transgenic fish with cell membrane localized EGFP |
Références
- Assheton, R. . Growth in length: Embryological Essays. , (1916).
- Gomez, C., et al. Control of segment number in vertebrate embryos. Nature. 454 (7202), 335-339 (2008).
- Westerfield, M. . The Zebrafish Book: a guide for the laboratory use of zebrafish (Danio rerio), 3rd edition. , (1995).
- Icha, J., Weber, M., Waters, J. C., Norden, C. Phototoxicity in live fluorescence microscopy, and how to avoid it. BioEssays. 39 (1700003), (2017).
- Simsek, M. F., Ozbudak, E. M. Spatial fold change of Fgf signaling encodes positional information for segmental determination in zebrafish. Cell Reports. 24 (1), 66-78 (2018).
- Dubrulle, J., Pourquié, O. fgf8 mRNA decay establishes a gradient that couples axial elongation to patterning in the vertebrate embryo. Nature. 427 (6973), 419-422 (2004).
- Diez del Corral, R., et al. Opposing FGF and Retinoid Pathways Control Ventral Neural Pattern, Neuronal Differentiation, and Segmentation during Body Axis Extension. Neuron. 40 (1), 65-79 (2003).
- Stern, H. M., Hauschka, S. D. Neural tube and notochord promote in vitro myogenesis in single somite explants. Developmental Biology. 167 (1), 87-103 (1995).
- Langenberg, T., Brand, M., Cooper, M. S. Imaging brain development and organogenesis in zebrafish using immobilized embryonic explants. Developmental Dynamics. 228 (3), 464-474 (2003).
- Picker, A., Roellig, D., Pourquié, O., Oates, A. C., Brand, M. Tissue micromanipulation in zebrafish embryos. Methods in molecular biology. 546 (11), 153-172 (2009).
- Manning, A. J., Kimelman, D. Tbx16 and Msgn1 are required to establish directional cell migration of zebrafish mesodermal progenitors. Developmental Biology. 406 (2), 172-185 (2015).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental Dynamics. 203 (3), 253-310 (1995).
- Kaufmann, A., Mickoleit, M., Weber, M., Huisken, J. Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 139, 3242-3247 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.