
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Eine 3D-Schwanz-Explant-Kultur zur Untersuchung der Wirbeltiersegmentierung bei Zebrafischen
In diesem Artikel
Zusammenfassung
Hier stellen wir das Protokoll für die 3D-Gewebekultur der hinteren Körperachse des Zebrafischs vor, das eine Live-Untersuchung der Wirbeltiersegmentierung ermöglicht. Dieses Explant-Modell bietet Kontrolle über die Achsenverlängerung, die Veränderung von Morphogenquellen und die Subzellularauflösung auf Gewebeebene.
Zusammenfassung
Wirbeltierembryonen strukturieren ihre Hauptkörperachse als sich wiederholende Somiten, die Vorläufer von Wirbeln, Muskeln und Haut. Somites segmentieren sich progressiv vom präsomitischen Mesoderm (PSM), da sich das Schwanzende des Embryos nach hinterde verlängert. Somites bilden sich mit regelmäßiger Periodizität und Skalierung in der Größe. Zebrafisch ist ein beliebter Modellorganismus, da er genetisch handhabbar ist und transparente Embryonen hat, die Eine Live-Bildgebung ermöglichen. Dennoch werden während der Somitogenese Fischembryonen um ein großes, abgerundetes Eigelb gewickelt. Diese Geometrie schränkt die Live-Bildgebung von PSM-Gewebe in Zebrafischembryonen ein, insbesondere bei höheren Auflösungen, die einen engen objektiven Arbeitsabstand erfordern. Hier stellen wir eine abgeflachte 3D-Gewebekulturmethode zur Live-Bildgebung von Zebrafischschwanz-Explantierungen vor. Schwanzentspannen ahmen intakte Embryonen nach, indem sie eine proportionale Verlangsamung der Achsenverlängerung und eine Verkürzung der rostrocaudalen Somitenlängen zeigen. Wir sind außerdem in der Lage, die Achsendehnungsgeschwindigkeit durch Explant-Kultur zu stoppen. Dies ermöglicht es uns erstmals, den chemischen Eingang von Signalgradienten vom mechanistischen Eingang der axialen Dehnung zu entwirren. In zukünftigen Studien kann diese Methode mit einem mikrofluidischen Aufbau kombiniert werden, um zeitkontrollierte pharmazeutische Störungen oder ein Screening der Wirbeltiersegmentierung ohne Bedenken hinsichtlich der Arzneimittelpenetration zu ermöglichen.
Einleitung
Die metamere Segmentierung von Organismen ist in der Natur weit verbreitet. Wiederholte Strukturen sind essentiell für die Funktionalität von Seitenorganen wie Wirbeln, Muskeln, Nerven, Gefäßen, Gliedmaßen oder Blättern in einem Körperplan1. Als Ergebnis solcher physiologischen und geometrischen Einschränkungen der axialen Symmetrie weisen die meisten Phyla von Bilateria - wie Anneliden, Arthropoden und Chordaten - eine Segmentierung ihres embryonalen Gewebes (z. B. Ektoderm, Mesoderm) antero-posterior auf.
Wirbeltierembryonen segmentieren ihr paraxiales Mesoderm entlang der Hauptkörperachse sequenziell in Somiten mit artspezifischen Intervallen, Zählungen und Größenverteilungen. Trotz dieser Robustheit bei einzelnen Embryonen innerhalb einer Art ist die somite Segmentierung zwischen Wirbeltierarten vielseitig. Die Segmentierung erfolgt in einem riesigen Regime von Zeitintervallen (von 25 min bei Zebrafischen bis 5 h beim Menschen), Größen (von ~20 μm bei Schwanz somiten von Zebrafischen bis zu ~200 μm bei Stamm somites von Mäusen) und Anzahl (von 32 bei Zebrafischen bis ~300 bei Kornschlangen)2. Interessanterweise können sich Fischembryonen in einem weiten Temperaturbereich entwickeln (von ~ 20,5 ° C bis zu 34 ° C für Zebrafische), während sie ihre Somiten mit den richtigen Größenverteilungen intakt halten, indem sie sowohl Segmentierungsintervalle als auch axiale Dehnungsgeschwindigkeiten ausgleichen. Über diese interessanten Merkmale hinaus bleibt Zebrafisch ein nützlicher Modellorganismus, um die Segmentierung bei Wirbeltieren aufgrund der äußeren, synchronen und transparenten Entwicklung einer Fülle von Geschwisterembryonen sowie ihrer zugänglichen genetischen Werkzeuge zu untersuchen. Aus mikroskopischer Sicht entwickeln sich Teleost-Embryonen auf einem sperrigen kugelförmigen Eigelb, dehnen und runden das gastrulierende Gewebe um es herum (Abbildung 1A). In diesem Artikel stellen wir eine abgeflachte 3D-Gewebe-Explant-Kultur für Zebrafischschwänze vor. Dieses Explant-System umgeht die sphärischen Einschränkungen der Dottermasse und ermöglicht den Zugang zu hochauflösenden Live-Imagings von Fischembryonen für die somite Musterung.

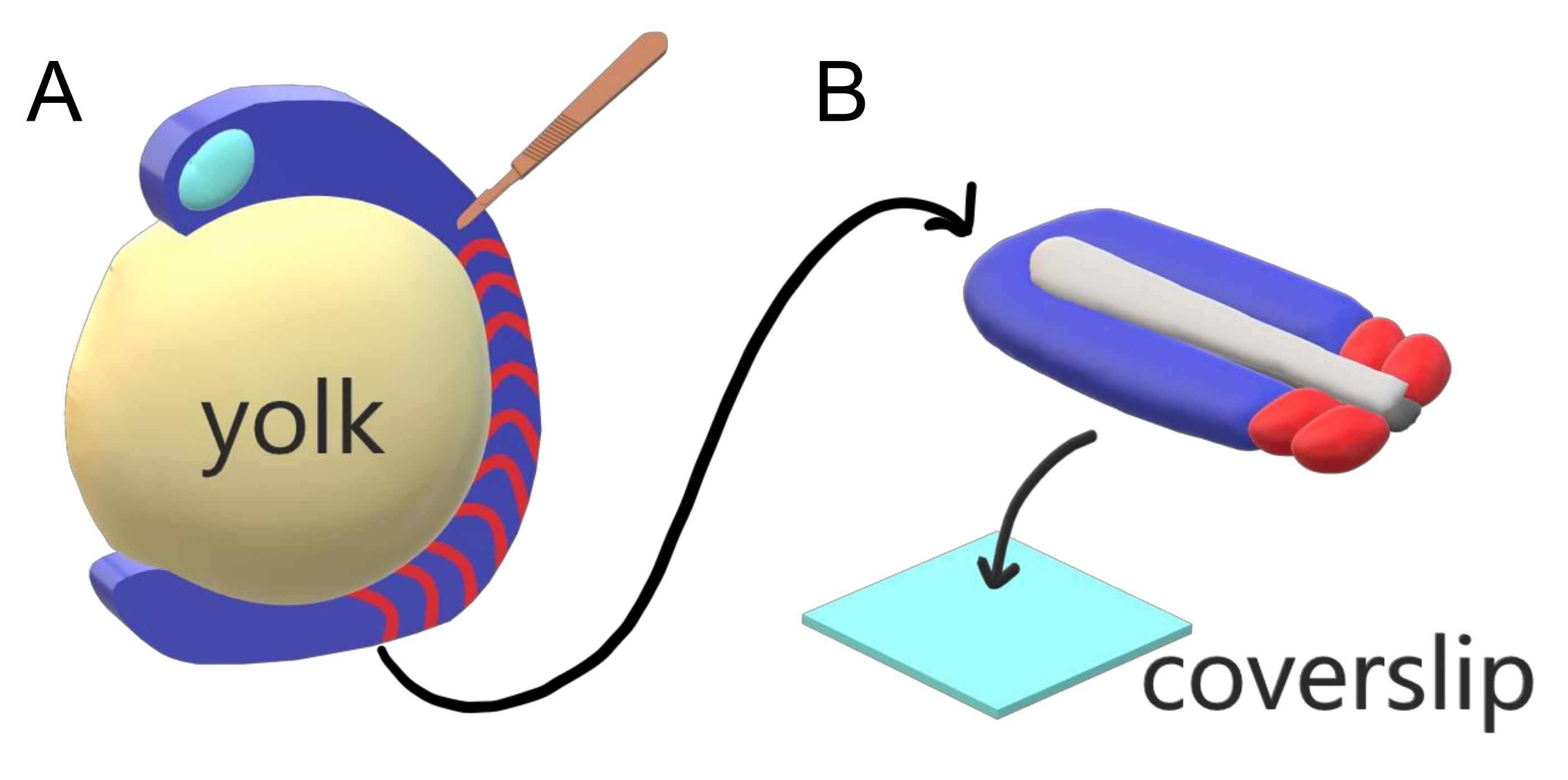
Abbildung 1: Diakammer-Explant-System für Zebrafischembryonen. (A) Zebrafischembryonen haben Vorteile für die Live-Bildgebung, wie die Transparenz von gastrulierendem embryonalem Gewebe (blau), aber das Gewebe bildet sich um eine sperrige kugelförmige Eigelbmasse (gelb), die eine nahezu objektive, hochauflösende Bildgebung in intakten Embryonen verhindert. Schwanzentstellungen können seziert werden, beginnend mit einem mikrochirurgischen Messer (braun), das aus dem Gewebe vor somites (rot) geschnitten wird und an der Grenze mit dem Eigelb posterior fortgesetzt wird. (B) Sezierte Schwanzausscheidungen können dorsoventral auf einen Abdeckungsrutsch (hellblau) gelegt werden; hält Neuralgewebe (hellgrau) oben und Notochord (dunkelgrau) unten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Protokoll
Dieses Protokoll beinhaltet die Verwendung von lebenden Wirbeltierembryonen, die jünger als 1 Tag nach der Befruchtung sind. Alle Tierversuche wurden nach den ethischen Richtlinien des Cincinnati Children's Hospital Medical Center durchgeführt; Tierprotokolle wurden vom Institutional Animal Care and Use Committee überprüft und genehmigt (Protokoll Nr. 2017-0048).
1. Embryonenentnahme
- Stellen Sie In der Nacht vor dem Embryonensammeltag Zebrafischpaare in Kreuzungsbecken auf. Für eine präzise Staging-Kontrolle der Embryonalentwicklung verwenden Sie Barrieren zwischen Paarungspaaren.
- Heben Sie Barrieren vor der bevorzugten Laichzeit an und sammeln Sie Eier innerhalb von 15 Minuten in einer 100 mm Petrischale.
- Reinigen Sie die Trümmer aus der Petrischale. Wenn mehr als 50 Embryonen aus einem einzigen Gelege entnommen werden, teilen Sie das Gelege entsprechend in mehrere Petrischalen auf.
- Inkubieren Sie Embryonen im Fischsystemwasser bei 28 °C, bis sie ein Epibolystadium von 50% erreichen (5 Stunden nach der Befruchtung). Anstelle des Aquariensystems Wasser bis Schritt 3.2 kann auch ein standardisiertes Embryowachstumsmedium wie E3 verwendet werden.
- Entfernen Sie unbefruchtete Eizellen unter einem Stereoskop und bewegen Sie die Embryonen über Nacht in einen 23,5 °C-Inkubator (O/N). Die Embryonen sollten am Morgen nach dem Entnahmetag im Stadium von 8-10 Somites sein.
2. Werkzeugvorbereitung
- Sterilisieren Sie die mikrochirurgische Messerklinge, die Nadelspitzen (zum Sezieren des Gewebes) und die Pasteur-Pipette aus Glas, indem Sie sie in 100% Ethanol (EtOH) und Feuerglasur einweichen.
- Verwenden Sie zwei Schichten transparentes Klebeband (~100-120 μm Dicke) auf Mikroskopobjektträgern von 25 mm x 75 mm. Schneiden Sie ~ 18 mm x 18 mm quadratische Vertiefungen in der Mitte der Bandabdeckung jedes Dias mit einem Skalpell.
- Wischen Sie die vorbereiteten Gleitkammern mit 70% EtOH ab. Diese Vertiefungen halten ~ 40 μL Medium.
3. Probenvorbereitung
- Enthorionieren Sie Embryonen mit der Spitze von zwei Nadelspritzen unter einem Stereoskop. Übertragen Sie die Embryonen in eine separate Petrischale mit Fischsystemwasser zum Spülen.
- Übertragen Sie die Embryonen mit einer feuerglasierten sterilen Pasteur-Pipette in eine 6 cm große Petrischale mit Dissektionsmedium (Leibovitz-15-Zellkulturmedium mit L-Glutamin ohne Phenolrot, 0,8 mM CaCl2 und 1× antibiotisch-antimykotische Lösung).
HINWEIS: Verwenden Sie weiterhin eine sterilisierte Glaspipette für alle Transfers nach diesem Schritt.- Verwenden Sie eine Glas-Petrischale für die Explanzierung, um Polystyrol-Chips während der Dissektion zu vermeiden.
- 50 μL Gewebewachstumsmedium (Dissektionsmedium und 10% FBS) in die Objektträgerkammer geben.
- Stabilisieren Sie einen Embryo zur Dissektion unter dem Stereoskop mit einer Nadelspitze an der Eigelb-Gewebe-Kreuzung in der Nähe des Hinterhirns.
- Halten Sie das embryonale Gewebe mit einer Nadel stabil, verwenden Sie das mikrochirurgische Messer mit der Klinge, die bei 45° gehalten wird, um das Gewebe vor dem Hinterhirn auseinander zu schneiden und das Eigelb vom embryonalen Gewebe zu schälen, beginnend mit dem vorderen Und in Richtung der Schwanzknospe (Abbildung 1A).
HINWEIS: Achten Sie darauf, das Hautgewebe beim Reinigen des Eigelb nicht zu verlieren. Die Haut würde sich während der Dissektion leicht als flankierendes einschichtiges elastisches Gewebe um den Embryo ablösen, so dass es leicht zu erkennen ist. - Sobald das Eigelb vollständig aus dem embryonalen Körper entfernt ist, schneiden Sie das flankierende Hautgewebe aus der Schwanzknospe. Halten Sie die zuletzt gebildeten 3-4 Somiten intakt, schneiden Sie das vordere Gewebe aus (Vollachsen-Explant).
- Das Eigelb sollte sich bei diesem Verfahren weitgehend intakt abziehen. Im Falle eines gerissenen Eigelbs können signifikante Eigelbkörner an der ventralen Oberfläche des Gewebes haften bleiben. Wenn ja, verwenden Sie ein Wimpernwerkzeug, um verbleibendes Eigelbgranulat vorsichtig zu reinigen.
HINWEIS: Ein Ungleichgewicht des Hautgewebes an den seitenseitigen Seiten einer Explantat würde es dem Gewebe nicht ermöglichen, eine gerade Wachstumsrichtung beizubehalten. Die Explant wird sich stattdessen zur Seite der gestreckten Haut beugen. Dieses Ungleichgewicht kann unter dem Stereoskop korrigiert werden, indem die Hautschicht mit Hilfe eines mikrochirurgischen Messers gerissen wird. - Bei hautlosen Explanten mit der Nadel auf eine Spitze der Hautschicht drücken und das gewebeausgeplante Gewebe mit dem mikrochirurgischen Messer abziehen. Diese Explanten verlängern ihre Körperachse in der Kultur nicht.
- Zusätzlich zu den Vollachsen-Explanten können in diesem Schritt alternative Explants vorgenommen werden. Sezieren Sie beispielsweise bereits segmentierte Somiten mit dem mikrochirurgischen Messer (Full-PSM-Explants) oder sezieren Sie das PSM in seinen halben Anteroposterior (Halb-PSM-Explants). Eine Anwendung solcher alternativen Explanten ist Abschnitt 5.1 zu entnehmen.
- Das Eigelb sollte sich bei diesem Verfahren weitgehend intakt abziehen. Im Falle eines gerissenen Eigelbs können signifikante Eigelbkörner an der ventralen Oberfläche des Gewebes haften bleiben. Wenn ja, verwenden Sie ein Wimpernwerkzeug, um verbleibendes Eigelbgranulat vorsichtig zu reinigen.
- Übertragen Sie die sezierte Explant sofort auf eine 22 mm x 22 mm große Abdeckung, auf der die Bildgebung durchgeführt wird.
- Ordnen Sie die Explant auf der dorsoventralen Achse flach an, wobei die ventrale Seite den Abdeckungsrutsch berührt (Abbildung 1B). Entfernen Sie vorsichtig die überschüssigen Medien um die Gewebeexplantisation mit einer 20 μL gefilterten Spitzenpipette.
HINWEIS: Eine verzögerte Übertragung von sezierten Explantaten auf den Coverslip führt zu Verformungen des Gewebes, da es von den geometrischen Einschränkungen des Eigelbs befreit wird.
- Ordnen Sie die Explant auf der dorsoventralen Achse flach an, wobei die ventrale Seite den Abdeckungsrutsch berührt (Abbildung 1B). Entfernen Sie vorsichtig die überschüssigen Medien um die Gewebeexplantisation mit einer 20 μL gefilterten Spitzenpipette.
- Drehen Sie den Abdeckrlip mit der Explant schnell und vorsichtig über die mit dem Wachstumsmedium gefüllte Gleitkammer.
- Um Blasenbildung zu verhindern, legen Sie eine Seite des quadratischen Abdecks auf die Bandkammer und lassen Sie die andere Seite vorsichtig los. Achten Sie darauf, die Explant in diesem Schritt nicht zu bewegen/zu verformen.
- Entfernen Sie überschüssige Medien, die aus der Kammer ausbluten, vorsichtig, indem Sie die Objektträgerkammer auf ein Laborgewebe drücken. Der Deckdeckel sitzt stabil auf der Diakammer für Live-Imaging, aufgrund der Oberflächenspannung des flüssigen Mediums ohne Abdichtung.
- Für die Langzeitkultur (>6 Stunden) verwenden Sie eine größere Kammer. 22 mm x 50 mm rechteckige Abdecklippen zusammen mit zwei parallelen Bahnen von Bandschichten auf Dias können in solchen Fällen verwendet werden. Ein ~ 1 mm breiter Spalt kann zwischen zwei Bandbahnen belassen werden, um den Luftzugang in das Wachstumsmedium zu erleichtern.
- Wiederholen Sie die Schritte 3.3-3.8, um weitere Explants vorzubereiten. Vorbereitete Explantierungen verlängern ihre A-P-Körperachse mit einer durchschnittlichen Geschwindigkeit von ~30 μm/h und segmentieren ihre Somiten mit ~40-min-Intervallen bei 25 °C (Abbildung 2A, Video 1).
- Bei nicht länglichen Explantierungen sanften Druck auf die Seiten des Objektträgers ausüben, der die Probe in Schritt 3.8.2 hält, während Sie die überschüssigen Medien auf ein Laborgewebe aussaugen. Alternativ können Explants in Einbandschicht-Gleitkammern kultiviert werden. Auch die chemische Aktivierung der Objektträgerkammeroberfläche mit Kollagen Typ I führt zu nicht länglichen Explantierungen (Abbildung 2B, Video 2).
- Führen Sie die Beschichtung der Kammer mit Kollagen Typ I im Voraus durch, indem Sie die Objektträgerkammern mit 15-20 ml vorverdünnter Kollagenlösung bei Raumtemperatur für 1 h vollständig abdecken. Verwenden Sie für dieses Protokoll eine Laminar-Flow-Haube, um die Sterilität aufrechtzuerhalten. Spülen Sie die Kammern vorsichtig mit Dissektionsmedium am Ende aus.
- Bei Embryonen, die älter als 15 Jahre sind, montieren Sie das Schwanzentscheidegewebe seitlich anstelle einer flachen (dorsoventralen) Montierung (Video 3). Um Muskelzuckungen vorzubeugen, 0,004% ige Tricainlösung in die Nährmedien als Anästhetikumaufnehmen 3.
- Bei nicht länglichen Explantierungen sanften Druck auf die Seiten des Objektträgers ausüben, der die Probe in Schritt 3.8.2 hält, während Sie die überschüssigen Medien auf ein Laborgewebe aussaugen. Alternativ können Explants in Einbandschicht-Gleitkammern kultiviert werden. Auch die chemische Aktivierung der Objektträgerkammeroberfläche mit Kollagen Typ I führt zu nicht länglichen Explantierungen (Abbildung 2B, Video 2).
4. Live-Bilderfassung
- Bildproben entweder auf einem Präparierbereich für die Weitfeld-Durchlichtbildgebung von somiten Segmentierungsgrößen und -perioden oder mit strukturierter Beleuchtung / Konfokal- / Lichtblattmikroskopie unter Verwendung transgener Reporterfischlinien.
- Die Temperatur der Gewebeerregungen mit der bildgebenden Raumtemperatur für mindestens 15 min ausgleichen.
- Für eine präzisere Temperaturregelung verwenden Sie ein kommerzielles Temperaturkontrollsystem, das auf einem invertierten Mikroskop montiert ist.
- Stellen Sie die Bilderfassungsrahmenintervalle auf 2 - 10 minuten ein, abhängig vom biologischen Prozess, der sie interessiert.
HINWEIS: Die Segmentierung von Zebrafisch-Somite ist ein schneller Prozess, der zwischen 20 und 55 Minuten bei lebensfähigen Temperaturen von 30 °C bis 21,5 °C in ganzen Embryonen liegt. Explanten verlängern und segmentieren ~ 30% langsamer als die ganzen Embryonen.- Achten Sie darauf, genügend Verzögerung zwischen den Kanalakquisitionen zu lassen, um eine mögliche Phototoxizität für lebendes Gewebe zu vermeiden. Setzen Sie das Gewebe nicht länger als die Hälfte der Bildgebungsdauer dem Anregungsstrahl aus und verringern Sie die Strahlintensität so weit wie möglich.
HINWEIS: Die Ansammlung von reaktiven Sauerstoffspezies (ROS) ist im Allgemeinen die Hauptursache für Phototoxizität in lebenden Proben4. Ascorbinsäure als ROS-Fänger kann in einer Konzentration von 4 mM zum Wachstumsmedium ergänzt werden, um die ROS-Aktivität zu puffern und die Phototoxizität zu lindern. Nebenwirkungen der Phototoxizität können während der Live-Bildgebung schwer zu bemerken sein. Schwanzentplants sind in diesem Aspekt vorteilhaft, da einige visuelle Marker der Phototoxizität wie mitotischer Arrest, behinderte Gewebeentwicklung (dh Bildung von Somiten, Schwanzdehnung) und zerfallendes Gewebe leichter zu bemerken sind. Bitte beachten Sie die angegebene Referenz4 für eine detaillierte Diskussion.
- Achten Sie darauf, genügend Verzögerung zwischen den Kanalakquisitionen zu lassen, um eine mögliche Phototoxizität für lebendes Gewebe zu vermeiden. Setzen Sie das Gewebe nicht länger als die Hälfte der Bildgebungsdauer dem Anregungsstrahl aus und verringern Sie die Strahlintensität so weit wie möglich.
- Verwenden Sie RNA-injizierte Embryonen im Einzelzellstadium, um 4-D-Bilder zu erhalten, die in zellulärer Auflösung segmentiert und analysiert werden sollen.
- Verwenden Sie 300 pg RNA aus in vitro transkribierten Membranen und kernfluoreszierenden Reportermarkerplasmiden wie pCS-membrane-ceruleanFP (Addgene plasmid #53749) oder pCS-memb-mCherry (Addgene plasmid #53750) in Kombination mit pCS2+ H2B-mTagBFP2 (Addgene plasmid #99267) oder pCS2+ H2B-TagRFP-T (Addgene plasmid #99271) in Injektionen. Einen Beispielfilm mit Zellmembran- und Kernmarkern finden Sie in Video 4.
HINWEIS: Die durchschnittliche Zellgröße von PSM-Gewebe beträgt etwa ~5 μm Durchmesser, von denen Kerne ~3 - 4 μm umfassen. Für die korrekte Zellsegmentierung sollten eine Pixelgröße von ~0,5 μm und ein Z-Schnitt von ~1 μm aufgezeichnet werden.
- Verwenden Sie 300 pg RNA aus in vitro transkribierten Membranen und kernfluoreszierenden Reportermarkerplasmiden wie pCS-membrane-ceruleanFP (Addgene plasmid #53749) oder pCS-memb-mCherry (Addgene plasmid #53750) in Kombination mit pCS2+ H2B-mTagBFP2 (Addgene plasmid #99267) oder pCS2+ H2B-TagRFP-T (Addgene plasmid #99271) in Injektionen. Einen Beispielfilm mit Zellmembran- und Kernmarkern finden Sie in Video 4.
5. Immunfärbung von Schwanzentbordungen
HINWEIS: Gewebe, die nach verschiedenen Dissektionsszenarien (länglich, nicht länglich, Schwanzknospen seziert, halbe PSM usw.) als flach montierte Schwanzentstellungen5 gezüchtet wurden, können aus Objektträgerkammern für weitere immunanregende Quantifizierungen von Proteinen von Interesse gewonnen werden. Hier stellen wir das Protokoll vor, das für die diphosphorylierte extrazelluläre signalregulierte Kinase (ppERK) Färbung von Explanten als FGF-Signalgradientenanzeige verwendet wird.
- Nach der Bildung der Somiten bis zur gewünschten Stufe vorsichtig den Deckenschlupf auf halbem Weg in die Ecke der Gleitkammer verschieben, ohne ihn anzuheben.
- Mit Hilfe von ~100 μL zusätzlichem Dissektionsmedium in einer Pasteur-Pipette aus Glas gewinnen Sie die Explantate vom Objektträger zurück und werden in eine 64-Well-Zellkulturplatte überführen.
HINWEIS: Ab diesem Schritt können alle Lösungsersatzungen unter einem Sezierbereich mit einer separaten Glaspipette für feste Proben durchgeführt werden. Dadurch wird sichergestellt, dass explantiertes Gewebe in Vertiefungen nicht verloren geht oder zwischendurch übertragen wird. - Nachdem Sie alle Explanten übertragen haben, saugen Sie das überschüssige Medium einzeln aus den Vertiefungen und geben Sie 100 μL 4% Paraformaldehyd in PBS (PFA) in jede Vertiefung.
ACHTUNG: PFA ist eine toxische Lösung mit krebserregender Wirkung. Bei der Handhabung sollte eine geeignete PSA verwendet werden. - Fixieren Sie die Explants in einer 64-Well-Platte bei Raumtemperatur für 1 h auf einem Shaker.
- Gewebeentpflanzungen sind empfindlicher gegenüber Verformungen als ganze Embryonen. Passen Sie die Shakergeschwindigkeit entsprechend an.
- Waschen Sie das Fixiermittel dreimal mit 150 μL PBS-Tw (0,1% Tween20 in PBS) aus. Sammeln Sie die erste Wäsche in einem speziellen "PFA Waste" -Behälter.
- Dehydrieren Sie Explantierungen in 4×5-Minuten-Schritten, indem Sie jedes Mal ~ 40 μL Lösung durch 100% Methanol (MeOH) ersetzen.
ACHTUNG: MeOH ist eine giftige Chemikalie, die flüchtig und brennbar ist. Arbeiten Sie in einem gut belüfteten Raum und verwenden Sie geeignete PSA für die Handhabung. - Als letzten Schritt der Dehydrierung entfernen Sie die gesamte Lösung aus den Vertiefungen und ersetzen Sie sie durch 100 μL MeOH. Bei -20 °C 15 min inkubieren.
HINWEIS: Verwenden Sie einen bestimmten "MeOH Waste"-Behälter, um die Lösungen bis Schritt 5.11 zu sammeln. - 50 μL MeOH hinzufügen und bei Raumtemperatur 5 min schütteln.
- Rehydrieren Sie Explantierungen in 4×5-Minuten-Schritten, indem Sie jedes Mal ~ 40 μL Lösung durch PBS-T (0,1% Triton-X 100 in PBS) ersetzen. Verwenden Sie einen speziellen "MeOH Waste" -Behälter, um die Lösungen zu sammeln.
- Als letzten Schritt der Rehydratation entfernen Sie die gesamte Lösung aus den Vertiefungen und ersetzen Sie sie durch 100 μL PBS-T.
- Zur Gewebepermeabilisation explantierte Explanten mit 1,5% Triton-X 100 in PBS für 20 min bei Raumtemperatur auf dem Shaker behandeln.
- Waschen Sie die Proben mit MAB-D-T (0,1% Triton-X 100 Reinigungsmittel und 1% Dimethylsulfoxid (DMSO) in 150 mM NaCl 100 mM Maleinsäurepuffer pH 7,5) 3×5 min.
ACHTUNG: DMSO ist brennbar und ein giftiges Mutagen. Bei der Handhabung sollte eine geeignete PSA verwendet werden. - Inkubieren Sie Explantierungen in 100 μL/Well-Serumblocklösung (2% fetales Rinderserum in MAB-D-T) für 2 Stunden bei Raumtemperatur.
- Ersetzen Sie die gesamte Blockierungslösung durch 50-100 μL / Well primäre Antikörperlösung (1:1000 Verdünnung des monoklonalen Mausantikörpers gegen ppERK im Serumblock). Inkubieren Sie proben O/N (>16 h) bei 4 °C auf dem Shaker.
- Waschen Sie die primäre Antikörperlösung mit MAB-D-T 5×5 min.
- Inkubieren Sie Proben in sekundärer Antikörperlösung (Alexa Fluor 597 Ziegen-Anti-Maus-IgG2b (1:200) und Hoechst 33342 (1:5000) in MAB-D-T) O/N bei 4 °C auf einem Shaker oder für 3 h bei Raumtemperatur.
HINWEIS: Decken Sie ab diesem Schritt die 64-Well-Platte mit Aluminiumfolie ab, um eine Lichtexposition von sekundären Antikörper-behandelten Proben zu vermeiden.
ACHTUNG: Hoechst 33342 ist ein potenzielles Karzinogen. Bei der Handhabung sollte eine geeignete PSA verwendet werden. - Waschen Sie die sekundäre Antikörperlösung mit PBS-Tw 3×5 min.
- Fixieren Sie die Proben mit PFA für 15 Minuten bei Raumtemperatur.
- Waschen Sie das Fixiermittel mit PBS-Tw und gleichen Sie die Proben innerhalb von 60% Glycerin aus. Montieren Sie Explants auf Objektträgern mit Nagellack und 60% Glycerin für die Bildgebung. Repräsentative Ergebnisse der Immunfärbung finden Sie in Abbildung 3.
Ergebnisse
Dieses Protokoll ermöglicht die flache geometrische Kultivierung von lebenden Zebrafischschwanz-Explantierungen. Die Gewebekultur bietet drei Hauptvorteile gegenüber ganzen Embryonen: 1) Kontrolle der Achsendehnungsgeschwindigkeit, 2) Kontrolle über verschiedene Signalquellen (Morphogenquellen) durch einfache Dissektion und 3) nahezu objektive, hohe Vergrößerung und hohe NA-Live-Bildgebung.
Chemisch unbehandelte Gleitkammern ermöglichen es dem Schwanzentscheider, seine Hauptachse (
Diskussion
Dieser Artikel stellt ein detailliertes Protokoll einer Gewebekultur-Explantierungstechnik vor, die wir entwickelt und kürzlich5 für Zebrafischembryonen verwendet haben. Unsere Technik baut auf den bisherigen Explantierungsmethoden in Küken8 undZebrafischen 9,10,11 Modellorganismen auf. Mit diesem Protokoll hergestellte Schwanzentspannungen können bis >12 h in einer einfachen G...
Offenlegungen
Die Autoren haben nichts offenzulegen und erklären keine Interessenkonflikte.
Danksagungen
Wir danken der AECOM Zebrafish Core Facility und Cincinnati Children's Veterinary Services für die Fischpflege, dem Cincinnati Children's Imaging Core für die technische Unterstützung, Didar Saparov für die Unterstützung bei der Videoproduktion und Hannah Seawall für die Bearbeitung des Manuskripts. Die in dieser Veröffentlichung berichtete Forschung wurde vom National Institute of General Medical Sciences der National Institutes of Health unter der Vergabenummer R35GM140805 an E.M.Ö. Der Inhalt liegt allein in der Verantwortung der Autoren und stellt nicht unbedingt die offiziellen Ansichten der National Institutes of Health dar.
Materialien
| Name | Company | Catalog Number | Comments |
| 1 mL Sub-Q Syringe with PrecisionGlide Needle | Becton, Dickinson and Co. | REF 309597 | for dechorionating embryos and manipulations |
| 200 Proof Ethanol, Anhydrous | Decon Labs | 2701 | for immunostaining |
| Antibiotic Antimycotic Solution (100×) | Sigma-Aldrich | A5955 | for tissue dissection media |
| Calcium Chloride Anhydrous, Powder | Sigma-Aldrich | 499609 | for tissue dissection media |
| Dimethylsulfoxide | Sigma-Aldrich | D5879 | for immunostaining |
| Disposable Scalpel, #10 Stainless Steel | Integra-Miltex | MIL4-411 | for preparing tape slide wells |
| Ethyl 3-aminobenzoate methanesulfonate salt (Tricaine) | Sigma-Aldrich | 886-86-2 | (optional) for anesthesizing tissues older than 20 somites stage |
| Fetal Bovine Serum (FBS) | ThermoFisher | A3160601 | additional for tissue culture media |
| Goat anti-Mouse IgG2b, Alexa Fluor 594 | Invitrogen | Cat#A-21145; RRID: AB_2535781 | secondary antibody for immunostaining |
| L-15 Medium with L-Glutamine w/o Phenol Red | GIBCO | 21083-027 | for tissue dissection media |
| Methanol | Sigma-Aldrich | 179337 | for immunostaining |
| Microsurgical Corneal Knife 2.85 mm Angled Tip Double Bevel Blade | Surgical Specialties | 72-2863 | for tissue dissection |
| Mouse monoclonal anti-ppERK | Sigma-Aldrich | Cat#M8159; RRID:AB_477245 | for ppERK immunostaining |
| NucRed Live 647 ReadyProbes Reagent | Invitrogen | R37106 | (optional) for live staining of cell nuclei |
| Paraformaldehyde Powder, 95% | Sigma-Aldrich | 158127 | for fixation of samples for immunostaining |
| Rat Tail Collagen Coating Solution | Sigma-Aldrich | 122-20 | (optional) for chemically activating slide chambers |
| Stage Top Incubator | Tokai Hit | tokai-hit-stxg | (optional) for temperature control during live imaging |
| Transparent Tape 3/4'' | Scotch | S-9782 | for preparing tape slide wells |
| Triton X-100 | Sigma-Aldrich | X100 | for immunostaining |
| Tween 20 | Sigma-Aldrich | P1379 | for immunostaining |
| Zebrafish: Tg(actb2:2xMCP-NLS-EGFP) | Campbell et al., 2015 | ZFIN: ZDB-TGCONSTRCT-150624-4 | transgenic fish with nuclear localized EGFP |
| Zebrafish: Tg(Ola.Actb:Hsa.HRAS-EGFP) | Cooper et al., 2005 | ZFIN: ZDB-TGCONSTRCT-070117-75 | transgenic fish with cell membrane localized EGFP |
Referenzen
- Assheton, R. . Growth in length: Embryological Essays. , (1916).
- Gomez, C., et al. Control of segment number in vertebrate embryos. Nature. 454 (7202), 335-339 (2008).
- Westerfield, M. . The Zebrafish Book: a guide for the laboratory use of zebrafish (Danio rerio), 3rd edition. , (1995).
- Icha, J., Weber, M., Waters, J. C., Norden, C. Phototoxicity in live fluorescence microscopy, and how to avoid it. BioEssays. 39 (1700003), (2017).
- Simsek, M. F., Ozbudak, E. M. Spatial fold change of Fgf signaling encodes positional information for segmental determination in zebrafish. Cell Reports. 24 (1), 66-78 (2018).
- Dubrulle, J., Pourquié, O. fgf8 mRNA decay establishes a gradient that couples axial elongation to patterning in the vertebrate embryo. Nature. 427 (6973), 419-422 (2004).
- Diez del Corral, R., et al. Opposing FGF and Retinoid Pathways Control Ventral Neural Pattern, Neuronal Differentiation, and Segmentation during Body Axis Extension. Neuron. 40 (1), 65-79 (2003).
- Stern, H. M., Hauschka, S. D. Neural tube and notochord promote in vitro myogenesis in single somite explants. Developmental Biology. 167 (1), 87-103 (1995).
- Langenberg, T., Brand, M., Cooper, M. S. Imaging brain development and organogenesis in zebrafish using immobilized embryonic explants. Developmental Dynamics. 228 (3), 464-474 (2003).
- Picker, A., Roellig, D., Pourquié, O., Oates, A. C., Brand, M. Tissue micromanipulation in zebrafish embryos. Methods in molecular biology. 546 (11), 153-172 (2009).
- Manning, A. J., Kimelman, D. Tbx16 and Msgn1 are required to establish directional cell migration of zebrafish mesodermal progenitors. Developmental Biology. 406 (2), 172-185 (2015).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental Dynamics. 203 (3), 253-310 (1995).
- Kaufmann, A., Mickoleit, M., Weber, M., Huisken, J. Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 139, 3242-3247 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten