
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Extraktion und Reinigung von FAHD1-Protein aus Schweineniere und Mausleber
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt, wie das domänenhaltige Fumarylacetoacetat-Hydrolase-haltige Protein 1 (FAHD1) aus Schweineniere und Mausleber extrahiert werden kann. Die aufgeführten Methoden können an andere Proteine von Interesse angepasst und für andere Gewebe modifiziert werden.
Zusammenfassung
Fumarylacetoacetathydrolase-Domänen-haltiges Protein 1 (FAHD1) ist das erste identifizierte Mitglied der FAH-Superfamilie in Eukaryoten und wirkt als Oxalacetat-Decarboxylase in Mitochondrien. Dieser Artikel stellt eine Reihe von Methoden zur Extraktion und Reinigung von FAHD1 aus Schweineniere und Mausleber vor. Abgedeckte Methoden sind die Ionenaustauschchromatographie mit schneller Proteinflüssigkeitschromatographie (FPLC), die präparative und analytische Gelfiltration mit FPLC und proteomische Ansätze. Nach der Gesamtproteinextraktion wurden die Ammoniumsulfatfällung und die Chromatographie des Ionenaustauschs untersucht, und FAHD1 wurde über eine sequentielle Strategie unter Verwendung der Ionenaustausch- und Größenausschlusschromatographie extrahiert. Dieser repräsentative Ansatz kann an andere Proteine von Interesse angepasst werden (in signifikanten Mengen exprimiert) und für andere Gewebe modifiziert werden. Gereinigtes Protein aus Gewebe kann die Entwicklung hochwertiger Antikörper und/oder potenter und spezifischer pharmakologischer Inhibitoren unterstützen.
Einleitung
Das eukaryotische FAH-domänenhaltige Protein 1 (FAHD1) wirkt als bifunktionelle Oxalacetat (OAA)-Decarboxylase (ODx)1 und Acylpyruvathydrolase (ApH)2. Es ist in Mitochondrien 2 lokalisiert und gehört zur breiten FAH-Superfamilie der Enzyme 1,2,3,4,5,6. Während seine ApH-Aktivität nur von geringer Relevanz ist, ist die ODx-Aktivität von FAHD1 an der Regulation des TCA-Zyklusflusses 1,7,8,9 beteiligt. OAA wird nicht nur für die zentrale Citratsynthase-Reaktion im Tricarbonsäurezyklus benötigt, sondern wirkt auch als kompetitiver Inhibitor der Succinat-Dehydrogenase als Teil des Elektronentransportsystems und als kataplerotischer Metabolit. Die Herunterregulierung der FAHD1-Genexpression in menschlichen Nabelvenendothelzellen (HUVEC) führte zu einer signifikanten Verringerung der Zellproliferation10 und einer signifikanten Hemmung des mitochondrialen Membranpotentials, verbunden mit einem gleichzeitigen Wechsel zur Glykolyse. Das Arbeitsmodell bezieht sich auf den mitochondrialen Dysfunktions-assoziierten Seneszenz (MiDAS)11-ähnlichen Phänotyp 8, bei dem die mitochondrialen OAA-Spiegel durch die FAHD1-Aktivität 1,8,9 streng reguliert werden.
Rekombinantes Protein ist leichter durch Expression und Reinigung von Bakterien12 als von Gewebe zu erhalten. Ein in Bakterien exprimiertes Protein kann jedoch durch mögliches Fehlen posttranslationaler Modifikationen verzerrt sein oder einfach problematisch sein (d. H. Aufgrund von Plasmidverlust, bakteriellen Stressreaktionen, verzerrten / ungeformten Disulfidbindungen, keiner oder schlechter Sekretion, Proteinaggregation, proteolytischer Spaltung usw.). Für bestimmte Anwendungen muss Protein aus Zelllysat oder Gewebe gewonnen werden, um solche Modifikationen einzuschließen und/oder mögliche Artefakte auszuschließen. Gereinigtes Protein aus Gewebe unterstützt die Entwicklung hochwertiger Antikörper und/oder potenter und spezifischer pharmakologischer Inhibitoren für ausgewählte Enzyme, wie z.B. für FAHD113.
Dieses Manuskript stellt eine Reihe von Methoden zur Extraktion und Reinigung von FAHD1 aus Schweineniere und Mausleber vor. Die beschriebenen Methoden erfordern eine schnelle Proteinflüssigkeitschromatographie (FPLC), verwenden aber ansonsten gängige Laborgeräte. Alternative Methoden können an anderer Stellegefunden werden 14,15,16,17. Nach der Gesamtproteinextraktion umfasst das vorgeschlagene Protokoll eine Testphase, in der Teilprotokolle für die Ammoniumsulfatfällung und die Chromatographie des Ionenaustauschs diskutiert werden (Abbildung 1). Nach der Definition dieser Subprotokolle wird das interessierende Protein über eine sequentielle Strategie unter Verwendung von Ionenaustausch und Größenausschlusschromatographie mit FPLC extrahiert. Basierend auf diesen Richtlinien kann das endgültige Protokoll individuell für andere Proteine von Interesse angepasst werden.

Abbildung 1: Die Gesamtstrategie dieses Protokolls. Von oben nach unten: Protein wird aus Geweben extrahiert. Gewebehomogenat wird hergestellt, zentrifugiert und filtriert. Für jedes Paar von überstehenden und aus Pellets gewonnenen Proben müssen Tests zur Ammoniumsulfatfällung und zur Ionenaustauschchromatographie (FPLC) durchgeführt werden, um optimale Bedingungen zu untersuchen. Nach der Festlegung dieser Subprotokolle kann das Protein durch ein sequentielles Verfahren der Ammoniumsulfatfällung, der Ionenaustauschchromatographie und der repetitiven Größenausschlusschromatographie (FPLC) bei unterschiedlichen pH- und Salzkonzentrationen extrahiert werden. Alle Schritte müssen von Western Blot kontrolliert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
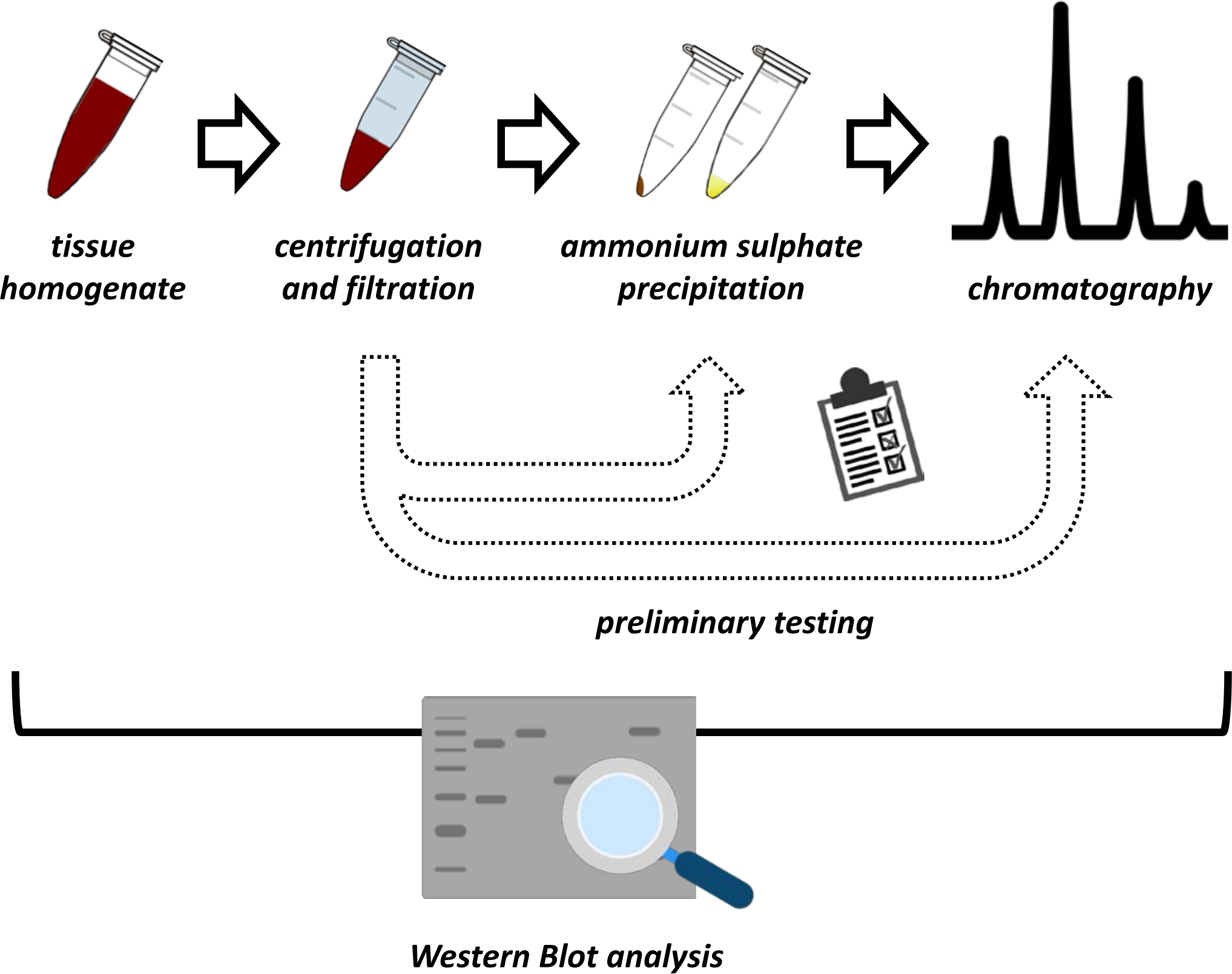{kind=link}
Protokoll
Alle Experimente wurden in Übereinstimmung mit den institutionellen Richtlinien durchgeführt. Die Schweineniere wurde frisch aus dem örtlichen Supermarkt bezogen. Lebergewebe wurde von C57BL6-Wildtyp-Mäusen entnommen, die am Institut für Biomedizinische Alternsforschung der Universität Innsbruck, Rennweg 10, 6020 Innsbruck, Österreich, unter der Aufsicht von Univ.-Doz aufbewahrt wurden. Dr. Pidder Jansen-Dürr, 2013 als Projektleiter ethisch zugelassen (BMWF-66.008/0007-II/3b/2013). Die Wartung und Verwendung der Mäuse für das Projekt ist durch die ethische Genehmigung Nr. 2020-0.242.978 vom 5. Mai 2020 des österreichischen Bundesministeriums für Bildung, Wissenschaft und Forschung (BMBWF) abgedeckt.
1. Vorbereitungen
HINWEIS: Bevor das Protokoll beginnt, müssen mehrere Dinge vorbereitet werden, d.h. der Proteinlysepuffer, die Rohgewebeprobe und ein spezifischer Antikörper, neben allgemeinen Chemikalien und Materialien.
- Bereiten Sie 250 ml Proteinlysepuffer pro 100 g Nettogewicht des Gewebes vor: 250 ml 1x PBS mit 50 mM NaF, 1 mM PMSF, 2 μg/ml Aprotinin und 1 mM aktiviertem Orthovanadat (siehe Tabelle 1). Filtern Sie die Lösung mit einer 0,22 μm Spritzenfiltereinheit.
HINWEIS: Die Aktivierung von Orthovanadat ist vor der Anwendung erforderlich, um es in einen stärkeren Inhibitor von Proteintyrosinphosphatasenumzuwandeln 18. Aktiviertes Orthovanadat kann von kommerziellen Anbietern bezogen, aber auch wie folgt hergestellt werden.- Es wird eine 200 mM Stammlösung von (Natrium-) Orthovanadat in ddH2O hergestellt. Zur Herstellung von 10 ml Lösung 368 mgNa3VO4 auf 9 ml Wasser zugeben und durch Rühren auflösen. Nach der Auflösung das Volumen auf 10 ml mit ddH2O auffüllen.
HINWEIS: Der Ausgangs-pH-Wert der Natriumorthovanadat-Lösung kann je nach Materialquelle variieren, und der pH-Wert muss in einem sich wiederholenden Ansatz wie folgt auf 10 eingestellt werden. - Abhängig vom anfänglichen pH-Wert der Lösung stellen Sie den pH-Wert mit NaOH oder HCl auf 10 ein. Bei einem pH-Wert > 10 hat die Lösung eine gelbe Farbe. Kochen Sie die Lösung, bis sie farblos wird, kühlen Sie sie auf Raumtemperatur ab und überprüfen Sie den pH-Wert. Wenn der pH-Wert >10 beträgt, fügen Sie ein kleines Volumen HCl hinzu, um den pH-Wert auf 10 einzustellen. An diesem Punkt kann die Lösung wieder gelb werden.
- Wiederholen Sie das Kochen und Abkühlen, bis die Lösung farblos bleibt und sich der pH-Wert bei 10 (ca. 5-7 mal) stabilisiert. An diesem Punkt führt die Zugabe von HCl zu einem schwachen Erscheinungsbild der gelben Farbe in der Lösung. Lagern Sie aktiviertes Orthovanadat in 1 ml Aliquots bei -20 °C.
- Es wird eine 200 mM Stammlösung von (Natrium-) Orthovanadat in ddH2O hergestellt. Zur Herstellung von 10 ml Lösung 368 mgNa3VO4 auf 9 ml Wasser zugeben und durch Rühren auflösen. Nach der Auflösung das Volumen auf 10 ml mit ddH2O auffüllen.
- Bereiten Sie Röhrchen mit 2 ml Lysepuffer pro Gramm Gewebe vor und legen Sie sie auf Eis.
HINWEIS: Dieses Protokoll verwendete acht 50-ml-Röhrchen, die jeweils mit insgesamt 30 ml Lysepuffer für eine Schweineniere (ca. 100-150 g) gefüllt waren, und zwei Röhrchen, die jeweils mit 40 ml Lysepuffer für insgesamt 20 Mauslebern (jeweils 1-2 g) gefüllt waren. - Bereiten Sie das Gewebe vor: Sezieren Sie das Gewebe auf einer vorgereinigten Glasplatte, die auf Eis in einer Styroporschaumbox platziert ist. Schneiden Sie Gewebestücke von jeweils etwa 100 mg, um sie für die anschließende Lyse leicht in die entsprechenden Röhrchen zu übertragen. Die Gewebestücke werden in die vorbereiteten Röhrchen überführt (Schritt 1.2).
- Bereiten Sie eine gesättigte Ammoniumsulfatlösung vor: 500 ml ddH 2 O auf70°C erhitzen und unter Rühren nach und nach Ammoniumsulfatpulver (siehe Materialtabelle) hinzufügen, bis kein Ammoniumsulfat mehr gelöst ist. Diese (über)gesättigte Lösung auf Raumtemperatur abkühlen und über Nacht bei 4 °C lagern.
2. Gesamte Proteinextraktion
HINWEIS Nach der Aufbereitung der Probe im kalten Proteinlysepuffer (siehe Schritt 1.3) wird das Gewebe durch Beschallung durch eine Ultraschallsonde oder mit einem elektrischen Homogenisator wie folgt so gut wie möglich homogenisiert.
- Homogenisierung von Geweben
- Im Falle einer Schweineniere beschallen Sie die Suspension vorzugsweise durch eine Ultraschallsonde, während Sie die Probe auf Eis halten (10 Zyklen von 15 s Puls, mit Intervallen von 30 s zwischen den Pulsen, um die Probe auf Eis zu kühlen, bei mittlerer Amplitude mit 50% Tastverhältnis).
- Im Falle von Mausorganen homogenisieren Sie die Suspension mit einem elektrischen Homogenisator (beginnend mit einer geringen Kraft und langsam beschleunigend auf mittlere Kraft), während Sie die Probe auf Eis halten. Waschen Sie den elektrischen Homogenisator regelmäßig in PBS, um organisches Material zu entfernen, das das Gerät verstopft.
- Nehmen Sie 20 μL aus den Proben und überprüfen Sie unter dem Mikroskop, ob die Zellen des homogenisierten Gewebes ordnungsgemäß zerstört werden; Andernfalls wiederholen Sie die Homogenisierung.
- Zentrifugieren Sie die Röhrchen in einer Tischzentrifuge bei 10.000 x g für 30 min bei 4 °C.
HINWEIS: Optional zentrifugieren Sie den Überstand ein zweites Mal bei 20.000 x g für 30 min bei 4 °C, um kleine Bruchteile des ursprünglichen Pellets zu eliminieren, die möglicherweise übertragen wurden. Dies vereinfacht die anschließende Filtration in Schritt 2.3. - Sammeln Sie den Überstand in einer frischen Tube und legen Sie ihn auf Eis. Filtern Sie den Überstand nacheinander mit 0,45 μm und 0,22 μm Spritzenfiltereinheiten. Aliquotieren Sie den Überstand in 10 ml-Chargen und frieren Sie sie bei -20 °C für eine kurzfristige Lagerung oder bei -80 °C für eine längere Lagerung ein.
HINWEIS: Die Vorfilterung mit 0,45 μm entfernt den Großteil der Partikel, bevor ein zweiter Filterschritt mit 0,22 μm die feineren Partikel entfernt. Die direkte Verwendung des 0,22-μm-Filters kann das Risiko einer Verstopfung der Filter verursachen. - Bereiten Sie eine 50-μL-Probe für die SDS-PAGE/Western-Blot-Analyse vor, indem Sie 10 μL 5x SDS-Probenpuffer (siehe Tabelle 1) in 40 μL des Überstands geben und dann bei 95 °C für 10 min sieden.
- Optional werden etwa 100 μL Pellet, das in Schritt 2.2 in 900 μLddH2Oerhalten wurde, resuspendiert und eine Probe für die SDS-PAGE/Western-Blott-Analyse wie oben beschrieben vorbereitet.
HINWEIS: Die Einbeziehung von aus Pellets gewonnenen Proben in die Western-Blot-Analyse zeigt zusätzlich zur Positivkontrolle an, ob die Expression des Proteins niedrig oder der Antikörper problematisch ist.
- Optional werden etwa 100 μL Pellet, das in Schritt 2.2 in 900 μLddH2Oerhalten wurde, resuspendiert und eine Probe für die SDS-PAGE/Western-Blott-Analyse wie oben beschrieben vorbereitet.
3. SDS-PAGE und Western Blot Analyse
HINWEIS: Eine Western-Blot-Analyse ist erforderlich, um die Proteinlöslichkeit zu überprüfen. Im Folgenden wird ein Protokoll für das Elektroblotting unter Verwendung eines Nass-/Tank-Blotting-Systems beschrieben (siehe Materialtabelle). Ein alternatives Protokoll für SDS-PAGE finden Sie an anderer Stelle19.
- Bereiten Sie ein diskontinuierliches 12,5% Polyacrylamid-SDS-PAGE-Gel gemäß den Anweisungen des Herstellers vor (d. h. ein Stapelgel auf einem auflösenden Gel; siehe Tabelle 1). Führen Sie die zuvor in Schritt 2 vorbereiteten Beispiele aus (ähnlich den Schritten 4, 5 und 6; siehe unten).
- Laden Sie eine Proteinmarkerleiter in die erste Vertiefung (siehe Materialtabelle). 5 ng rekombinantes hFAHD1-Protein (gewonnen aus Bakterien12; siehe Tabelle 1) als Positivkontrolle in das zweite Well.
- Laden Sie anschließend 20 μL der zu analysierenden Probe und füllen Sie alle verbleibenden Vertiefungen mit 20 μL vorbereitetem SDS-PAGE 1x Probenpuffer (d.h. 5x Probenpuffer verdünnt mit ddH2O). Lassen Sie die SDS-PAGE-Gele mit dem SDS-Laufpuffer bei 125 V laufen (siehe Tabelle 1).
- Nachdem SDS-PAGE abgeschlossen ist, führen Sie eine Western-Blot-Analyse durch und untersuchen Sie die Membranen mit dem verfügbaren Antikörper, der gegen FAHD1 erhoben wird (siehe Tabelle 1).
HINWEIS: Da die Proben aus rohem Gewebehomogenat entnommen werden, ist die Qualität der SDS-PAGE und der Western-Blot-Analyse an dieser Stelle in der Regel beeinträchtigt. Es ist jedoch wichtig zu überprüfen, ob das zu extrahierende Protein im Überstand löslich ist. Das folgende Protokoll wurde auf Schweineniere und verschiedene Mausorgane, einschließlich Leber, Herz, Gehirn und Niere, getestet.- Bereiten Sie den 10-fachen Western Blot-Transferpuffer vor (siehe Tabelle 1). Bereiten Sie den 1x Western Blot Transferpuffer vor (siehe Tabelle 1) und kühlen Sie ihn auf 4 °C ab.
- Aktivieren Sie eine PVDF-Membran für 2 min in Methanol. Waschen Sie die Membran in ddH 2 O für2min. Ausgleich der Membran für 15 min in 1x Western Blot Transfer Buffer.
- Waschen Sie das SDS-Gel mit 1x PBS für 10 min während des Schüttelns, um den SDS-laufenden Puffer zu entfernen, und inkubieren Sie dann das Gel in 1x Western Blot Transfer Buffer für 10 min zur Gleichgewichtung. Montieren Sie die Elektroblotting-Kassette (d. h. kombinieren Sie die aktivierte PVDF-Membran und die Gele) gemäß den Anweisungen des Herstellers.
- Führen Sie den Blot per Elektroblotting bei 300 mA für 1 h in einer mit Eis gefüllten Polystyrolschaumbox oder im Kühlraum (4 °C) aus. Übertragen Sie die PVDF-Membran in ein 50-ml-Rohr, dessen freiliegende Seite der Innenseite des Rohrs zugewandt ist. Inkubieren Sie die Membran in 20 ml Western Blot-Blocking-Puffer (siehe Tabelle 1) über Nacht bei 4 °C, während Sie auf einer Rohrwalze rollen (siehe Materialtabelle).
- Waschen Sie die Membran am nächsten Tag 5 Minuten lang mit 20 ml Western Blot-Waschpuffer (PBS mit 0,1% (v/v) Tween 20) im selben Rohr während des Rollens. Inkubieren Sie die Membran im selben Röhrchen mit dem primären Antikörper2 (der auf FAHD1 abzielt; siehe Tabelle 1) verdünnt 1:500 im Western Blot-Blocking-Puffer für 1 h bei Raumtemperatur während des Rollens.
- Waschen Sie die Membran im selben Schlauch dreimal für jeweils 10 Minuten mit 20 ml Western Blot Waschpuffer während des Walzens. Inkubieren Sie die Membran für 30 min bei Raumtemperatur mit HRP-konjugiertem Sekundärantikörper (siehe Materialtabelle), verdünnt 1:3000 in 5 ml Western Blot Blocking Buffer.
- Waschen Sie die Membran im selben Schlauch dreimal für jeweils 10 min mit 20 ml Western Blot Waschpuffer und zweimal für jeweils 5 min mit 1x PBS. Trocknen Sie die Membran, indem Sie sie vorsichtig mit einer Pinzette an einer Kante halten und ein Stück Zellulose oder ein Stück Whatman-Papier mit dem gegenüberliegenden (unteren) Rand der Membran berühren. Legen Sie die Membran (freiliegende Seite nach oben) auf eine gereinigte Glasplatte.
- Bedecken Sie die gesamte Membran vorsichtig mit 1 ml vorbereitetem ECL-Western-Blot-Substrat mit einer Pipette und achten Sie darauf, keine Luftblasen zu erzeugen. Lassen Sie die ECL-Lösung 3 min inkubieren und entwickeln Sie die Membran sofort mit Röntgenfilm oder mit einem Bildgebungssystem.
HINWEIS: Wenn das Protein in keiner der Proben, sondern nur in der Positivkontrolle nachgewiesen wurde, kann dies darauf hindeuten, dass das Protein unlöslich ist oder nicht in ausreichenden Mengen vorhanden ist, um vom Antikörper nachgewiesen zu werden. Wenn nur Nanogramm der Positivkontrolle geladen würden, ist das erste Szenario wahrscheinlicher. Wenn überhaupt kein Protein nachgewiesen wurde, überprüfen Sie die Qualität des Antikörpers und wechseln Sie möglicherweise zu einem polyklonalen Antikörper anstelle eines monoklonalen Antikörpers. In seltenen Fällen, z. B. bei einigen hydrophoben Proteinen, kann das Protein nach der Zentrifugation nachweisbar sein, jedoch nicht nach der Filtration. In einem solchen Fall empfiehlt es sich, spezielle Filtereinheiten für hydrophobe Proteine zu verwenden.
- Optional färben Sie die PVDF-Membranen nach Western Bplot, um den erfolgreichen Transfer des Proteins vom SDS-PAGE-Gel auf die PVDF-Membran zu kontrollieren.
HINWEIS: Die Coomassie-Färbung wird für die Fehlerbehebung, Methodenentwicklung und Dokumentation empfohlen, aber beachten Sie, dass nach der Anwendung dieses Protokolls Membranen für weitere westliche Fleckenanalysen verloren gehen. Die Ponceau S-Färbung führt zu einer schwächeren Färbung, kann jedoch verwendet werden, wenn die Membranen erneut untersucht werden sollen.- Bereiten Sie kleine Schalen vor, die die Färbelösungen (Coomassie oder Ponceau S) und Entfärbungslösungen enthalten.
- Mit einer Pinzette die Membran in die Färbelösung geben und vorsichtig schütteln, bis die Membran gut gefärbt ist (5-10 min).
- Die Membran in die Färbelösung geben und schütteln, bis die Lösung gesättigt ist (5-10 min). Wiederholen Sie den Färbungsschritt, bis die Proteinbänder auf der Membran beobachtet werden können; Wenn überhaupt keine Bänder beobachtet werden, wiederholen Sie die Färbung mit einer längeren Inkubationszeit. Trocknen Sie die Membran, indem Sie sie mit einer Pinzette auf eine Glasplatte legen.
4. Prüfung: Ausfällung von Ammoniumsulfat
HINWEIS: Die Ausfällung von Ammoniumsulfat ist eine Methode der Proteinreinigung durch Veränderung der Löslichkeit des Proteins. In einem vorläufigen Experiment wird die Ammoniumsulfatkonzentration sequentiell auf einen Wert erhöht, der eine maximale Menge an Proteinverunreinigungen ausfällt, während FAHD1 in Lösung bleibt. Die Löslichkeit des Proteins wird erneut mittels Western-Blot-Analyse untersucht.
- Fahren Sie mit Schritt 2.3 fort: Tauen Sie entweder ein Aliquot der Probe auf oder fahren Sie direkt nach der Proteinextraktion fort (d. h. ohne die Probe einzufrieren). Filtern Sie die Probe mit einer 0,22 μm Filtereinheit, um mögliche Ausfällungen nach dem Auftauen auszuschließen. Bereiten Sie sechs 1,5-ml-Röhrchen auf Eis vor und übertragen Sie 250 μL Probe in jedes Röhrchen.
- Bereiten Sie eine Verdünnungsreihe von 5%, 10%, 15%, 20%, 25% und 30% Ammoniumsulfat in den oben hergestellten Röhrchen vor und machen Sie das Endvolumen auf 1000 μL mit Proteinlysepuffer aus. Inkubieren Sie die Proben bei 4 °C über Nacht auf einem Röhrchenrotator (siehe Materialtabelle).
- Mit einer Tischzentrifuge bei 10.000 x g für 30 min bei 4 °C zentrifugieren und alle Überstände vorsichtig in separate Röhrchen überführen. Die resultierenden Pellets an der Luft trocknen und jedes von ihnen in 1000 μL von ddH2O resuspendieren.
- Für jedes Paar resuspendiertes Pellet und Überstand aus dem vorherigen Schritt 40 μL mit 10 μL 5x SDS-Probenpuffer mischen und bei 95 °C mit offenen Deckeln kochen, bis der größte Teil der Flüssigkeit verdampft ist. Dann suspendieren Sie das Pellet in einer Mischung von 50% DMSO in ddH2O.
- Führen Sie SDS-PAGE (Schritt 3) durch, aber lassen Sie die Gele bei 80 V für 3 h laufen. Für jede Konzentration von Ammoniumsulfat werden die aus dem resuspendierten Pellet und dem Überstand (Schritt 4.3) gewonnenen Proben paarweise geladen. Führen Sie eine Western-Blot-Analyse durch (Schritt 3).
- Überprüfen Sie auf die höchste Konzentration von Ammoniumsulfat, bei der das zu reinigende Protein (dh FAHD1) in der aus dem Überstand gewonnenen Probe verbleibt. Basierend auf den Ergebnissen definieren Sie ein Ammoniumsulfat-Fällungsprotokoll für das interessierende Protein, das in zukünftigen Experimenten verwendet werden soll.
HINWEIS: Es ist bekannt, dass Ammoniumsulfat SDS-PAGE und Western Blot verzerrt. Wenn die Konzentration von Ammoniumsulfat zunimmt, wird die Qualität der Western Blot-Analyse beeinträchtigt. Wie bei Schritt 3 zuvor wird diese Analyse jedoch verwendet, um die Löslichkeit des interessierenden Proteins bei bestimmten Konzentrationen von Ammoniumsulfat zu überprüfen. Dieses Protokoll zielt darauf ab, andere Proteine auszufällen, während das zu reinigende Protein löslich bleiben muss.
5. Testen: Ionenaustauschchromatographie mit FPLC
HINWEIS: Moleküle mit geladenen funktionellen Gruppen sind für die FPLC an eine Kieselsäurepartikelsäule gebunden, was die Differenzierung von Proteinen nach ihrer Oberflächenladung ermöglicht. Führen Sie diesen Schritt zweimal aus, indem Sie die kationische Austauschspalte und die anionische Austauschspalte verwenden (siehe Tabelle der Materialien). Die Protokollschritte sind für die kationische oder anionische Austauschchromatographie gleich, aber die zu verwendenden Puffer sind unterschiedlich (siehe Tabelle 1); sowohl mit "salzarmen" 15 mM NaCl als auch mit "salzreichen" 1 M NaCl-Bedingungen. Für die verwendeten Säulen wird ein Durchfluss von 1 ml/min empfohlen.
- Richten Sie das FPLC-System mit der anionischen oder kationischen Austauschsäule ein. Waschen Sie die Säule mit 5 Säulenvolumina (CVs) von 20% EtOH (in H2 O), gefolgt von 5 CVs von ddH2O. Alternativ waschen Sie die Säule mit 1 CV niedrigem Salzpuffer, hohem Salzpuffer und wieder niedrigem Salzpuffer in der Reihenfolge, bis keine Peaks mehr im Chromatogramm beobachtet werden, aber waschen Sie mindestens einmal.
- Nach der Bestimmung des optimalen Protokolls für die Ammoniumsulfatfällung auf kleiner Skala (Schritt 4) wenden Sie das Fällungsprotokoll auf 10 ml ursprüngliches Gewebehomogenat an (Schritt 2). Optional dialysieren Sie die Probe gegen den niedrigen Salzpuffer.
- Tragen Sie die Probe auf die Säule auf (z. B. durch Injektion oder mit einer Probenpumpe) und sammeln Sie den Durchfluss. Waschen Sie die Säule mit 1 CV des salzarmen Puffers.
- Richten Sie eine lineare Gradientenelution von 100% niedrigem Salzpuffer / 0% hohem Salzpuffer zu 0% niedrigem Salzpuffer / 100% hohem Salzpuffer innerhalb von 3 CVs ein. Sammeln Sie kontinuierlich 1 ml Fraktionen. Nachdem der Gradient beendet ist, laufen Sie mit dem hohen Salzpuffer weiter, bis keine proteinassoziierten Peaks (UV-Absorption bei 280/255 nm) mehr im Chromatogramm über den Bereich von 1 CV nachgewiesen werden.
- Tragen Sie 1 ml 25% SDS auf, gelöst in 0,5 M NaOH (in ddH2O), um die Säule zu reinigen. Waschen Sie die Säule nacheinander mit 3 CVs von ddH 2 O und3 CVs von 20% EtOH (in ddH2O).
- Sammeln Sie SDS-PAGE-Proben aller Peak-Fraktionen und des Durchflusses und untersuchen Sie sie über Western Blot auf das Vorhandensein des interessierenden Proteins (Schritt 3). Die gesammelten Fraktionen in flüssigem Stickstoff einfrieren und bei -80 °C lagern.
- Nachdem die Western Blot-Analyse abgeschlossen ist, tauen und sammeln Sie die Fraktionen, die das interessierende Protein enthalten, und verwerfen Sie die anderen. Wiederholen Sie die Schritte 5.1-5.5 mit der alternativen Spalte (d. h. kationische oder anionische Austauschspalte).
- Nachdem beide Säulen untersucht wurden, definieren Sie ein FPLC-Protokoll für das interessierende Protein, das in zukünftigen Experimenten verwendet werden soll. Reduzieren Sie das Volumen der Proteinlösung mit Ultrazentrifugationsfiltereinheiten (10 kDa, siehe Materialtabelle) auf 2 ml.
HINWEIS: Es gibt zwei erwartete Ergebnisse dieser Versuchsreihe. Entweder hat sich das interessierende Protein an eine der Säulen gebunden, und die Proteinlösung ist nach der Elution bereits recht rein, oder das Protein verblieb in beiden Fällen im Durchfluss. Im letzteren Szenario könnte der Reinigungseffekt dieses Schritts, obwohl sich das Protein im Durchfluss befindet, immer noch signifikant sein. In einem solchen Fall, wie bei FAHD1 in Schweineniere und Mausleber, wird dieser Schritt des Ionenaustauschs noch durchgeführt. Wenn weder die kationische noch die anionische Austauschsäule einen angemessenen Reinigungseffekt erzielen kann, kann versucht werden, den pH-Wert des Lysats und des Puffers zu ändern und die Probe vor der Anwendung auf FPLC gegen den laufenden Puffer zu dialysieren.
6. Proteinextraktion unter Verwendung definierter Subprotokolle für Ammoniumsulfatfällung und FPLC
HINWEIS: Poröse Partikel in einer Kieselgelsäule für FPLC (siehe Materialtabelle) ermöglichen die Differenzierung von Proteinen nach ihrem hydrodynamischen Radius. Die beschriebenen Schritte sind mit einem FPLC-System unter Verwendung der Größenausschlusschromatographie (SEC) durchzuführen. Für die verwendete SEC-Spalte (siehe Materialtabelle) wird ein Durchfluss von 0,3 ml/min empfohlen.
- Bereiten Sie alle erforderlichen Materialien vor (siehe Schritt 1) und extrahieren Sie das Gesamtprotein aus dem Gewebe (siehe Schritt 2). Führen Sie eine Ammoniumsulfatfällung mit dem gesamten Gewebehomogenat durch, das nicht zum Testen verwendet wurde (siehe Schritt 4). Bei größeren Volumina konzentrieren Sie das Lysat mit Ultrazentrifugationsfiltereinheiten (10 kDa; siehe Materialtabelle) auf ein kleineres Volumen von 50 ml oder weniger.
- Führen Sie einen ersten Reinigungsschritt mittels Ionenaustauschchromatographie durch (siehe Schritt 5).
- Bereiten Sie Proben für Western Blot vor, wie in den vorherigen Schritten beschrieben. Führen Sie eine Western-Blot-Analyse durch und legen Sie alle FAHD1-haltigen Fraktionen aus der Ionenaustauschchromatographie zusammen.
- Reduzieren Sie das Volumen der Proteinlösung mit Ultrazentrifugationsfiltereinheiten (10 kDa) auf bis zu 2 ml. Filtern Sie die Lösung nacheinander mit 0,45 μm und 0,22 μm Spritzenfiltereinheiten, um Mikrofällungen zu entfernen.
- Gleichen Sie die SEC-Spalte mit 1 CV des laufenden SEC-Puffers aus (siehe Tabelle 1), der 1 mM DTT enthält. Laden Sie die Probe auf die Säule und führen Sie die Chromatographie durch, bis alle Proteine eluiert sind (1-2 CV).
- Sammeln Sie Fraktionen von 1 ml des Durchflusses, der signifikanten Peaks im Chromatogramm entspricht (UV-Absorption bei 280/255 nm) und bereiten Sie 50 μL-Proben jeder gesammelten Fraktion für die SDS-PAGE- und Western-Blot-Analyse vor, wie in den vorherigen Schritten beschrieben. Alle Fraktionen mit flüssigem Stickstoff einfrieren und bei -80 °C lagern.
- Waschen Sie die SEC-Säule nacheinander mit 1 CV von ddH 2 O und1 CV von 20% EtOH (in ddH2O). Führen Sie eine Western-Blot-Analyse durch und bündeln Sie alle FAHD1-haltigen Fraktionen. Reduzieren Sie das Volumen der Proteinlösung auf 2 ml mit Ultrazentrifugationsfiltereinheiten (10 kDa, siehe Materialtabelle).
- Beurteilen Sie die Proteinkonzentration mit einem kommerziellen BCA-Assay-Kit (siehe Materialtabelle).
HINWEIS: Der pH- und Salzgehalt der mobilen Phase kann das Elutionsprofil von globulären Proteinenbeeinflussen 20. Saure oder basische Bedingungen können dazu führen, dass Peaks weniger definiert sind und Protein-Matrix-Wechselwirkungen zunehmen, was zu einer teilweisen Retention von Protein auf der Spalte20 führt. Dieser Effekt kann für eine weitere Proteinreinigung ausgenutzt werden. Eine Wiederholung von Schritt 6 mit unterschiedlichen Flussraten, pH-Wert und Salzkonzentrationen kann die Reinheit des Proteins20 verbessern.
7. Silberfärbung
HINWEIS: Die Silberfärbungsanalyse von SDS-PAGE-Gelen ist erforderlich, um nach Proteinkontaminationen zu suchen, die bei der Coomassie-Färbung möglicherweise nicht beobachtet werden. Das folgende Protokoll ist eine von vielen Versionen, die in der Literatur21 zu finden sind. Führen Sie alle Inkubationsschritte durch, indem Sie in einer sauberen Glasschale schütteln. Sammeln Sie alle silber- und formaldehydhaltigen Flüssigkeiten in einem speziellen Abfallbehälter und entsorgen Sie sie ordnungsgemäß.
- Inkubieren Sie die SDS-PAGE-Gele in silberfarbener Färbefixierlösung (siehe Tabelle 1) über Nacht im Kühlraum. Die Gele werden in silberfärbender Inkubationslösung (siehe Tabelle 1) für 3 h bei Raumtemperatur inkubiert. Optional fügen Sie Glutaraldehyd hinzu (siehe Tabelle 1), um die Erkennung schwacher Banden zu verbessern. Waschen Sie die Gele viermal in ddH2O für jeweils 10 min.
- Die Gele werden in silberfärbender Silberlösung (siehe Tabelle 1) für 1 h inkubiert.
HINWEIS: Beachten Sie, dass von nun an alle Flüssigkeiten und das Gel selbst Silber und Formaldehyd enthalten, die giftig sind. - Die Gele werden in einer Silberfärbungs-Entwicklerlösung (siehe Tabelle 1) mit starkem Schütteln inkubiert, bis die Bänder deutlich sichtbar sind. Um die Reaktion zu stoppen, verwerfen Sie die Entwicklerlösung und inkubieren Sie die Gele sofort in Silberflecken-Stopplösung (siehe Tabelle 1) für mindestens 10 min.
HINWEIS: Bänder, die in den Schritten 7.2 und 7.3 gefärbt sind, werden ständig weiterentwickelt. Die Zugabe von mehr Formaldehyd zur Lösung als angegeben kann erforderlich sein, wenn die Färbung schwach ist.
Ergebnisse
Das FAHD1-Protein wurde mit dem vorgestellten Protokoll aus Schweineniere und Mausleber extrahiert. Für Mausgewebe sind mehrere Organe erforderlich, um nach dem letzten Reinigungsschritt mehrere μg zu erhalten. Aus diesem Grund konzentriert sich dieser Artikel auf die Extraktion von FAHD1 aus Schweinenieren, was ein viel exemplarischeres Experiment ist. Die Extraktion von FAHD1 aus der Mausleber wird durchgeführt, um die Schwierigkeiten und möglichen Fallstricke dieses Protokolls darzustellen. Es wird allgemein empfo...
Diskussion
Kritische Schritte im Protokoll
Die Einhaltung gemeinsamer Richtlinien für den Umgang mit Proteinen ist unerlässlich, wie z.B. die Arbeit auf Eis und bei moderaten pH- und Salzbedingungen. Die Verwendung von Proteasehemmern ist für die Methode von Vorteil, während die Verwendung von Proteasom-Inhibitoren dringend empfohlen wird. Das Einfrieren und Auftauen der Probe kann immer zu einer Proteinausfällung (zumindest teilweise) führen, daher sollte jedes aufgetaute Aliquot des anfänglichen Protein...
Offenlegungen
Die Autoren haben keine konkurrierenden finanziellen Interessen.
Danksagungen
Die Autoren sind sehr dankbar für die technische Unterstützung durch Ayse Öztürk und Eva Albertini. Mäuse, die zur Erzeugung von Lebergewebe verwendet wurden, wurden unter der Aufsicht von Univ.-Doz gepflegt. Dr. Pidder Jansen-Dürr (Institut für Biomedizinische Alternsforschung an der Universität Innsbruck, Rennweg 10, 6020 Innsbruck, Österreich).
Materialien
| Name | Company | Catalog Number | Comments |
| 0.22 µm filter units | MERCK | SLGP033RS | Millex-HP, 0.22 µm, PES 33 mm, not steril |
| 0.45 µm filter units | MERCK | SLHP033NS | Millex-HP, 0.45 µm, PES 33 mm, not steril |
| 15 mL Falcon tubes | VWR | 734-0451 | centrifugal tubes |
| 50 mL Falcon tubes | VWR | 734-0448 | centrifugal tubes |
| 96-Well UV Microplate | Thermo-Fischer | 8404 | UV/VIS transparent flat-bottom 96 well plates |
| Acrylamide/Bis Solution (40%, 29:1 ratio) | BIO-RAD | #1610147 | 40% acrylamide/bis-acrylamide, 29:1 (3.3% crosslinker) solution for casting polyacrylamide gels |
| ÄKTA FPLC system | GE Healthcare Life Sciences / Cytiva | - | using the FPLC system by GE Healthcare; different custom versions exist; this work used the "ÄKTA pure" system |
| Amicon Ultra-15, PLGC Ultracel-PL Membran, 10 kDa | MERCK | UFC901024 | centrifigal filters for protein enrichment; 10 kDa molecular mass filter; 15 mL |
| Amicon Ultra-4, PLGC Ultracel-PL Membran, 10 kDa | MERCK | UFC801024 | centrifigal filters for protein enrichment; 10 kDa molecular mass filter; 4 mL |
| Ammonium sulfate powder | MERCK | A4418 | ammonium sulphate for molecular biology, ≥99.0% |
| Ammoniumpersulfat reagent grade, 98% | MERCK | 215589 | Catalyst for acrylamide gel polymerization. |
| Coomassie Brilliant blue R 250 | MERCK | 1125530025 | Coomassie Brilliant blue R 250 (C.I. 42660) for electrophoresis Trademark of Imperial Chemical Industries PLC. CAS 6104-59-2, pH 6.2 (10 g/l, H2O, 25 °C) |
| Dialysis tubing cellulose membrane | MERCK | D9277 | Cellulose membranes for the exchange of buffers via dialysis. |
| Eppendof tubes 1.5 mL | VWR | 525-1042 | microcentrifugal tubes; autoclaved |
| HiLoad 26/600 Superdex 75 pg | GE Healthcare Life Sciences / Cytiva | 28989334 | HiLoad Superdex 75 pg prepacked columns are for high-resolution size exclusion chromatography of recombinant proteins |
| Immun-Blot PVDF Membrane | BIO-RAD | #1620177 | PVDF membranes are protein blotting membranes optimized for fluorescent and multiplex fluorescent applications. |
| Mini Trans-Blot Electrophoretic Transfer Cell | BIO-RAD | #1703930 | Use the Mini Trans-Blot Cell for rapid blotting of Mini-PROTEAN precast and handcast gels. |
| Mini-PROTEAN Tetra Vertical Electrophoresis Cell for Mini Precast Gels | BIO-RAD | #1658004 | 4-gel vertical electrophoresis system, includes electrode assembly, companion running module, tank, lid with power cables, mini cell buffer dam. |
| Mono Q 10/100 GL | GE Healthcare Life Sciences / Cytiva | 17516701 | Mono Q columns are strong anion exchange chromatography columns for protein analysis or small scale, high resolution polishing of proteins. |
| Mono S 10/100 GL | GE Healthcare Life Sciences / Cytiva | 17516901 | Mono S columns are strong cation exchange chromatography columns for protein analysis or small scale high resolution polishing of proteins. |
| PageRuler Prestained Protein Ladder, 10 to 180 kDa | Thermo-Fischer | 26616 | A mixture of 10 blue-, orange-, and green-stained proteins (10 to 180 kDa) for use as size standards in protein electrophoresis (SDS-PAGE) and western blotting. |
| Pierce BCA Protein Assay Kit | Thermo-Fischer | 23225 | A two-component, high-precision, detergent-compatible protein assay for determination of protein concentration. |
| Sonifier 250; Ultrasonic Cell Disruptor w/ Converter | Branson | - | New models at https://www.emerson.com/documents/automation/brochure-sonifier-sfx250-sfx550-cell-disruptors-homogenizers-branson-en-us-168180.pdf |
| Swine Anti-Rabbit Immunoglobulins/HRP (affinity isolated) | Agilent Dako | P0399 | The antibody used for horseradish peroxidase conjugation reacts with rabbit immunoglobulins of all classes. |
| TEMED, 1,2-Bis(dimethylamino)ethane, TMEDA | MERCK | T9281 | TEMED (N,N,N′,N′-Tetramethylethylenediamine) is molecule which allows rapid polymerization of polyacrylamide gels. |
| Tube Roller | - | - | A general tube rotator roller; e.g. a new model at https://labstac.com/de/Mixer/Roller/c/71 |
| Tube Rotator | - | - | A general tube rotator wheel; e.g. a new model at https://labstac.com/de/Tube-Roller/p/MT123 |
| ULTRA-TURRAX; T 25 digital | IKA | 0003725000 | New models at https://www.ika.com/de/Produkte-Lab-Eq/Dispergierer-Dipergiergeraet-Homogenisierer-Homogenisator-csp-177/T-25-digital-ULTRA-TURRAX-cpdt-3725000/ |
Referenzen
- Pircher, H., et al. Identification of FAH domain-containing protein 1 (FAHD1) as oxaloacetate decarboxylase. Journal of Biological Chemistry. 290 (11), 6755-6762 (2015).
- Pircher, H., et al. Identification of human Fumarylacetoacetate Hydrolase Domain-containing Protein 1 (FAHD1) as a novel mitochondrial acylpyruvase. Journal of Biological Chemistry. 286 (42), 36500-36508 (2011).
- Kang, T. -. W., et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 479 (7374), 547-551 (2011).
- Hong, H., Seo, H., Park, W., Kim, K. K. -. J. Sequence, structure and function-based classification of the broadly conserved FAH superfamily reveals two distinct fumarylpyruvate hydrolase subfamilies. Environmental Microbiology. 22 (1), 270-285 (2020).
- Timm, D. E., Mueller, H. A., Bhanumoorthy, P., Harp, J. M., Bunick, G. J. Crystal structure and mechanism of a carbon-carbon bond hydrolase. Structure. 7 (9), 1023-1033 (1999).
- Bateman, R. L., et al. Mechanistic inferences from the crystal structure of Fumarylacetoacetate Hydrolase with a bound phosphorus-based inhibitor. Journal of Biological Chemistry. 276 (18), 15284-15291 (2001).
- Weiss, A. K. H., et al. Structural basis for the bi-functionality of human oxaloacetate decarboxylase FAHD1. Biochemical Journal. 475 (22), 3561-3576 (2018).
- Etemad, S., et al. Oxaloacetate decarboxylase FAHD1 - a new regulator of mitochondrial function and senescence. Mechanisms of Ageing and Development. 177, 22-29 (2019).
- Weiss, A. K. H., et al. Regulation of cellular senescence by eukaryotic members of the FAH superfamily - A role in calcium homeostasis. Mechanisms of Ageing and Development. 190, 111284 (2020).
- Petit, M., Koziel, R., Etemad, S., Pircher, H., Jansen-Dürr, P. Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Experimental Gerontology. 92, 7-12 (2017).
- Wiley, C. D., et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metabolism. 23 (2), 303-314 (2016).
- Weiss, A. K. H., et al. Expression, purification, crystallization, and enzyme assays of Fumarylacetoacetate Hydrolase Domain-containing proteins. Journal of Visualized Experiments: JoVE. (148), e59729 (2019).
- Weiss, A. K. H., et al. Inhibitors of Fumarylacetoacetate Hydrolase Domain Containing Protein 1 (FAHD1). Molcules. 26 (16), 5009 (2021).
- Mizutani, H., Kunishima, N. Purification, crystallization and preliminary X-ray analysis of the fumarylacetoacetase family member TTHA0809 from Thermus thermophilus HB8. Acta Crystallographica Section F Structural Biology and Crystallization Communications. 63 (9), 792-794 (2007).
- Lee, C. H. A simple outline of methods for protein isolation and purification. Endocrinology and Metabolism. 32 (1), 18-22 (2017).
- Amer, H. E. A. Purification of proteins: Between meaning and different methods). Proteomics Technologies and Applications. , (2019).
- Niu, L., Yuan, H., Gong, F., Wu, X., Wang, W. Protein extraction methods shape much of the extracted proteomes. Frontiers in Plant Science. 9, 802 (2018).
- Gordon, J. A. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods in Enzymology. 201, 477-482 (1991).
- Gallagher, S. R. SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Current Protocols in Essential Laboratory Techniques. , (2012).
- . Effect of pH on Protein Size Exclusion Chromatography Available from: https://www.agilent.com/cs/library/applications/5990-8138EN.pdf (2011)
- Sørensen, B. K., et al. Silver staining of proteins on electroblotting membranes and intensification of silver staining of proteins separated by polyacrylamide gel electrophoresis. Analytical Biochemistry. 304 (1), 33-41 (2002).
- Fagerberg, L., et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & Cellular Proteomics. 13 (2), 397-406 (2014).
- . Cytiva Life Fundamentals of size exclusion chromatography Available from: https://www.cytivalifesciences.com/en/us/solutions/protein-research/knowledge-center/protein-purification-methods/size-exclusion-chromatography (2022)
- Rosano, G. L., Ceccarelli, E. A. Recombinant protein expression in Escherichia coli: advances and challenges. Frontiers in Microbiology. 5, 172 (2014).
- Rosano, G. L., Morales, E. S., Ceccarelli, E. A. New tools for recombinant protein production in Escherichia coli: A 5-year update. Protein Science: A Publication of the Protein Society. 28 (8), 1412-1422 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten