
Method Article
Monitoring Protein Aggregation Kinetics In Vivo using Automated Inclusion Counting in Caenorhabditis elegans
* These authors contributed equally
In This Article
Summary
Here, a method is presented for the analysis of protein aggregation kinetics in the nematode Caenorhabditis elegans. Animals from an age-synchronized population are imaged at different time points, followed by semiautomated inclusion counting in CellProfiler and fitting to a mathematical model in AmyloFit.
Abstract
Protein aggregation into insoluble inclusions is a hallmark of a variety of human diseases, many of which are age-related. The nematode Caenorhabditis elegans is a well-established model organism that has been widely used in the field to study protein aggregation and toxicity. Its optical transparency enables the direct visualization of protein aggregation by fluorescence microscopy. Moreover, the fast reproductive cycle and short lifespan make the nematode a suitable model to screen for genes and molecules that modulate this process.
However, the quantification of aggregate load in living animals is poorly standardized, typically performed by manual inclusion counting under a fluorescence dissection microscope at a single time point. This approach can result in high variability between observers and limits the understanding of the aggregation process. In contrast, amyloid-like protein aggregation in vitro is routinely monitored by thioflavin T fluorescence in a highly quantitative and time-resolved fashion.
Here, an analogous method is presented for the unbiased analysis of aggregation kinetics in living C. elegans, using a high-throughput confocal microscope combined with custom-made image analysis and data fitting. The applicability of this method is demonstrated by monitoring inclusion formation of a fluorescently labeled polyglutamine (polyQ) protein in the body wall muscle cells. The image analysis workflow allows the determination of the numbers of inclusions at different timepoints, which are fitted to a mathematical model based on independent nucleation events in individual muscle cells. The method described here may prove useful to assess the effects of proteostasis factors and potential therapeutics for protein aggregation diseases in a living animal in a robust and quantitative manner.
Introduction
The accumulation of misfolded proteins into insoluble deposits occurs in a wide range of diseases. Well-known examples are the aggregation of amyloid-β and tau in Alzheimer's disease, α-synuclein in Parkinson's disease, and huntingtin with expanded polyQ in Huntington's disease1,2. The misfolding of these polypeptides into amyloid fibrils is associated with toxicity and cell death by mechanisms that are still largely unclear. Elucidating the mechanisms of amyloid formation will be crucial to developing effective therapies, which are currently unavailable.
Detailed investigations of amyloid formation have been performed in vitro based on thioflavin T fluorescence measurements, leading to a mechanistic understanding of the aggregation process and the effect of inhibitory molecules3,4,5. However, it is not clear whether the same aggregation mechanisms hold true in the complex environment of living cells and organisms. The nematode worm Caenorhabditis elegans is a suitable model organism to study protein aggregation in vivo. It has a relatively simple anatomy but consists of multiple tissues, including muscle, intestine, and a nervous system. It is genetically well-characterized, and tools for genetic modification are readily available. Furthermore, it has a short generation time of ~3 days and a total lifespan of 2-3 weeks. As such, protein aggregation can be examined across the lifespan of the animal on an experimentally convenient timescale. Finally, the nematode is optically transparent, enabling the tracking of the aggregation of fluorescently labeled proteins in live animals.
These features of C. elegans have been previously exploited to investigate the aggregation of polyQ proteins as a model for Huntington's and other polyQ expansion diseases. Above the pathogenic threshold of 35-40 glutamine residues, the polyQ proteins labeled with yellow fluorescent protein (YFP) can be observed to form insoluble inclusions in the muscle tissue6,7, neurons8, and the intestine9,10. These features have been widely used to screen for genes11,12,13 and small-molecule modifiers14 of protein aggregation and toxicity.
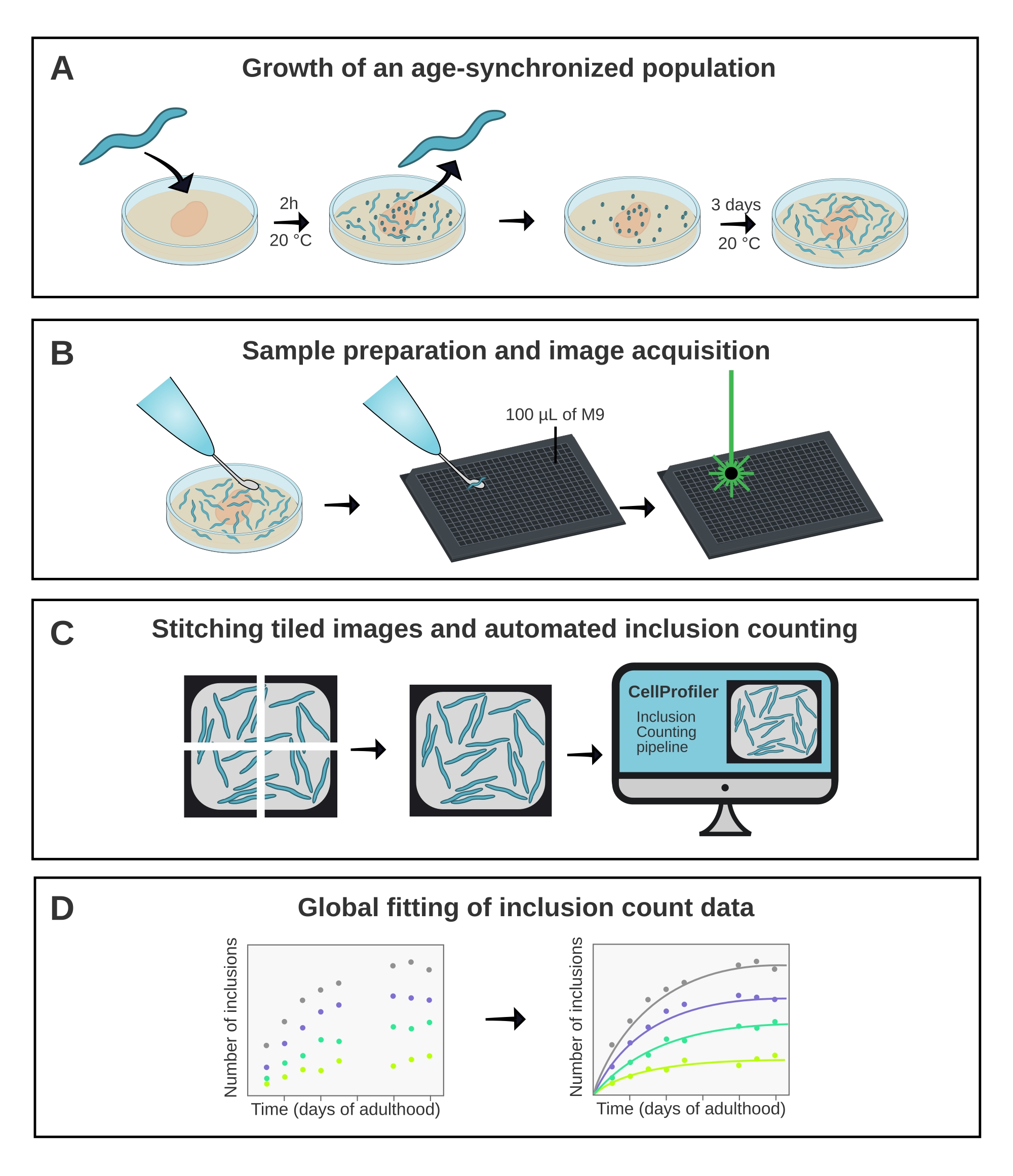
C. elegans has the potential to play an important role in bridging the gap between in vitro studies of protein aggregation and more complex disease models such as mice15. C. elegans is amenable to drug screening16 but can also be exploited to obtain a fundamental understanding of the molecular mechanisms of protein aggregation in vivo, as demonstrated recently17. However, for both applications, it is of prime importance to extract a quantitative and reproducible measure of protein aggregation. Here, this is achieved with the use of a high-throughput confocal microscope combined with a dedicated image analysis pipeline (Figure 1).
Protocol
1. Growth of an age-synchronized population of C. elegans
- Maintain the C. elegans strains on nematode growth medium (NGM) plates seeded with Escherichia coli OP50 at 20 °C according to standard procedures18.
- Perform a synchronized egg-lay by placing 10 adult nematodes onto a 6 cm seeded NGM plate with a platinum worm pick. Leave the adults to lay eggs for ~2 h at 20 °C before removing them. Prepare 1-4 plates per strain, depending on the fertility of the strain and the number of time points to be taken.
- Place the plates with eggs in the incubator at 20 °C. Monitor the development of the animals until they reach adulthood.
NOTE: The day on which the animals have reached adulthood is defined here as day 1. Typically, this is three days after the egg-lay. - Starting from day 1, transfer the animals to new seeded NGM plates daily to separate them from their offspring. To compensate for animals that die or are lost during transfer, transfer ~40 animals per strain times the number of points to be imaged (see step 2). Proceed until the animals have ceased to lay fertilized eggs (~day 6 of adulthood).
NOTE: Exclude animals with bagging or other developmental phenotypes. Bagging is commonly observed in strains expressing aggregation-prone proteins.
2. Sample preparation of C. elegans in a multiwell plate
NOTE: As the imaging procedure requires anesthetics that will eventually kill the animals, the same animals cannot be reused for subsequent time points. Instead, different animals from the same age-synchronized batch are imaged on different days. Even though most strains will have few inclusions at day 1, it is recommended to include this time point as a baseline.
- Prepare the 384-well plate by filling the required number of wells with 100 µL of M9 buffer supplemented with 25 mM NaN3 as an anesthetic. Fill one well per strain to be imaged.
NOTE: Sodium azide (NaN3) is toxic and should be handled with care. - For each strain, transfer 20 animals into one well using a platinum worm pick.
NOTE: The worms must be placed outside the bacterial lawn before placing them in the well. Bacteria make the animals adhere to the worm pick, which can prevent their release and will cloud the well contents. Generally, 20 is the optimal number of animals per well to prevent overlap between the worms while limiting unnecessary imaging of empty well space. - Cover the plate with the lid to prevent evaporation, and image the plate within 1 h after preparation.
- Repeat steps 2.1-2.3 daily until a steady plateau in the inclusion numbers is reached or until most animals have died. Perform the sample preparation and imaging at the same time every day to ensure intervals of 24 h.
3. Image acquisition on the high-throughput confocal microscope
NOTE: This experiment can also be set up on a regular spinning disk confocal microscope with a multiwell plate holder. A camera with a large field-of-view is beneficial to limit the number of tiles needed to be imaged to span the entire well. See the Table of Materials for details about the microscope and software used in this protocol.
- Switch on the instrument and open the software.
- Start a new protocol by going to Measurement Settings | New. Select the correct multiwell plate type and click Create a New Measurement Setting.
- Set up the channel for fluorescence by going to Ch 1. Set the objective to 10x. Select 488 nm as the light source and BP525/25 as the emission filter to image YFP. Set binning to 2x2 to reduce file size.
- Click Add Channel and select Brightfield as the method.
- To add a z-stack confocal fluorescence image to the measurement, choose 3D Fluorescence acquisition under Action List. Go to Select and choose Ch 1. To minimize file sizes, set Image Processing to Maximum so that the maximum projection image is saved rather than the full z-stack.
- Click BF/Ph Acquisition | Select | Ch 2 for the brightfield channel.
- Click on the play button (look for the right-pointing triangle symbol) next to Unload Well Plates and place the 384-well plate in the microscope.
- Under 3D Fluorescence Acquisition, click Test and select a well containing worms to determine the optimal shifting distance at which the worms are centered correctly. Set Ascending Distance to 50 µm, Descending Distance to -50 µm, and Slicing Interval to 2 µm to capture the entire thickness of the animals in the z-stack.
- Optimize the exposure time to get a good signal intensity for all four strains while avoiding saturation. Use the same exposure time for all strains and time points.
- Select the wells to be imaged under Well Plate Scan Setting. Select Tile and Acquire Whole Well.
- Save the Measurement Setting and start the experiment by clicking Start Measurement. For subsequent time points, open the same Measurement Setting and adjust the shifting distance and the wells to be imaged.
4. Stitching tiled images in ImageJ
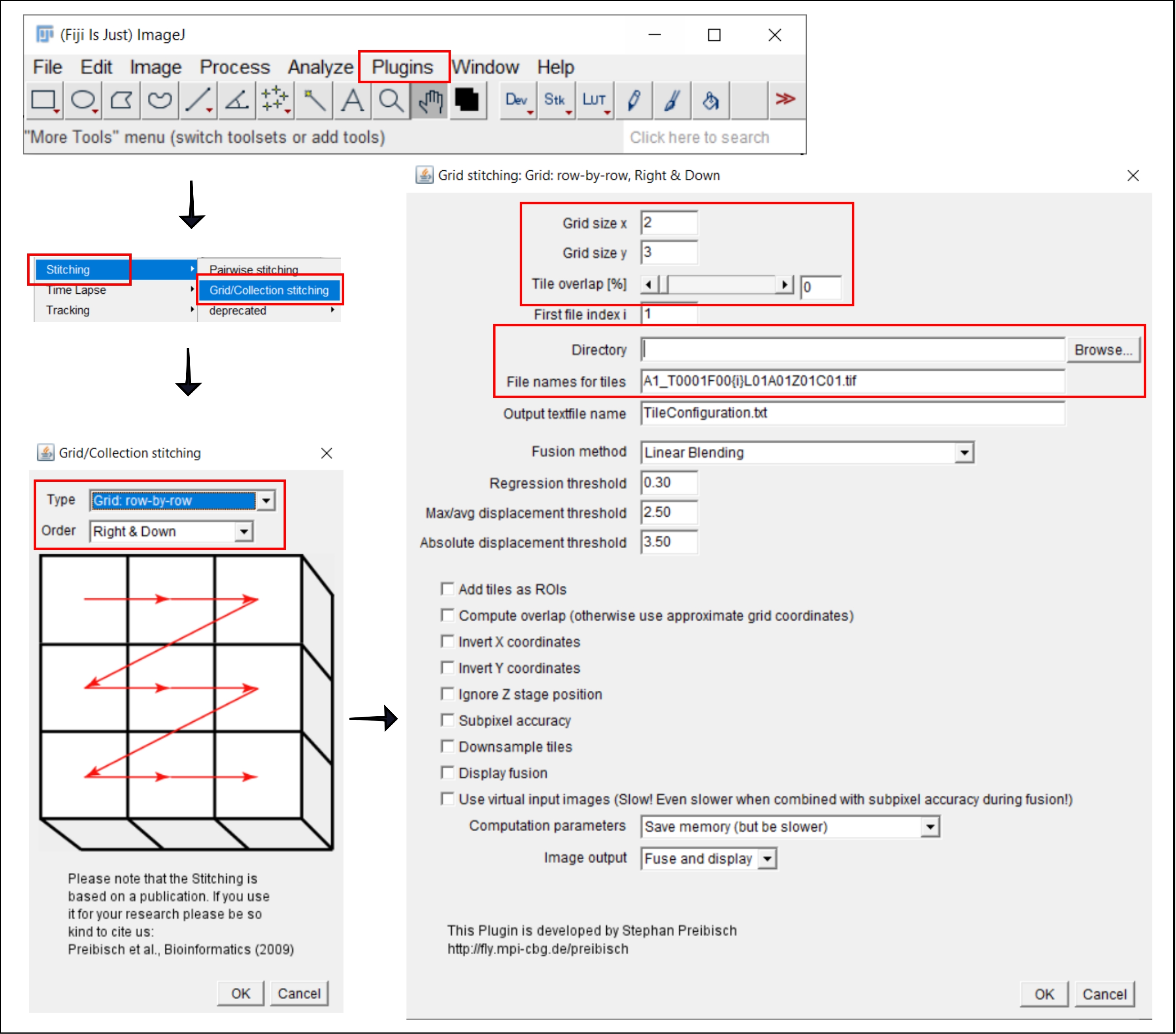
NOTE: This step is only required when using an objective larger than 4x, for which the image of each well is acquired as multiple tiles. In this analysis workflow, stitching of the tiles is performed using the free software FIJI/ImageJ19 (Figure 2). Depending on the instrument used in step 3, it may also be possible to perform stitching directly in the accompanying software.
- Download FIJI20 and open it.
- Go to Plugins | Stitching | Grid/Collection Stitching21.
- In the popup window, Grid/Collection Stitching, select the type and order by which the tiles were collected. Choose Grid: row-by-row and Right & Down.
- In the next window, Grid stitching: Grid: row-by-row, Right & Down, insert the number of tiles in x and y directions. For the 10x objective used here, choose 2 as the Grid size x, 3 as the Grid size y, and 0 as the Tile overlap.
- Click Browse and select the folder containing the TIFF images to be stitched.
- Insert the common file name under File names for tiles, using {i} at the position of the tile number in each file name.
- Untick all boxes below.
- Run the plugin.
- Save the resulting images as TIFF files for analysis in the next step.
5. Automated inclusion counting using CellProfiler22
- Download and install the open-source image analysis software, CellProfiler23. Download the InclusionCounting.cpproj pipeline from github.com/sinnigelab/aggregate-quantification.
- Open CellProfiler and drag the pipeline into the Drop a pipeline file here window. Click Yes to load the project.
- Click on the Images input module and drag the stitched images into the window Drop files and folder here.
- Click on the Metadata input module. Adjust Regular expression to extract from file name according to the names of the stitched images.
- Click on the NamesandTypes input module and adjust Select the rule criteria to match the channels in the file names.
NOTE: In the default settings of the pipeline, file names containing BF are recognized as brightfield images and are named Worms. File names containing YFP are recognized as fluorescence images and are named Fluorescence. - Click on View output settings to select a default folder to save the output from CellProfiler.
- Click on Start Test Mode to check the settings of the pipeline using the first imaging dataset. Click on Run to run through all modules in the pipeline or Step to run through the pipeline one module at a time. To adjust the worm outlines in the EditObjectsManually module, click on Help to see the instructions and click on Done to continue running the pipeline.
NOTE: The extracted measurements will not be exported while in test mode. The thresholding parameters to detect worms and inclusions may need to be adjusted based on the strains and magnification used. - Click on Exit Test Mode and Analyze Images.
- Open the output folder to view the output files. Open the images with the original file name followed by outlines to check whether the worms and inclusions were correctly overlaid.
NOTE: The number of inclusions per worm can be found in the file named ExpandedWormObjects. More information about the input images can be found in the file named Image. Additional output can be selected in the ExportToSpreadsheet module in the pipeline.
6. Global fitting of inclusion count data using AmyloFit5
NOTE: This step can only be performed when data for multiple protein concentrations are available. For Q40-YFP, a set of four strains with different levels of overexpression in the body wall muscle cells has been created previously17. In other cases, novel strains should be generated using plasmid microinjection and genomic integration24.
- Go to the free online fitting platform for aggregation kinetics AmyloFit25. Either register or log in with an existing account.
NOTE: An extensive manual on how to use AmyloFit can be accessed for additional help. See the link on the top-left of the webpage (after login) for more information. - To start using AmyloFit, name the project and click on Create project. Open the project by clicking on Open and create a session by giving it a name and clicking on Create & load session.
- Click Add Data and upload the file containing the average numbers of inclusions per animal, following the data format requirements shown in the left panel. Click Load New Data.
- Skip the preprocessing steps, which are not required for inclusion count data, by setting number of points to average over for zero-point offset and number of points to average over for plateau to 0. Click Submit. Repeat this step for each protein concentration (i.e., each column in the uploaded file).
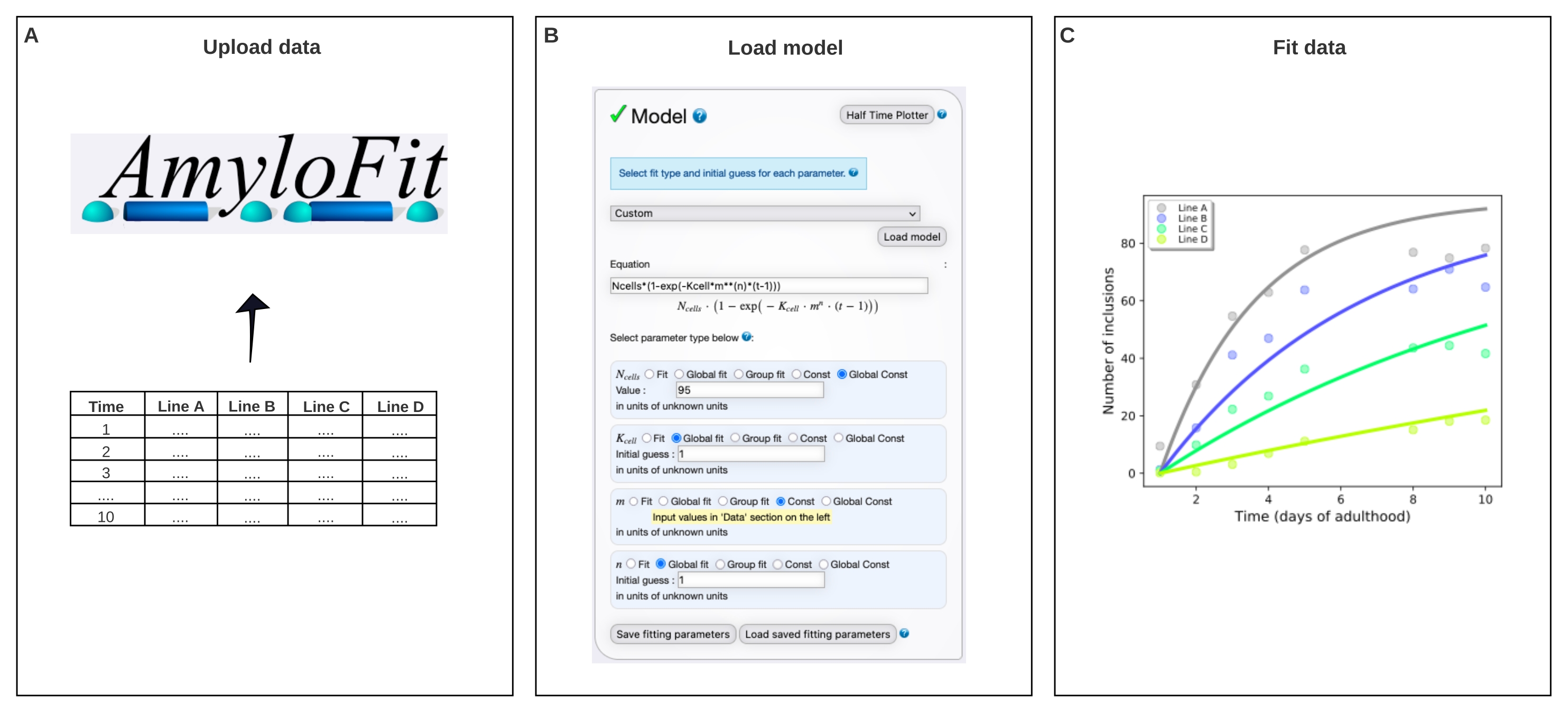
- Select Custom in the model panel, enter Ncells*(1-exp(-Kcell*m**(n)*(t-1))) in the equation box and click Load model.
NOTE: As AmyloFit was originally designed for the analysis of kinetic data from in vitro assays, a custom-made model must be loaded to analyze the inclusion count data of C. elegans. In the equation used here, Ncells is the number of cells in which inclusion formation takes place, Kcell is the nucleation rate constant, m the intracellular protein concentration, and n the reaction order of nucleation. - Set the parameter types to Global Const for Ncells, Global fit for Kcell and n, and Const for m. Set Value of Ncells to 95 for body wall muscle cells and Initial guess for Kcell and n to 1. Enter the values of m for the different strains in the left panel.
NOTE: Initial guesses are not relevant for the relatively simple model used here. For more complex models, it is beneficial to enter an estimate of the expected values to shorten the calculation time. - Leave the number of basin hops unchanged and click Fit in the fitting panel.
- Extract the fit by clicking Download Data and Fit.
NOTE: The parameters extracted by the global fit of the model will be listed in the bottom-right corner. A plot of the data and fit will be automatically generated in the top-right panel. This plot can be extracted by clicking Download pdf and customized by going to Display Plot Options. Kcell has units of molecules concentration-n time-1 cell-1. To compare values with different n, Kcell can be converted to the nucleation rate at a given protein concentration by multiplying it with mn.
Results
The method described here (Figure 1) was used to analyze the aggregation kinetics of a construct comprising 40 glutamines fused to YFP (Q40-YFP). The protein is expressed under the control of the unc-54 promoter, driving expression in the body wall muscle cells. As these are relatively large and easy to visualize, the use of a 10x objective is sufficient to resolve the inclusions formed by Q40-YFP in this tissue. Four strains (lines A-D) were previously developed expressing the protein to different extents to assess the concentration-dependence of polyQ aggregation in vivo17.
Age-synchronized populations of lines A-D were generated by a 2 h egg-lay, followed by daily transfer once the offspring reached adulthood. From day 1 to day 10 of adulthood, 20 animals from each of the four strains were imaged in a 384-well plate, using a high-throughput confocal microscope. The images of the wells were acquired as 6 tiles, which were stitched together using a plugin in ImageJ21 (Figure 2). The stitched images were subsequently analyzed using a custom-made CellProfiler22 pipeline (Figure 3) to quantify the average inclusion number per animal for each strain and time point.
The data were then fitted to a mathematical model in AmyloFit5 (Figure 4). The model is based on the assumption that each of the 95 body wall muscle cells independently acquires one inclusion by a rate-limiting nucleation event, followed by fast aggregate growth17. The fit yielded a nucleation rate constant of 9.9 × 105 molecules M-2.1 d-1 cell-1 and a reaction order of 2.1, corresponding to a nucleation rate of 0.38 molecules d-1 cell-1 at an intracellular protein concentration of 1 mM. Two independent biological replicates led to closely corresponding values for the nucleation rate and reaction order, which are in agreement with a previous study using a similar protocol17 (Table 1).

Figure 1: Schematic overview of the method. (A) Age-synchronized C. elegans populations are generated by a timed egg-lay. (B) Animals from the same population are imaged in a 384-well plate at different time points. (C) The tiles are stitched together to form images of the entire wells, which are analyzed in CellProfiler to quantify the inclusion numbers per animal. (D) The data are fitted to a mathematical model using AmyloFit. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Screenshots of the stitching procedure in ImageJ using the plugin Grid/Collection stitching21. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Schematic of the CellProfiler pipeline to quantify inclusions numbers. (A-C) The brightfield image (A) is used to identify the worms (B, close-up in C). (D-G) The fluorescence image (D, close-up in E) is used to identify the inclusions (F). The worms and inclusions are related to provide the number of inclusions for each worm in the well (G). The images shown are of Q40 line A animals at day 3 of adulthood. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Fitting the data to a mathematical model in AmyloFit. (A) Data are uploaded to AmyloFit. (B) A custom equation is entered to model inclusion formation, assuming independent nucleation events in each cell. (C) Fitting of the aggregation kinetics for C. elegans lines A-D expressing different levels of Q40-YFP. The data are representative of two independent biological replicates. Please click here to view a larger version of this figure.
{kind=link}
| Dataset 1 | Dataset 2 | Sinnige et al.17 | |
| n | 2.1 | 1.9 | 1.6 |
| Kcell (molecules M-n d-1 cell-1) | 9.9 x 105 | 1.4 x 105 | 3.1 x 104 |
| Nucleation rate at 1 mM (molecules d-1 cell-1) | 0.38 | 0.21 | 0.35 |
Table 1: Values of the nucleation rate and reaction order of Q40-YFP aggregation. Data for two independent biological replicates of the protocol and comparison with previously published data17.
Discussion
The method presented herein facilitates an unbiased and quantitative analysis of protein aggregation kinetics in the model organism C. elegans. It depends on four key elements (Figure 1): 1) maintaining an age-synchronized population of nematodes; 2) fluorescence microscopy in multiwell plates; 3) automated inclusion counting in CellProfiler; 4) data fitting in AmyloFit. Compared to manual counting of inclusions in freely moving animals or from saved images26, quantification in CellProfiler is both faster and more unbiased. The other key advancement of the protocol is the acquisition of kinetic data, rather than single timepoints, which provides quantitative insights into the aggregation mechanism upon fitting the data to a mathematical model.
The four elements of the protocol can be used as independent modules that can be modified depending on the application. Age-synchronized populations can also be maintained using 5-fluoro-2′-deoxyuridine (FUDR) to sterilize the animals. This compound affects lifespan and proteostasis24,25 and is highly carcinogenic to the experimenter; however, it precludes manual transfer of the worms, which can be labor-intensive when large numbers are handled. Other alternatives are the use of sterile mutants29 or filtration devices to separate offspring30.
The fluorescence microscopy step can also be adjusted, for example, using higher magnifications to monitor protein aggregation in neurons. Widefield microscopy may be sufficient to monitor polyQ aggregation in muscle cells when the relative difference between conditions is more important than the absolute numbers of inclusions. The CellProfiler pipeline can still be used in these cases, although the settings to recognize worms and inclusions will need to be adjusted by the user. The throughput of the technique is currently limited by the need for manual picking of the animals into the 384-well plate. This can potentially be remedied by the use of microfluidic devices16. Sodium azide is a relatively harsh anesthetic, which could be replaced by physical immobilization with hydrogels or beads28,29.
The analysis in AmyloFit presented here is based on an aggregation mechanism consisting of independent nucleation events in individual cells. In cases where this model does not fit, the user should consider an alternative such as the cooperative aggregation model developed previously17. A limitation of this approach is that strains expressing the protein of interest at different concentrations need to be available, although these can be generated using routine C. elegans methods24.
Altogether, this protocol provides the means to obtain high-quality data for protein aggregation kinetics in an in vivo model system, allowing for detailed analysis of aggregation mechanisms17. Although the method was demonstrated for polyQ aggregation in the C. elegans muscle tissue, future applications of the protocol may include other proteins and tissues and the effects of proteostasis factors and small molecules.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgements
We thank the Morimoto lab for C. elegans strains and Esmeralda Bosman for assistance on the high-throughput confocal microscope. This work was funded by a start-up grant from Utrecht University to T.S.
Materials
| Name | Company | Catalog Number | Comments |
| 384-well plate | Greiner | 781091 | Black with flat clear bottom |
| AmyloFit | Knowles lab | v2.0 | Access at www.amylofit.ch.cam.ac.uk |
| C. elegans Q40 line A | Morimoto lab | AM1228 | Genotype rmIs404[unc-54p::Q40::YFP] |
| C. elegans Q40 line B | Morimoto lab | AM1229 | Genotype rmIs404[unc-54p::Q40::YFP] |
| C. elegans Q40 line C | Morimoto lab | AM1230 | Genotype rmIs404[unc-54p::Q40::YFP] |
| C. elegans Q40 line D | Morimoto lab | AM1231 | Genotype rmIs404[unc-54p::Q40::YFP] |
| CellProfiler | Broad Institute | 4.1.3 | Downloaded from https://cellprofiler.org |
| E. coli OP50 | Caenorhabditis Genetics Center (CGC) | OP50 | |
| FIJI | Open-source | (Fiji Is Just) ImageJ v2.1/1.5.3j | Downloaded from https://imagej.net/software/fiji/ |
| High-throughput confocal microscope | Yokogawa | CellVoyager CV7000S | |
| M9 buffer | Home-made | 3 g/L KH2PO4, 6 g/L Na2HPO4, 0.5 g/L NaCl, 1 mM MgSO4 | |
| NGM plates | Home-made | 17 g/L agar, 2.5 g/L bacto-peptone, 3 g/L NaCl, 25 mM KPO4 buffer pH 6.0, 1 mM MgSO4, 1 mM CaCl2, 5 mg/L cholesterol | |
| Pasteur pipette | WU Mainz | 250 | To make worm pick, 150 mm length |
| Platinum iridium wire | Alfa Aesar | 39383 | To make worm pick, 0.25 mm diameter |
| Sodium azide | Sigma-Aldrich | S2002 | |
| Stereomicroscope | Leica | S9 |
References
- Knowles, T. P. J., Vendruscolo, M., Dobson, C. M. The amyloid state and its association with protein misfolding diseases. Nature Reviews. Molecular Cell Biology. 15 (6), 384-396 (2014).
- Chiti, F., Dobson, C. M. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annual Review of Biochemistry. 86 (1), 27-68 (2017).
- Knowles, T. P. J., et al. An analytical solution to the kinetics of breakable filament assembly. Science. 326 (5959), 1533-1537 (2009).
- Cohen, S. I. A., et al. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proceedings of the National Academy of Sciences of the United States of America. 110 (24), 9758-9763 (2013).
- Meisl, G., et al. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nature Protocols. 11 (2), 252-272 (2016).
- Satyal, S. H., et al. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 97 (11), 5750-5755 (2000).
- Morley, J. F., Brignull, H. R., Weyers, J. J., Morimoto, R. I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 99 (16), 10417-10422 (2002).
- Brignull, H. R., Moore, F. E., Tang, S. J., Morimoto, R. I. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. Journal of Neuroscience. 26 (29), 7597-7606 (2006).
- Moronetti Mazzeo, L. E., Dersh, D., Boccitto, M., Kalb, R. G., Lamitina, T. Stress and aging induce distinct polyQ protein aggregation states. Proceedings of the National Academy of Sciences of the United States of America. 109 (26), 10587-10592 (2012).
- Prahlad, V., Morimoto, R. I. Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proceedings of the National Academy of Sciences of the United States of America. 108 (34), 14204-14209 (2011).
- Nollen, E. A. A. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proceedings of the National Academy of Sciences of the United States of America. 101 (17), 6403-6408 (2004).
- Silva, M. C., et al. A genetic screening strategy identifies novel regulators of the proteostasis network. PLoS Genetics. 7 (12), 1002438(2011).
- Brehme, M., et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Reports. 9, 1-16 (2014).
- Calamini, B., et al. Small-molecule proteostasis regulators for protein conformational diseases. Nature Chemical Biology. 8 (2), 185-196 (2012).
- Sinnige, T., Stroobants, K., Dobson, C. M., Vendruscolo, M. Biophysical studies of protein misfolding and aggregation in in vivo models of Alzheimer's and Parkinson's diseases. Quarterly Reviews of Biophysics. 49, 22(2020).
- Mondal, S., et al. Large-scale microfluidics providing high-resolution and high-throughput screening of Caenorhabditis elegans poly-glutamine aggregation model. Nature Communications. 7, 13023(2016).
- Sinnige, T., et al. Kinetic analysis reveals that independent nucleation events determine the progression of polyglutamine aggregation in C. elegans. Proceedings of the National Academy of Sciences of the United States of America. 118 (11), 202188118(2021).
- Brenner, S. Caenorhabditis elegans. Methods. 77 (1), 71-94 (1974).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- FIJI/ImageJ. , Available from: https://imagej.net/downloads (2012).
- Preibisch, S., Saalfeld, S., Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 25 (11), 1463-1465 (2009).
- Lamprecht, M. R., Sabatini, D. M., Carpenter, A. E. CellProfiler: free, versatile software for automated biological image analysis. BioTechniques. 42 (1), 71-75 (2007).
- CellProfiler. Broad Institute. , Available from: https://cellprofiler.org/releases (2021).
- Mello, C. C., Kramer, J. M., Stinchcomb, D., Ambros, V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO Journal. 10 (12), 3959-3970 (1991).
- Knowles group, University of Cambridge. , Available from: https://amylofit.com/amylofitmain/login/ (2021).
- Lazaro-Pena, M. I., Cornwell, A. B., Samuelson, A. V. Quantifying tissue-specific proteostatic decline in Caenorhabditis elegans. Journal of Visualized Experiments: JoVE. (175), (2021).
- Feldman, N., Kosolapov, L., Ben-Zvi, A. Fluorodeoxyuridine improves Caenorhabditis elegans proteostasis independent of reproduction onset. PLoS One. 9 (1), 85964(2014).
- Brunquell, J., Bowers, P., Westerheide, S. D. Fluorodeoxyuridine enhances the heat shock response and decreases polyglutamine aggregation in an HSF-1-dependent manner in Caenorhabditis elegans. Mechanisms of Ageing and Development. 141-142, 1-4 (2014).
- David, D. C., et al. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biology. 8 (8), 47-48 (2010).
- Hunter, S., Maulik, M., Scerbak, C., Vayndorf, E., Taylor, B. E. Caenorhabditis sieve: A low-tech instrument and methodology for sorting small multicellular organisms. Journal of Visualized Experiments: JoVE. (137), (2018).
- Burnett, K., Edsinger, E., Albrecht, D. R. Rapid and gentle hydrogel encapsulation of living organisms enables long-term microscopy over multiple hours. Communications Biology. 1, 73(2018).
- Dong, L., et al. Reversible and long-term immobilization in a hydrogel-microbead matrix for high-resolution imaging of Caenorhabditis elegans and other small organisms. PLoS One. 13 (3), 0193989(2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved