
Method Article
Achieving Efficient Fragment Screening at XChem Facility at Diamond Light Source
* These authors contributed equally
In This Article
Summary
This paper describes the complete XChem process for crystal-based fragment screening, starting from applying for access and all subsequent steps to data dissemination.
Abstract
In fragment-based drug discovery, hundreds or often thousands of compounds smaller than ~300 Da are tested against the protein of interest to identify chemical entities that can be developed into potent drug candidates. Since the compounds are small, interactions are weak, and the screening method must therefore be highly sensitive; moreover, structural information tends to be crucial for elaborating these hits into lead-like compounds. Therefore, protein crystallography has always been a gold-standard technique, yet historically too challenging to find widespread use as a primary screen.
Initial XChem experiments were demonstrated in 2014 and then trialed with academic and industrial collaborators to validate the process. Since then, a large research effort and significant beamtime have streamlined sample preparation, developed a fragment library with rapid follow-up possibilities, automated and improved the capability of I04-1 beamline for unattended data collection, and implemented new tools for data management, analysis and hit identification.
XChem is now a facility for large-scale crystallographic fragment screening, supporting the entire crystals-to-deposition process, and accessible to academic and industrial users worldwide. The peer-reviewed academic user program has been actively developed since 2016, to accommodate projects from as broad a scientific scope as possible, including well-validated as well as exploratory projects. Academic access is allocated through biannual calls for peer-reviewed proposals, and proprietary work is arranged by Diamond's Industrial Liaison group. This workflow has already been routinely applied to over a hundred targets from diverse therapeutic areas, and effectively identifies weak binders (1%-30% hit rate), which both serve as high-quality starting points for compound design and provide extensive structural information on binding sites. The resilience of the process was demonstrated by continued screening of SARS-CoV-2 targets during the COVID-19 pandemic, including a 3-week turn-around for the main protease.
Introduction
Fragment-Based Drug Discovery (FBDD) is a widely-used strategy for lead discovery, and since its emergence 25 years ago, it has delivered four drugs for clinical use and more than 40 molecules have been advanced to clinical trials1,2,3. Fragments are small chemical entities usually with a molecular weight of 300 Da or less. They are selected for their low chemical complexity, which provide good starting points for development of highly ligand efficient inhibitors with excellent physicochemical properties. Their size means that they sample the binding landscape of proteins more thoroughly than libraries of larger drug- or lead-like compounds, and thus also reveal hot spots and putative allosteric sites. Combined with structural information, fragments provide a detailed map of the potential molecular interactions between protein and ligand. Nevertheless, reliably detecting and validating those entities, which tend to bind weakly to the target protein, requires an array of robust and sensitive biophysical screening methods such as Surface Plasmon Resonance (SPR), Nuclear Magnetic Resonance (NMR), or Isothermal Titration Calorimetry (ITC)4,5.
X-ray crystallography is an essential part of the FBDD toolkit: it is sensitive enough to identify weak binders and directly yields structural information about the interactions at a molecular level. It is complementary to other biophysics screens and usually essential for progressing fragment hits to lead compounds; it requires high quality crystal systems, meaning that crystallization is highly reproducible, and crystals ideally diffract to better than 2.8 Å resolution.
Historically, it has been very difficult to use crystallography as primary fragment screen6,7,8, whether in academia or in industry. In contrast, synchrotrons achieved order of magnitude improvements in robotics, automation9,10,11 and detector technology12,13, and combined with equally accelerated computing power and algorithms of data processing14,15,16, complete diffraction datasets can be measured in seconds and large numbers of them entirely unattended, as pioneered at LillyCAT7 and later MASSIF17,18 (European Synchrotron Radiation Facility (ESRF)). This led synchrotrons to develop highly streamlined platforms to make crystal-based fragment screening as primary screen accessible to a wide user community (XChem at Diamond; CrystalDirect at EMBL/ESRF19; BESSY at Helmholtz-Zentrum Berlin20; FragMax at MaxIV21).
This paper documents the protocols that constitute the XChem platform for fragment screening by X-ray crystallography, from sample preparation to the final structural results of 3D-modeled hits. The pipeline (Figure 1) required developing new approaches to crystal identification22, soaking23, and harvesting24, as well as data management software25 and an algorithmic approach to identifying fragments26 that is now widely used in the community. The crystal harvesting technology is now sold by a vendor (see Table of Materials), and the open availability of the tools has allowed other synchrotrons to adapt them to set up equivalent platforms21. Ongoing projects address data analysis, model completion, and data dissemination through the Fragalysis platform27. The sample preparation laboratory is adjacent to beamline I04-1, simplifying the logistics of transferring hundreds of frozen samples to the beamline and dedicated beamtime on I04-1 allows rapid X-ray feedback to guide the campaign.
XChem is an integral part of Diamond's user program, with two calls per year (early April and October). The peer-review process has been refined in consultation with experts in drug discovery from Academia and Industry. Along with a strong science case, the proposal process28 requires applicants to self-assess not only the readiness of the crystal system, but also their expertise in biochemical and orthogonal biophysical methods and capacity to progress screening hits through follow-up chemistry. The modes of access have also evolved to accommodate the multidisciplinary user community:
Tier 1 (single project) is for projects at the exploratory stage and hit validation tools (biophysics or biochemical tools) and follow-up strategies need not be in place. If accepted, the project is granted a reduced number of beamtime shifts, enough for proof of concept.
Tier 2 (single project) is for well-validated projects and requires downstream tools and follow-up strategies to be in place. If accepted, the project is allocated enough beamtime for a full fragment screening campaign. Single projects (Tier 1 or Tier 2) are to be completed within the 6 months of the allocation period (either April to September or October to March).
Block Allocation Group (BAG) is for a consortia of groups and projects, where a robust target selection and prioritization process is in place within the BAG, along with a clear follow-up pipeline. BAGs must have at least one fully XChem trained expert (superuser), who coordinates their activities with Diamond staff and trains the BAG members. The allocated number of beamtime shifts is defined by the number of scientifically strong projects in the BAG and is re-evaluated per allocation period based on the BAG's report. The access is available for 2 years.
The XChem experiment is divided into three stages, with a decision point for each of them: solvent tolerance test, pre-screen, and main screen (Figure 2). The solvent tolerance test helps define the soaking parameters, the amount of solvent (DMSO, ethylene glycol, or other cryoprotectants if needed) the crystal system can tolerate and for how long. Solvent concentrations typically range from 5%-30% over at least two time points. Diffraction data is collected and compared to the base diffraction of the crystal system; this will determine the soaking parameters for the following stage. For the pre-screen, 100-150 compounds are soaked using the conditions determined in the solvent test, and its purpose is to confirm that the crystals can tolerate the compounds in those conditions. If needed, the cryoprotectant is subsequently added to the drops already containing the fragments. The success criteria are that 80% or more of the crystals survive well enough to yield diffraction data of good and consistent quality; if this fails, soaking conditions are usually revised by altering the soak time or solvent concentration. Following a successful pre-screen, the rest of the compounds chosen for the experiment can be set up using the final parameters.
The DSI-poised library (see Table of Materials) was purposely designed to allow rapid follow-up progression using poised chemistry29 and has been the facility's workhorse library. It is available to users at a concentration of 500 mM in DMSO. Academic users can also access other libraries provided by collaborators (over 2,000 compounds in total) at concentrations of 100-500 mM in DMSO (a full list can be found on the website28). Much of the overall collection is also available in ethylene glycol, for crystal systems that do not tolerate DMSO. Users can also bring their own libraries, provided they are in plates compatible with the acoustic liquid handling system (see Table of Materials).
For all three steps of the experiment (solvent characterization, pre-screen or full screen), the following sample preparation procedures are identical (Figure 3): selection of the compound dispensing location through imaging and targeting of crystallization drops with TeXRank22; dispensing into drops using the acoustic liquid dispensing system for both solvent and compounds23; efficient harvesting of the crystals using the Crystal shifter24; and upload of sample information into the beamline database (ISPyB). The current interface for experiment design and execution is an Excel-based application (SoakDB), which generates the necessary input files for the different equipment of the platform, and tracks and records all results in an SQLite database. Barcode scanners are used at various stages throughout the process to help track samples and this data is added to the database.
Diffraction data are collected in unattended mode using dedicated beamtime on beamline I04-1. Two centering modes are available, namely, optical and X-ray based17. For needle and rod-shaped crystals, X-ray centering is advised, whereas chunkier crystals generally support optical mode, which is faster and, therefore, allows for more samples to be collected in the allotted beamtime. Depending on the resolution of the crystals (established before entering the platform) data collection can either be 60 s or 15 s total exposure. Data collection during the solvent test stage usually informs which combination will work best with the performance of beamline I04-1.
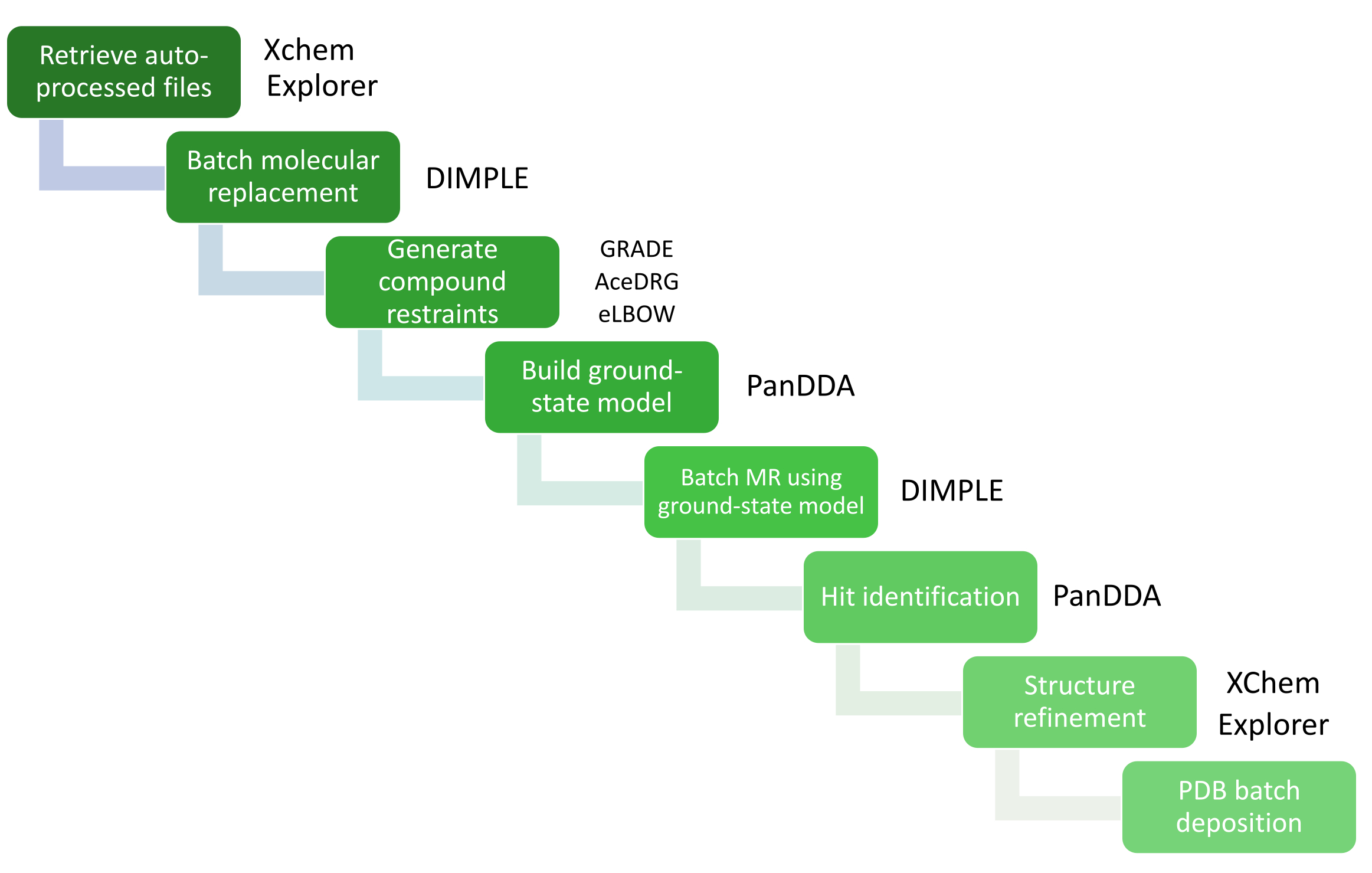
The large volume of data analysis is managed through XChemExplorer (XCE)25, which can also be used to launch the hit identification step using PanDDA26. XCE is a data management and workflow tool that supports large-scale analysis of protein-ligand structures (Figure 4); it reads any of the auto-processing results from data collected at Diamond Light Source (DIALS16, Xia214, AutoPROC30, and STARANISO31) and auto-selects one of the results based on data quality and similarity to a reference model. It is important that the model is representative of the crystal system used for XChem screening, and must include all waters or other solvent molecules, as well as all co-factors, ligands, and alternative conformations visible in crystals soaked with solvent only. The quality of this reference model will directly impact the amount of work required during the model building and refinement stage. PanDDA is used to analyze all the data and identify binding sites. It aligns structures to a reference structure, calculates the statistical maps, identifies events, and calculates event maps26,32. In the PanDDA paradigm, it is neither necessary nor desirable to build the full crystallographic model; what must be modeled is only the view of the protein where a fragment is bound (the bound-state model), so the focus need be only on building the ligand and surrounding residues/solvent molecules according to the event map32.
Protocol
1. Project proposal submission
- Proposal content: since the XChem program is oversubscribed, thorough and complete information in the proposal is critical for passing peer-review.
- Make the case! Present the importance of the target and put it in the broader context.
- Articulate the strategy after the fragment screening campaign: the orthogonal methods in place to validate the hits and how to progress them. Line up the collaborations, if needed.
- Due to the intense lab and data analysis part, it is highly recommended to assign an experienced crystallographer in advance.
- A robust crystal system is key to eliminate technical variation and users should address those essential points.
- Ensure that the crystallization conditions yield reproducible drops with similar diffracting quality crystals in plates suitable for use on the platform with a reservoir volume of 30 µL (or less) and a drop size between 200-500 nL. Ideally, more than 50% of drops in a plate will have crystals of at least 35 µm size33.
- Ensure consistent diffraction quality of crystals (2.6 Å or better).
- Check the suitability of the crystal system for fragment screening, including crystal packing and accessibility of known sites. Previous evidence of a molecule bound in those sites is often reassuring.
- Make the case! Present the importance of the target and put it in the broader context.
2. Preparation for the visit
- Transfer of crystallization protocols for on-site crystallization.
- Provide 2 x 50 mL of reservoir solution, ready to use.
- Provide the protein solution at the necessary concentration for crystallization, ready to use in aliquots of 30-50 µL.
- Provide 10 mL of the protein buffer solution.
- Provide seed stock (even if not needed in the crystallization protocol).
NOTE: Seeding favors crystallization reproducibility and speeds up the nucleation time33. - Complete the crystallization information form available on the XChem website28.
- Provide the storage information in the shipping form available on the XChem website28.
- Install NoMachine and set up a remote desktop to Diamond (https://www.diamond.ac.uk/Users/Experiment-at-Diamond/IT-User-Guide/Not-at-DLS/Nomachine.html).
- Generate and transfer a good reference model, in consultation with an expert crystallographer or XChem support staff.
3. Fragment screening experiment
- Defining the compound dispense location.
- Imaging crystallization plates.
- Image all the crystal plates (see Table of Materials) required for the experiment in the crystal plate imagers (see Table of Materials). Using the imager software, generate plate name(s) in the correct directory for the plate type in the following format Proposal Number_Plate Number.
- Print the barcodes (right click on plate name and select from the menu), place them on the opposite side of the plate from the row letters, put the plate(s) into the load port with the barcode facing away from the user.
- Use the imager control software, scan the load port, right click on plates, and then select Image Plates.
- Once imaging is complete, remove plates from the imager.
- Choosing crystals and compound location
NOTE: The images of the crystallization droplets are processed within the Luigi pipeline using TexRank's textons-based algorithm Ranker to rank the droplets by the likely presence of crystals22. This takes approximately 10 min and the images will then be available in TexRank.- Open TeXRank from a PC and select the crystal tray either from the list on the bottom right or by typing the barcode into the box at the top left.
- Select the correct imager format and the single well view. Move through the drop images and when there is a crystal that is suitable to use in an experiment, right click away from the crystal but inside the drop-the aim is to target where in the drop to add solvent/compounds, so do not want to directly hit the crystal23.
- Continue through the whole plate and once finished select the Echo 1 Target button; save in the crystal targets directory under the relevant visit. Do not change the file name.
- Repeat for any additional plates.
- Imaging crystallization plates.
- Compound dispensing
- Generating files for compound dispense
- In SoakDB, enter library selection or solvent information in the library/solvent table.
- Enter the drop volume and load in the list of targeted crystals.
- Generate the required batches.
- Enter the soak parameters. Click on Calculate and then on the Export Pending button. For solvent, add the various concentrations to the table. This generates the files for use in the acoustic dispenser.
- If using cryoprotectant, enter the concentration and create the files in the same way.
- Dispensing solutions using the acoustic dispenser (see Table of Materials)
- Take the source plate (compounds or solvent/cryoprotectant) and spin the plate in the centrifuge for 2 min at 1,000 x g.
- If dispensing solvent or cryoprotectant, pipette 30 µL into the relevant well on a 384PP plate; cover with a microseal film then centrifuge as above.
- Open the software; select New and choose the correct source well plate (384PP, 384LDV, or 1536LDV) and the liquid class (DMSO, CP, BP or GP). Ensure the correct plate type is selected as the destination plate. Then check the Custom box and continue.
- Select Import and choose the relevant batch file. Complete the import steps as prompted by the software.
- Use the plate maps to check the solution to dispense and the destination locations.
- Run the protocol, following the prompts as they come up. The solution(s) from the source plate will dispense into the chosen crystal drops.
- Store the plate in the incubator for the required time.
NOTE: These parameters are determined in the solvent characterization step, the temperature will be either 4 °C or 20 °C depending on the crystal growth temperature and the times are typically between 1 h and 3 h.
- Generating files for compound dispense
- Harvesting crystals using the semi-automatic crystal harvesting device (see Table of Materials).
NOTE: If cryoprotection is required, repeat step 3.2.2 for the addition of cryoprotectant solutions onto the crystal drops prior to harvesting the samples.- Preparation for harvesting
- Prepare the files required for harvesting in SoakDB. When asked, confirm that the soaks are done, and the batches completed.
- Scan out the number of pucks required for the experiment under the correct proposal number.
- Select a tray of the appropriately sized loops for the crystals (35 µm, 75 µm, or 150 µm). Importantly, choose a loop size that matches the size of the crystal as closely as possible to enable the automatic centering on the beamline to be more accurate, improve the data quality by reducing the background and to eliminate the need for cryoprotectant.
- Open the relevant software and open the workflow tab.
- Scan the pucks into the software and scroll back to the top of the list, highlighting the first puck.
- Place the pucks in a foam dewar and cool them down with liquid nitrogen.
- Choose Import File From SoakDB and select the batch to harvest; check to see whether the batch is assigned to the left-hand holder. A worklist appears.
- Take the crystal plate, remove the seal, and put in the left-hand holder; move the plate to the parking position.
- Harvesting crystals
- Get comfortable and press the Start Workflow button (the screen is a touch screen) to move to the first selected well position.
- If the crystal has survived, mount the crystal in the loop and plunge into the liquid nitrogen placing it in position 1 in the first puck in the list.
- Select the appropriate description for the crystal from the interface (normal, melted, cracked, jelly, or colored).
- If the drop is a compound soak, record the description of the compound state (clear, crystalline, precipitated, bad dispense, or phase separation).
- If the crystal has been successfully mounted, select Mounted otherwise select Fail.
- The plate will move to the next selected well. Fill all the puck positions consecutively (do not leave a gap if a crystal has failed). Carry on until the end of the workflow.
- At the end of the workflow, load any additional batches and continue to fill the pucks in order. There is no need to start a new puck for a new batch.
- Barcode-tracking of the harvesting results
- Once all the crystals have been harvested, take the pucks to the barcode scanner, place one at a time in the holder to scan the puck and pin barcodes.
- When this is completed, put the lids on the pucks and store in a liquid nitrogen storage dewar.
- Load the output file into the SoakDB interface.
- Recording sample information into ISPyB34,35,36
- Upload sample data into ISPyB
- In SoakDB, update the beamline visit Update for ISPyB and click on Export to create the file to upload into ISPyB.
- Open putty. Login and browse to the following directory dls/labxchem/data/year/lbXXXX-1/processing/lab36/ispyb.
- Run the script csv2ispyb (csv2ispyb lbXXXX-1-date.csv)
NOTE: The samples are now loaded into ISPyB.
- Record the puck location and data collection strategy.
- Record the details and the location of the pucks
NOTE: It is important to record the details and location of the pucks so they can be located and loaded onto the beamline.- In SoakDB, open the second tab labeled Pucks.
- Fill in the details in the boxes along the top. Specifically, location of pucks (storage dewar and canes), data collection parameters, including expected resolution and proposal number.
- Click on the Save button and a list of all the pucks will appear in the table. Copy the recently filled pucks.
- Open the XChem queue spreadsheet (shortcut on desktop) and paste in the information. Fill in any additional relevant information.
- Record the details and the location of the pucks
- Upload sample data into ISPyB
- Preparation for harvesting
4. Data collection
NOTE: Data is collected in an unattended mode and managed by the XChem/beamline team.
- Recollecting mis-centered samples.
NOTE: These are required when there have been issues with the data collection for certain samples, most likely caused when pins have not centered correctly.- Look at the sample changer view in ISPyB, select Rank by AP to grade the samples by auto-processed resolution in a color graduation from green to red.
- Click on the samples to check for any red or yellow samples.
NOTE: This will bring up the data collection. - Check the crystal snapshots to see whether the crystal has centered.
- Make a note of all those that have not centered and send to the local contact who will recollect the missing samples.
5. Data analysis
- Retrieving and analyzing Diamond's auto-processing results through XChemExplorer (XCE)25.
- In a terminal, go to the subfolder Processing: cd /dls/labxchem/data/year/visit/processing or for XChem BAGs: cd /dls/labxchem/data/year/visit/processing/project/processing/.
- Use the alias xce to open XChemExplorer.
- Select the Update Tables From Datasource button.
- Under the Overview tab, there is a summary of the experimental data. Add additional categories with the Select Columns to Show option in the Datasource menu.
- Under the Settings tab, select the data collection directory (/dls/i04-1/data/year/visit/).
- Open the Datasets tab, choose the target from Select Target drop-down menu, select Get New Results from Auto processing from Datasets drop-down menu, and click on Run.
NOTE: XCE will now parse the data collection visit for auto-processing results. This may take some time the first time it is run, depending on the number of datasets/directories that are being parsed. - Check consistency and quality of data by checking resolution, space group, and Rmerge. Exclude data lower than 2.8 Å resolution.
NOTE: By default, dataset selection is based on a score calculated from I/sigI, completeness and number of unique reflections but other processing results can be selected for use25. - To select a different processing result for individual datasets, if preferred, click on Sample ID and choose the desired program/run. To change the processing pipeline for all datasets select Edit preferences from the Preferences menu and change Dataset Selection Mechanism.
- If needed, reprocess the data through ISPyB37.
- If no processed data for a sample is acceptable, label as Failed to exclude from further analysis.
- When complete, click on Update Tables From Datasource to add data to subsequent tables.
- Calculating initial maps using DIMPLE38.
- Open the Maps tab, choose the reference model from the drop-down menu and select the desired datasets followed by Run DIMPLE on selected MTZ files.
- XCE runs numerous DIMPLE jobs simultaneously on the cluster at Diamond. Find the status of these jobs under the Dimple Status column and refresh using the Update Tables from Datasource button or using the qstat command in Linux.
- Once complete, check whether the Dimple Rcryst, Dimple Rfree, and Space Group values are acceptable. If necessary (high Rfree/wrong space group/large difference in unit cell volume), change the auto processing results as described previously and repeat map generation for these datasets.
- Generating ligand restraints using Grade39, AceDRG40, or phenix.eLBOW41.
- Select the desired program (Preferences, Edit preferences, Restraints generation program) and then select datasets under the Maps tab followed by running Create CIF/PDB/PNG file of SELECTED compounds from the Maps & Restraints dropdown.
- Refresh the status of these jobs found under the Compound Status column using the Update Tables from Datasource button.
- Building the ground state model (Pre-run)
NOTE: The term ground-state model represents the structure of the protein in its ligand-free form, as observed in 100 datasets (this number is chosen arbitrarily). Since the ground-state model is used as the reference for building ligand-bound state, it is critical to build an accurate ground state model, including all solvent and water molecules, prior to the analysis of the entire fragment screening campaign. In this step, the hundred first highest resolution datasets marked by PanDDA as lacking interesting events (and thus likely ligand-free) are used to generate the ground-state mean map while the dataset with lowest Rfree is selected for the refinement. The ground-state mean map is not a crystallographic map, however, it is important to only use this map for the building of the ground-state model.- Open the PanDDAs tab and update tables from datasource if necessary.
- Define the Output Directory (/dls/labxchem/data/year/visit/processing/analysis/panddas).
- Select Pre-run for Ground State Model and click on Run.
NOTE: Datasets with high Rfree and unexpected space groups should automatically be excluded from the analysis. - To manually exclude datasets with high Rfree and unexpected space groups, select Ignore Completely.
- Check the status of the pre-run job using qstat in a terminal window.
- Once complete, select Build ground state model and click on Run.
NOTE: This will open Coot with the PanDDA mean map and a reference model/2Fo-Fc/Fo-Fc maps from the best quality dataset for re-modeling and refinement using Coot. It is of utmost importance that only the PanDDA mean map is used for modeling.
- Identifying hits using PanDDA26
- PanDDA analysis
NOTE: It can take some time to run on the cluster if there are lots of datasets, the unit cell is large, and there are multiple copies of the protein in the asymmetric unit.- Repeat the previously described steps for Analyse DLS Auto-processing Results and Initial Map Calculation. For the map calculation, use the ground-state model as a reference: Refresh Reference File List > Set New Reference and generate Ligand Restraints as necessary for the new data (steps 6.1-6.3).
- Under the PanDDAs tab ensure the output directory is set as before and run pandda.analyse from Hit Identification drop-down menu.
- Check the status of the job in the Linux terminal using the qstat command.
- PanDDA inspect - checking/building binding events
- Under the PanDDAs tab in XCE, run pandda.inspect from the Hit Identification drop-down menu to open Coot42 with the PanDDA control panel.
NOTE: The pandda.inspect control panel provides a summary of PanDDA statistics and allows users to navigate through binding events/sites. A summary HTML file of the results is also generated and can be updated during inspection by selecting Update HTML. - To model a ligand, click on Merge Ligand With Model and Save Model before navigating to another event to avoid losing any changes to the bound-state model.
NOTE: Only models that have been updated and saved will be exported for refinement at a later stage. - Use the Event Comment field to annotate the binding event and the Record Site Information to annotate binding sites.
- Load average and 2mFo-DFc maps (from DIMPLE) for comparison with the event map and model.
- Once all viable ligands have been modeled, merged, and saved based on the event map, close pandda.inspect.
- Under the PanDDAs tab in XCE, run pandda.inspect from the Hit Identification drop-down menu to open Coot42 with the PanDDA control panel.
- PanDDA export and refinement
NOTE: Following PanDDA inspect models are exported back into the project directory and an initial round of refinement is launched. There are currently two available pipelines to do so under the PANDDAs tab in XCE.- Export NEW/ALL/SELECTED PANDDA models generates an ensemble of the bound and unbound models for refinement and generates occupancy restraint parameters for Refmac43.
NOTE: The ensemble model will be used for refinement but only the bound-state model will be updated in Coot and deposited in the PDB. This pipeline is best used for datasets with low occupancy fragments and significant changes to the protein model. - Refine NEW/ALL bound-state models with BUSTER refines the bound-state only with Buster44.
NOTE: This is best used with high occupancy ligands/datasets with minimal changes to the protein model.
- Export NEW/ALL/SELECTED PANDDA models generates an ensemble of the bound and unbound models for refinement and generates occupancy restraint parameters for Refmac43.
- PanDDA analysis
- Refining the hits (all datasets selected for refinement will now be visible in the Refinement tab). Select Open COOT - BUSTER Refinement or Open COOT - REFMAC Refinement from the Refinement drop down menu to open Coot with the XCE Refinement control panel.
- Select the status of the samples to be refined from the Select Samples drop down (usually 3 - in refinement) and click on GO.
NOTE: The XCE control panel provides a summary of the number of datasets for that category and allows navigation between datasets while providing a summary of refinement statistics. - Annotate the ligand confidence in the XCE control panel: 0 - no ligand present- Fragment has not bound; 1 - Low Confidence- Fragment has possibly bound but is not particularly convincing; 2 - Correct ligand, weak density- User is confident fragment has bound but it is low occupancy/there are some issues with the maps; 3 - Clear density, unexpected ligand- Maps clearly indicate ligand binding that does not correlate to provided chemical structure; 4 - High confidence- Ligand is unambiguously bound.
- Make any necessary changes to the model at this stage and initiate further refinement using the Refine button.
- Use Show MolProbity To-Do List button to access MolProbity45 analysis run on all refinement cycles.
- If required, add refinement parameters, e.g., for anisotropic temperature factors, twinned data, or occupancy refinement by selecting the Refinement Parameters button.
NOTE: Data processing statistics are also provided in XCE under the Refinement tab and if refinement is performed with the Buster pipeline, Buster-reports, including MOGUL analysis46, are provided. - Change the status of a dataset as it progresses through refinement in both the main XCE window under the Refinement tab or in the Coot XCE control panel. When satisfied that the model is accurate around the ligand and suitable to be shared for further analysis, change the status to CompChem Ready. When the refinement is complete and the model ready for upload to the PDB, change the status to Deposition ready.
- Select the status of the samples to be refined from the Select Samples drop down (usually 3 - in refinement) and click on GO.
6. Depositing the data
NOTE: All datasets from a fragment screen and the ground-state model used to generate the PanDDA event maps can be deposited in the PDB using group depositions.
- Convert all PanDDA event maps to MTZ format by running Event Map ->SF from the Hit Identification menu.
- Provide additional metadata such as authors and methods by selecting Deposition > Edit information. Fill out all the required items and click on Save to Database and then save this information for deposition of the ground-state model. Do this after the model status has been changed to Deposition Ready.
- In the Deposition tab, select the Prepare mmcif button to generate structure factor mmcif files for all Deposition Ready datasets. The following message will appear in the terminal window when this is complete: Finished Preparing mmcif Files for wwPDB Deposition.
- Select the Copy mmcif button to copy all these files to a single bzipped tar archive in the Group Deposition Directory of the visit.
- Go to https://deposit-group-1.rcsb.rutgers.edu/groupdeposit; login with username: grouptester and password: !2016rcsbpdb. Create a session and upload the ligand-bound.tar.bz2 file from the group deposition directory.
- After successful submission of the ligand-bound structures, an e-mail is sent with the PDB codes. Select Update DB with PDB Codes from the Deposition menu; copy and paste the information from this e-mail into the pop-up window and click on Update Database to add PDB IDs.
- In order to deposit the ground-state model used by PanDDA, select the relevant PanDDA directory in XCE and run apo->mmcif from the Hit Identification menu.
NOTE: XCE will arbitrarily select a high-resolution structure with low Rfree as the model for the deposition bundle and then compile all structure factor mmcif files into a single file. - In the Deposition tab, select the Add to Database button below the Group Deposition of Ground-State Model section.
- Enter the metadata for the ground state model (again by selecting Deposition > Edit Information), load the previous file and Save to Database.
- Prepare the ground-state mmcif file by running Prepare mmcif from the Group Deposition of Ground-State Model section and when complete, copy the mmcif to the Group Deposition directory by selecting the Copy mmcif button from the same section.
- As before, go to https://deposit-group-1.rcsb.rutgers.edu/groupdeposit; login with username: grouptester and password: !2016rcsbpdb. Create a session and upload the ground_state_structures.tar.bz2 file from the group deposition directory.
Results
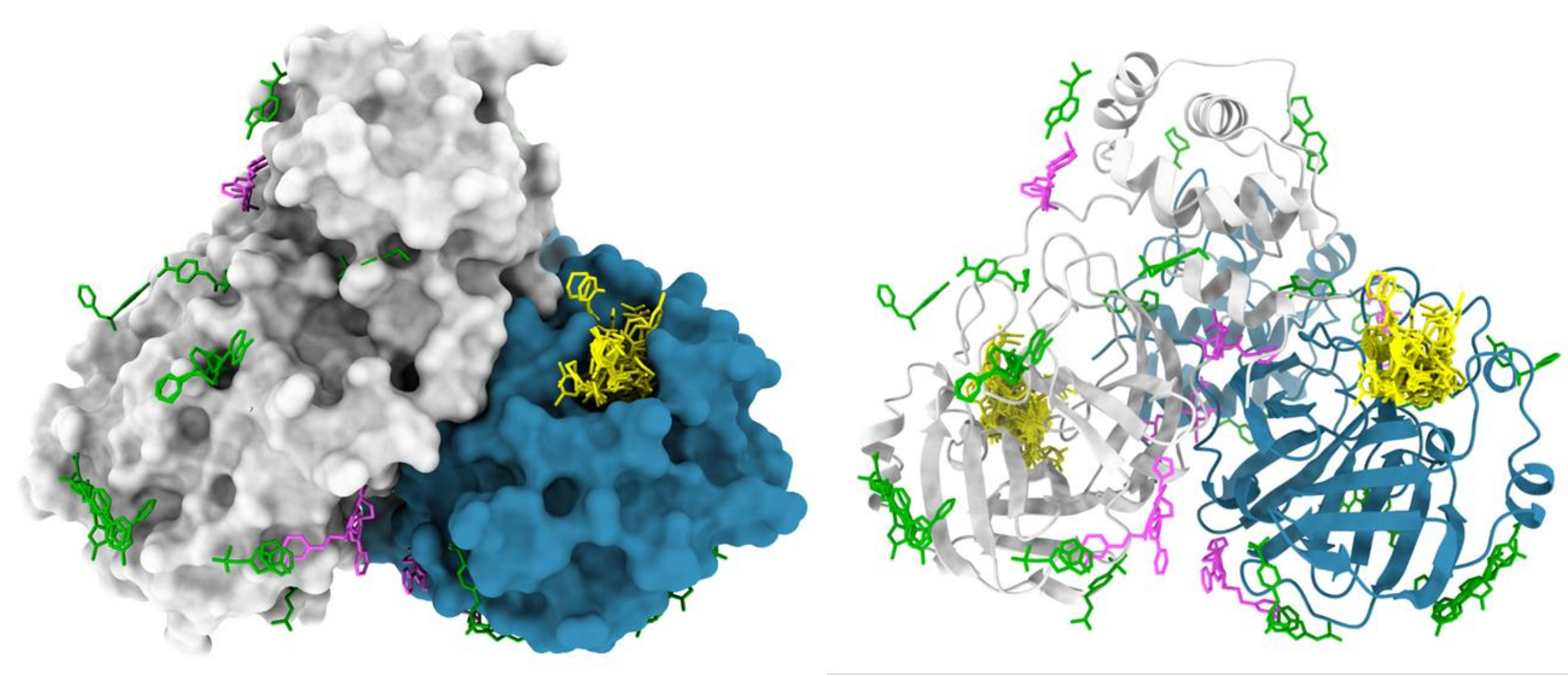
The XChem pipeline for fragment screening by X-Ray crystallography has been extensively streamlined, enabling its uptake by the scientific community (Figure 5). This process has been validated on over 150 of screening campaigns with a hit rate varying between 1% and 30%47,48,49,50,51,52 and by many repeat users. Crystal systems that are not suitable (low resolution, inconsistent in crystallization or in diffraction quality) or cannot tolerate either DMSO or ethylene glycol are eliminated early in the process, saving time, effort, and resource. Successful campaigns provide a three-dimensional map of potential interaction sites on the target protein; a typical outcome is the XChem screen of the main protease of SARS-CoV-2 (Figure 6). Typically, fragment hits are found in: (a) known sites of interest, such as enzyme active sites and sub-pockets48; (b) putative allosteric sites, for example, in protein-protein interactions53; (c) crystal packing interfaces, generally considered as false positives (Figure 6). This structural data generally provides a basis for merging, linking, or growing fragment hits into lead-like small molecules1,3.

Figure 1: The XChem pipeline. The platform is represented schematically from project proposal through sample preparation, data collection, and hit identification. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Screening strategy. The workflow indicates the purpose of each milestone, the experiment's requirements, and the decision points. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Sample preparation workflow. Critical steps for the sample preparation are represented with information from each step being recorded in an SQLite database. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Data analysis using XCE. Critical steps in the data analysis are represented by a workflow diagram with the relevant software packages. Please click here to view a larger version of this figure.
{kind=link}

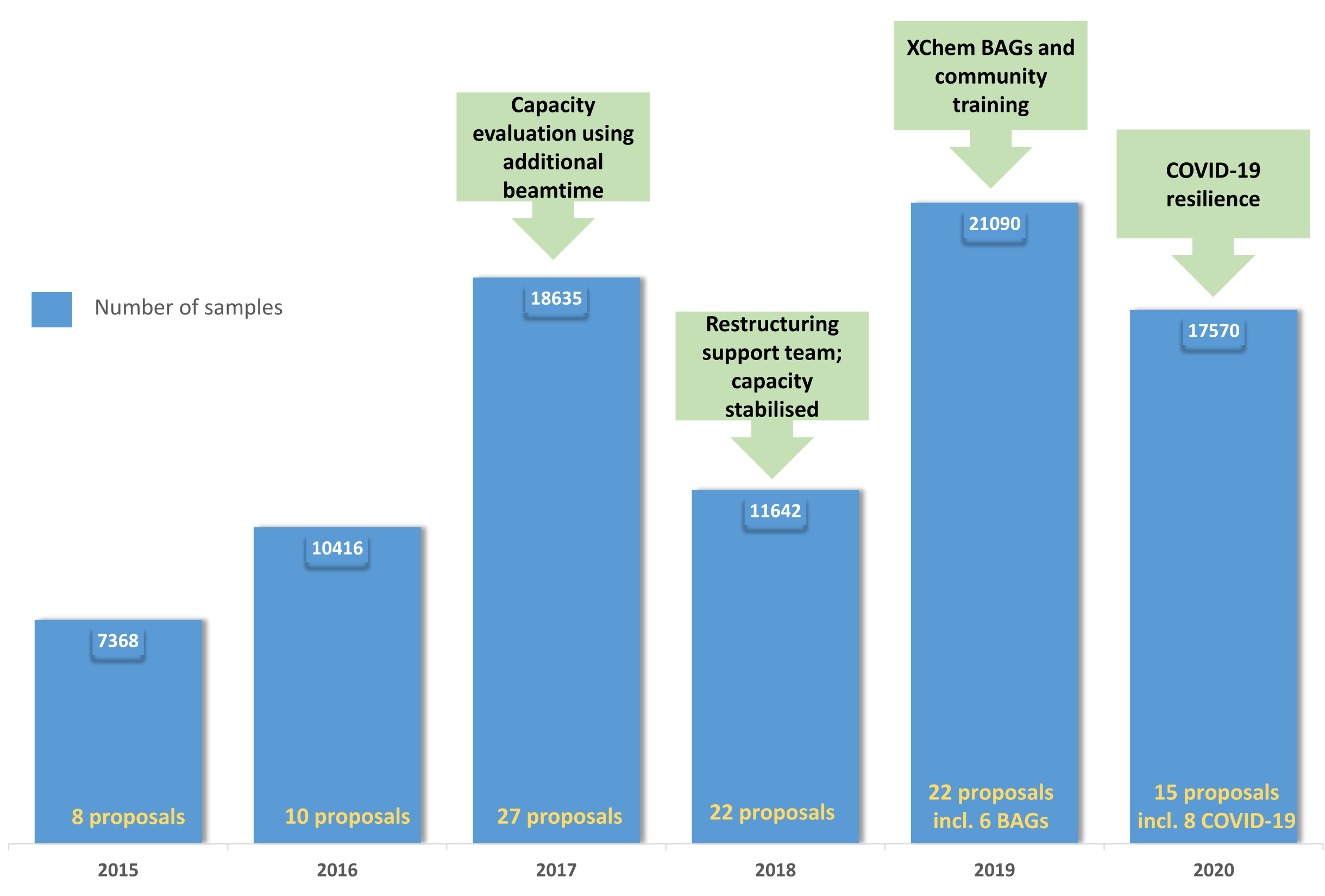
Figure 5: Evolution of the XChem user program: The chart demonstrates the uptake and consolidation of the user program from 2015 through to 2019 with the creation of BAGs in 2019 and the resilience of the platform through the COVID-19 pandemic in 2020. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Representative results of XChem fragment screen. SARS-CoV2 main protease (Mpro) dimer is represented in surface with active site hits shown in yellow, putative allosteric hits shown in magenta, and surface/crystal-packing artefacts shown in green. The figure was made using Chimera and Mpro PDB entries from group deposition G_1002156. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The process outlined in this paper has been extensively tested by the user community and the adaptability of the protocols described here is key for handling the wide variety of projects typically encountered on the platform. However, a few pre-requisites of the crystal system are necessary.
For any fragment screening campaign carried out using X-ray crystallography, a reproducible and robust crystal system is critical. As the standard XChem protocol involves addition of the fragment directly to the crystal drop, optimization should focus on the number of drops containing high-quality crystals rather than the total number of crystals. If drops contain multiple crystals, then they are effectively redundant although may alleviate the harvesting process. Furthermore, transferring the crystallization protocol from the home institute to onsite facilities can be challenging. This is generally best achieved using crystal seeding to promote reproducible nucleation54, and, therefore, a good practice is for users to provide seed stocks along with their protein and crystallization solutions.
To ensure good compound solubility and support, the high soaking concentrations intended to drive binding of weak fragments, fragment libraries are provided in organic solvents, specifically DMSO and ethylene glycol. Provision of two different solvents gives users an alternative for crystals which do not tolerate DMSO at all, or where it occludes the binding of fragments in a site of interest. Users can supply alternative libraries in aqueous buffer: compounds will dispense well provided they are completely dissolved and formatted in plates compatible with the liquid dispensing robot.
For projects where it is not possible to find an appropriate organic solvent that would both solubilize the library and be tolerated by the crystal system, an alternative procedure is to use dried compounds as established at BESSY55.
In the community, there is a long-standing question about being able to soak compounds into crystals grown in crystallization conditions containing high salt concentrations. Practically, more precipitation of the compounds and rapid formation of salt crystals at the harvesting stage is observed, which is reduced by applying a humid environment around the harvesting area. Generally, screening campaigns in crystal systems from high salt crystallization conditions give a comparable hit rate to low salt conditions.
The initial stages of the XChem process (solvent tolerance testing and pre-screen) are relatively small-scale and quick experiments but allow clear go/no go decision for the project. Most painfully, alternative crystal systems will need to be found if neither solvent is tolerated, or the pre-screen results in a very low hit rate. In contrast, if they are successful, the results directly inform the soaking condition to use for the screening experiment, and the best strategy for data collection. Since quality of the data, especially the resolution, will affect the quality of the electron density for hit identification and analysis, the aim is to soak at the highest possible compound concentration that does not have a deleterious effect on diffraction quality (with the majority of datasets (~80%) diffracting to a resolution of 2.8 Å or better).
The data analysis process is streamlined within XChemExplorer, which relies on the PanDDA software for the detection of weak binders and allows users to quickly visualize and review the outcomes of the screening campaign. XChemExplorer imports data processing results from the packages available at Diamond (DIALS16, autoPROC30, STARANISO31, and Xia214) with resolution limits determined by the standard method for each package (i.e., CC1/2 = 0.3). By default, dataset selection is based on a score calculated from I/sigI, completeness, and a number of unique reflections, but specific processing results can be selected for use both globally or for individual samples25. Data is also excluded from analysis by PanDDA based on criteria including resolution, Rfree, and difference in unit cell volume between reference and target data (defaults are 3.5 Å, 0.4, and 12% respectively), so that poorly diffracting, mis-centered, or mis-indexed crystals do not affect the analysis.
The PanDDA algorithm takes advantage of the substantial number of datasets collected during a fragment campaign to detect partial occupancy ligands that are not visible in standard crystallographic maps. Initally, PanDDA uses data collected during the solvent tolerance testing and pre-screen steps to prepare an average density map which is then used to create a ground-state model. As this model will be used for all subsequent analysis steps, it is vital that it accurately represents the un-liganded protein under the conditions used for the fragment screen. PanDDA then uses a statistical analysis to identify bound ligands, generating an event map for the bound state of the crystal. An event map is generated by subtracting the unbound fraction of the crystal from the partial-occupancy dataset and presents what would be observed if the ligand was bound at full occupancy. Even fragments that appear clear in conventional 2mFo-DFc maps might be mismodeled if the event maps are not consulted32. While PanDDA is a powerful method for identifying datasets that differ from the average maps (which is usually indicative of fragment binding) and metrics such as RSCC, RSZD, B-factor ratio, and RMSD during refinement are provided for the users benefit, the user is ultimately responsible for deciding whether the observed density accurately depicts the expected ligand and the most suitable conformation.
Following data analysis and refinement, it is possible for all users to simultaneously deposit multiple structures in the Protein Data Bank (PDB) using XChemExplorer. For each fragment-screen, two group depositions are made. The first deposition contains all fragment-bound models, with coefficients for calculating PanDDA event maps included in MMCIF files. The second deposition provides the accompanying ground-state model, along the measured structure factors of all datasets of the experiment: this data can be used to reproduce the PanDDA analysis, and for developing future algorithms. As for the structures of the hits, when fragment occupancy is low, refinement is better behaved if models are a composite of the ligand-bound and confounding ground-state structures32; nevertheless, the practice is to deposit only the bound-state fractions, since the full composite models are in general complex and difficult to interpret. As a result, some quality indicators recalculated by the PDB (in particular, R/Rfree) are sometimes slightly elevated. It is also possible to provide all raw data using platforms such as Zenodo56, although this is not currently supported by the XChem pipeline.
Overall, since its operation in 2016, fragment ligands could be identified in over 95% of the targets using this procedure. Experience from the many projects that XChem has supported was distilled into best practice for crystal preparation33, while a fragment library was evolved that implemented the poised concept for aiding fragment progression29, also helping establish the practice of making library composition public. The platform has demonstrated the importance of well-maintained infrastructure and documented processes, detailed here, and made it possible to evaluate other fragment libraries57,58, to compare libraries48, and to inform the design of the collaborative EUOpenscreen-DRIVE library59,60.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgements
This work represents a large joint effort between the Diamond Light Source and the Structure Genomic Consortium. The authors would like to acknowledge Diamond's various support groups and MX group for their contribution to the automation of i04-1 beamline and for providing streamlined data collection and auto-processing pipelines, which are commonly run across all MX beamlines. They would also like to thank the SGC PX group for their resilience being the first users to test the setup and Evotec for being the first serious industrial user. This work was supported by iNEXT-Discovery (Grant 871037) funded by Horizon 2020 program of the European Commission.
Materials
| Name | Company | Catalog Number | Comments |
| DSI-poised library | Enamine | DSI-896 | fragment library |
| Echo 550 and 650 series | Beckman-Coulter | acoustic dispensing system | |
| Echo microplates | Beckman-Coulter | 001-12380; 001-8768; 001-6025 | 1536-well and 384-well microplates |
| Shifter | Oxford Lab Technology | harvesting device | |
| Microplate centrifuge with a swing-out rotor | Sigma | model 11121 | microplate centrifuge |
| 3-drops crystallisation plates | Swissci | 3W96T-UVP | Crystallisation plates |
| Formulatrix plate imager and Rockmaker software | Formulatrix | Crystallisation plates imaging device |
References
- Erlanson, D. A., Fesik, S. W., Hubbard, R. E., Jahnke, W., Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nature Reviews Drug Discovery. 15 (9), 605-619 (2016).
- Jacquemard, C., Kellenberger, E. A bright future for fragment-based drug discovery: what does it hold. Expert Opinion on Drug Discovery. 14 (5), 413-416 (2019).
- Jahnke, W., et al. Fragment-to-lead medicinal chemistry publications in 2019. Journal of Medicinal Chemistry. 63 (24), 15494-15507 (2019).
- Li, Q. Application of fragment-based drug discovery to versatile targets. Frontiers in Molecular Biosciences. 7, 180 (2020).
- Kirsch, P., Hartman, A. M., Hirsch, A. K. H., Empting, M. Concepts and core principles of fragment-based drug design. Molecules. 24 (23), 4309 (2019).
- Patel, D., Bauman, J. D., Arnold, E. Advantages of crystallographic fragment screening: functional and mechanistic insights from a powerful platform for efficient drug discovery. Progress in Biophysics and Molecular Biology. 116 (2-3), 92-100 (2014).
- Wasserman, S., et al. Automated synchrotron crystallography for drug discovery: the LRL-CAT beamline at the APS. Acta Crystallographica Section A Foundations of Crystallography. 67 (1), 46-47 (2011).
- Hartshorn, M. J. Fragment-based lead discovery using X-ray crystallography. Journal of Medicinal Chemistry. 48 (2), 403-413 (2005).
- Arzt, S., et al. Automation of macromolecular crystallography beamlines. Progress in Biophysics and Molecular Biology. 89 (2), 124-152 (2005).
- Beteva, A. High-throughput sample handling and data collection at synchrotrons: Embedding the ESRF into the high-throughput gene-to-structure pipeline. Acta Crystallographica Section D, Biological Crystallography. 62, 1162-1169 (2006).
- Papp, G., et al. FlexED8: The first member of a fast and flexible sample-changer family for macromolecular crystallography. Acta Crystallographica. Section D, Structural Biology. 73, 841-851 (2017).
- Casanas, A., et al. EIGER detector: Application in macromolecular crystallography. Acta Crystallographica Section D, Structural Biology. 72, 1036-1048 (2016).
- Henrich, B., et al. PILATUS: A single photon counting pixel detector for X-ray applications. Nuclear Instruments and Methods in Physics Research, Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 607 (1), 247-249 (2009).
- Winter, G., Lobley, C. M. C., Prince, S. M. Decision making in xia2. Acta Crystallographica Section D, Biological Crystallography. 69, 1260-1273 (2013).
- Winter, G., McAuley, K. E. Automated data collection for macromolecular crystallography. Methods. 55 (1), 81-93 (2011).
- Winter, G., et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallographica Section D, Structural Biology. 74, 85-97 (2018).
- Bowler, M. W. MASSIF-1: A beamline dedicated to the fully automatic characterization and data collection from crystals of biological macromolecules. Journal of Synchrotron Radiation. 22 (6), 1540-1547 (2015).
- Von Stetten, D., et al. ID30A-3 (MASSIF-3) - A beamline for macromolecular crystallography at the ESRF with a small intense beam. Journal of Synchrotron Radiation. 27, 844-851 (2020).
- Cipriani, F., et al. CrystalDirect: a new method for automated crystal harvesting based on laser-induced photoablation of thin films. Acta Crystallographica. Section D, Biological Crystallography. 68, 1393-1399 (2012).
- . Helmholtz Zentrum Berlin Available from: https://www.helmholtzberlin.de/forschung/oe/np/gmx/fragment-screening/index_en.html (2021)
- Lima, G. M. A., et al. FragMAX: the fragment-screening platform at the MAX IV Laboratory. Acta crystallographica. Section D, Structural biology. 76 (8), 771-777 (2020).
- Ng, J. T., Dekker, C., Kroemer, M., Osborne, M., Von Delft, F. Using textons to rank crystallization droplets by the likely presence of crystals. Acta Crystallographica. Section D, Biological Crystallography. 70, 2702-2718 (2014).
- Collins, P. M., et al. Gentle, fast and effective crystal soaking by acoustic dispensing. Acta Crystallographica. Section D, Structural Biology. 73, 246-255 (2017).
- Wright, N. D., et al. The low-cost Shifter microscope stage transforms the speed and robustness of protein crystal harvesting. Acta Crystallographica. Section D, Structural Biology. 77, 62-74 (2021).
- Krojer, T., et al. The XChemExplorer graphical workflow tool for routine or large-scale protein-ligand structure determination. Acta Crystallographica. Section D, Structural Biology. 73, 267-278 (2017).
- Pearce, N. M., et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nature Communications. 8, 15123 (2017).
- . Fragalysis Available from: https://fragalysis.diamond.ac.uk (2021)
- . Diamond Light Source Ltd Available from: https://www.diamond.ac.uk/Instruments/Mx/Fragment-Screening.html (2021)
- Cox, O. B., et al. A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chemical Science. 7 (3), 2322-2330 (2016).
- Vonrhein, C., et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallographica. Section D, Biological Crystallography. 67, 293-302 (2011).
- Vonrhein, C., et al. Advances in automated data analysis and processing within autoPROC , combined with improved characterisation, mitigation and visualisation of the anisotropy of diffraction limits using STARANISO. Acta Crystallographica Section A: Foundations and Advances. 74 (1), 360 (2018).
- Pearce, N. M., Krojer, T., Von Delft, F. Proper modelling of ligand binding requires an ensemble of bound and unbound states. Acta Crystallographica. Section D, Structural Biology. 73, 265-266 (2017).
- Collins, P. M., et al. Achieving a good crystal system for crystallographic x-ray fragment screening. Methods in Enzymology. 610, 251-264 (2018).
- Delageniere, S., et al. ISPyB: an information management system for synchrotron macromolecular crystallography. Bioinformatics. 27 (22), 3186-3192 (2011).
- Fisher, S. J., Levik, K. E., Williams, M. A., Ashton, A. W., McAuley, K. E. SynchWeb: a modern interface for ISPyB. Journal of Applied Crystallography. 48, 927-932 (2015).
- Ginn, H. M., et al. SynchLink: an iOS app for ISPyB. Journal of Applied Crystallography. 47, 1781-1783 (2014).
- . Diamond Light Source Ltd Available from: https://www.diamond.ac.uk/Instruments/Mx/Common/Common-Manual/Data-Analysis/Reprocessing-in-ISPyB.html (2021)
- Wojdyr, M., Keegan, R., Winter, G., Ashton, A. DIMPLE - a pipeline for the rapid generation of difference maps from protein crystals with putatively bound ligands. Acta Crystallographica. Section A, Foundations of Crystallography. 69, 299 (2013).
- Long, F., et al. AceDRG: A stereochemical description generator for ligands. Acta Crystallographica. Section D, Structural Biology. 73, 112-122 (2017).
- Moriarty, N. W., Grosse-Kunstleve, R. W., Adams, P. D. Electronic ligand builder and optimization workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallographica. Section D, Biological Crystallography. 65, 1074-1080 (2009).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta Crystallographica. Section D, Biological Crystallography. 66, 486-501 (2010).
- Murshudov, G. N., Vagin, A. A., Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica. Section D, Biological Crystallography. 53, 240-255 (1997).
- Bricogne, G., et al. Buster version 2.10.3. Global Phasing Ltd. , (2017).
- Chen, V. B., et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallographica. Section D, Biological Crystallography. 66, 12-21 (2010).
- Bruno, I. J., et al. Retrieval of crystallographically-derived molecular geometry information. Journal of Chemical Information and Computer Sciences. 44 (6), 2133-2144 (2004).
- Delbart, F., et al. An allosteric binding site of the α7 nicotinic acetylcholine receptor revealed in a humanized acetylcholine-binding protein. TheJournal of Biological Chemistry. 293, 2534-2545 (2018).
- Douangamath, A., et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nature Communications. 11 (1), 5047 (2020).
- Guo, J., et al. In crystallo-screening for discovery of human norovirus 3C-like protease inhibitors. Journal of Structural Biology: X. 4, 100031 (2020).
- Keedy, D. A., et al. An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering. eLife. 7, 36307 (2018).
- McIntyre, P. J., et al. Characterization of three druggable hot-spots in the aurora-a/tpx2 interaction using biochemical, biophysical, and fragment-based approaches. ACS Chemical Biology. 12 (11), 2906-2914 (2017).
- Thomas, S. E., et al. Structure-guided fragmentbased drug discovery at the synchrotron: Screening binding sites and correlations with hotspot mapping. Philosophical Transactions. Series A, Mathematical, Physical and Engineering Sciences. 377 (2147), 20180422 (2019).
- Nichols, C., et al. Mining the PDB for tractable cases where x-ray crystallography combined with fragment screens can be used to systematically design protein-protein inhibitors: Two test cases illustrated by IL1β-IL1R and p38α-TAB1 complexes. Journal of Medicinal Chemistry. 63 (14), 7559-7568 (2020).
- D'Arcy, A., Bergfors, T., Cowan-Jacob, S. W., Marsh, M. Microseed matrix screening for optimization in protein crystallization: What have we learned. Acta Crystallographica. Section F, Structural Biology Communications. 70, 1117-1126 (2014).
- Wollenhaupt, J., et al. F2X-Universal and F2X-Entry: Structurally diverse compound libraries for crystallographic fragment screening. Structure. 28 (6), 694-706 (2020).
- . Zenodo Available from: https://zenodo.org (2021)
- Foley, D. J., et al. Synthesis and demonstration of the biological relevance of sp(3) -rich scaffolds distantly related to natural product frameworks. Chemistry. 23 (60), 15227-15232 (2017).
- Kidd, S. L., et al. Demonstration of the utility of DOS-derived fragment libraries for rapid hit derivatisation in a multidirectional fashion. Chemical Science. 11 (39), 10792-10801 (2020).
- . EU-openscreen ERIC Available from: https://www.eu-openscreen.eu/ (2021)
- Schuller, M., et al. Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking. bioRxiv. 393405, (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved