Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Préparation d’énoprures aziridines non activées et synthèse de biémamide B, D et d’épiallo-isomuscarine
Dans cet article
Résumé
Dans cette étude, nous préparons les deux énantiomères de l’aziridine-2-carboxylate, qui sont utilisés dans la synthèse asymétrique d’alcaloïdes, y compris le biémamide B et D, et la (-)-épiallo-isomuscarine.
Résumé
Les hétérocycles aziridines contenant de l’azote sont synthétiquement très précieux pour la préparation de molécules azacycliques et acycliques. Cependant, il est très difficile et laborieux de fabriquer des aziridines sous des formes optiquement pures à grande échelle pour appliquer une synthèse asymétrique de composés d’aza. Heureusement, nous avons réussi à obtenir à la fois des énantiomères (2R) et (2S)-aziridine-2-carboxylates avec le groupe α-méthylbenzyle donneur d’électrons au niveau de l’azote en anneau sous forme d’aziridines non activées. Ces aziridines de départ ont deux groupes fonctionnels distincts: un anneau à trois membres hautement réactif et un carboxylate polyvalent. Ils sont applicables dans l’ouverture de l’anneau ou la transformation de l’anneau avec l’aziridine et dans la transformation du groupe fonctionnel à d’autres à partir du carboxylate. Ces deux énantiomères ont été utilisés dans la préparation de composés aminés acycliques et/ou aza-hétérocycliques biologiquement importants de manière asymétrique. Plus précisément, ce rapport décrit la première synthèse asymétrique opportune des deux énantiomères de produits naturels marins de type 5, 6-dihydrouracile, le biémamide B et D, en tant qu’inhibiteurs potentiels du TGF-β. Cette synthèse consistait en une réaction d’ouverture de l’anneau régio- et stéréosélective de l’aziridine-2-carboxylate et de la formation ultérieure de 4-aminotérastérahydropyrimidine-2,4-dione. Un autre exemple dans ce protocole portait sur une réaction de Mukaiyama hautement stéréosélective d’aziridine-2-carboxylate et d’éther d’énol de silyle, suivant l’ouverture intramoléculaire de l’anneau aziridine pour fournir un accès facile et facile à l'(-)-épiallo-isomuscarine.
Introduction
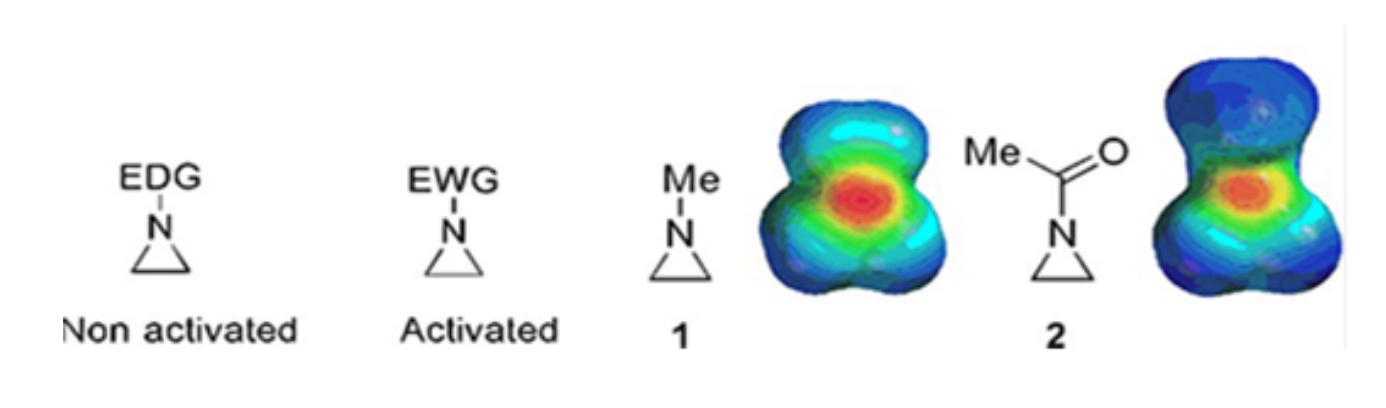
De petits anneaux constitués de cyclopropanes, d’oxiranes et d’aziridines se trouvent dans divers composés tels que les produits naturels et les médicaments 1,2. Ils sont principalement utilisés comme matières premières exploitant leur souche annulaire. Parmi les composés à trois anneaux, l’aziridine a été étudiée de manière moins approfondie en raison de son instabilité et de sa réactivité incontrôlable3. Comme le montrent les cartes de potentiel électrostatique (Figure 1), un groupe attaché à l’azote-cycle aziridine, qu’il donne ou attire des électrons, rend la basicité de l’azote différente. Cette différence offre un contraste frappant avec la réactivité et la sélectivité des aziridines correspondantes.

Figure 1 : Structures chimiques des aziridines « activées » et « non activées » et cartes du potentiel électrostatique de leurs exemples représentatifs N-méthylaziridine et N-acétylaziridine4. Cette figure a été modifiée avec la permission de Ranjith et al.4. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Lorsque l’azote annulaire a un groupe de retrait d’électrons, tel que le sulfonate, le phosphonate et le carbamate, nous l’appelons aziridine « activée ». Ceci est facilement réactif avec les nucléophiles pour compenser son instabilité avec une portée limitée de régiochimie. Ces aziridines activées sont préparées par diverses méthodes catalytiques et utilisées comme matière première. Une grande partie de la chimie récente de l’aziridine a traité de ces aziridines activées. Cependant, les aziridines activées souffrent de certaines restrictions résultant de leur instabilité et de la portée de réaction limitée de l’ouverture de l’anneau. D’autre part, les aziridines portant des substituants donneurs d’électrons, comme les groupes alkyle ou alkyle substitué, au niveau de l’azote en anneau appelé « non activé»4, sont relativement stables dans la plupart des circonstances et peuvent être laissées sur le banc pendant une longue période sans décomposition significative. Les réactions nucléophiles d’ouverture de cycle de l’aziridine non activée se produisent via la formation d’ions aziridinium. La plupart des réactions d’ouverture de cycle d’aziridine et de transformations d’anneaux se déroulent de manière hautement régiochimique. Cependant, très peu de rapports de littérature traitent de la préparation d’aziridines optiquement pures non activées avec des substituants aux positions C2 ou C3 5,6.
Cet article montre la préparation réussie de dérivés chiraux d’aziridine-2-carboxylate contenant du groupe α-méthylbenzyle, en particulier des dérivés de (-)-mentholyl(1R)-phényléthylaziridine-2-carboxylates comme mélange diastéréomérique, à partir de la réaction du 2,3-dibromopropionate et de la (1R)-phényléthylamine. À partir de ce mélange diastéréomérique, des énantiopures (1R)-phényléthyl-(2R)- et (2S)-aziridine-2-carboxylates sous forme d’esters (-)-mentholyliques ont été obtenus sous des formes optiquement pures par recristallisation sélective à partir de MeOH et de n-pentane sur des échelles de plusieurs centaines de kilos (Figure 1)7. Ces esters (-)-mentholyliques peuvent être facilement convertis en leurs esters éthyliques ou méthyliques par transestérification en présence de carbonate de magnésium ou de potassium7. Ces composés peuvent également être préparés facilement à l’échelle du laboratoire à partir des réactions des alkyl 2,3-dibromopropionates ou du triflate vinylique du α-cétoester avec la 2-phényléthylamine chirale suivie de la séparation du mélange diastéréomérique par simple chromatographie sur colonne flash8.
Une fois que nous avons énantiopure chiral aziridine-2-carboxylate, nous pouvons synthétiser diverses molécules cibles cycliques et acycliques contenant de l’azote biologiquement importantes sur la base de transformations de groupes fonctionnels de carboxylate et de réactions d’ouverture de cycle aziridine hautement régio- et stéréosélectives 6,9,10. La première synthèse asymétrique expéditive a été appliquée aux deux énantiomères de produits naturels marins de type 5, 6-dihydrouracile, le biémamide B et D, en tant qu’inhibiteurs potentiels du TGF-β11,12. Deuxièmement, la synthèse diastéréosélective des β-(aziridin-2-yl)-β-hydroxycétones a été réalisée par réaction d’aldol mukaiyama de 1-(1-phényléthyl)-aziridine-2-carboxaldéhyde optiquement pure et de divers silanes énols en présence de ZnCl2, à haut rendement (>82%) avec une stéréosélectivité presque parfaite (98:2 dr) via un état de transition contrôlé par chélation. Ceux-ci ont été utilisés pour la synthèse asymétrique des alcaloïdes épiallo-isomuscarine 13,14,15.
Protocole
1. Synthèse du mélange diastéréomérique de dérivé de l’ester chiral d’aziridine (-)-mentholyl (1)
- Ajouter l’ester 1a (5,0 g, 13,58 mM, 1,0 équiv) et une barre d’agitation magnétique de 250 mL séchée au four sous atmosphère d’azote (N2).
- Ajouter l’acétonitrile anhydre (60 mL) à la fiole réactionnelle à l’aide d’une seringue hermétique.
- Refroidir ensuite le mélange réactionnel à 0 °C à l’aide d’un bain de glace et remuer le mélange réactionnel pendant 5 min.
- Ajouter le carbonate de potassium (5,6 g, 40,74 mM, 3,0 équiv) dans le mélange réactionnel à la même température et laisser agiter pendant 30 min.
- Ajouter (2R)-phényléthylamine (2,0 mL, 16,29 nM, 1,2 équiv) goutte à goutte à température ambiante (RT) et laisser remuer le mélange réactionnel pendant 12 h.
- Surveiller la progression de la réaction à l’aide de la chromatographie sur couche mince à l’aide d’hexane:acétate d’éthyle 9:1 v/v (EtOAc; Rf = 0,4) comme éluant.
- Une fois la réaction terminée, filtrer le mélange sur du papier filtre (taille des pores 70 mm).
- Ensuite, ajoutez de l’eau (30 mL) dans le filtrat organique et extrayez la couche organique avec Et2O (2 x 50 mL) deux fois à l’aide d’un entonnoir de séparation.
- Sécher les extraits organiques combinés sur 7,5 g de Na2 SO4anhydre et les concentrer sous vide (<15 mbar) à l’aide d’un évaporateur rotatif.
REMARQUE: On obtient maintenant un mélange brut d’aziridine chirale diastéréomérique contenant à la fois des isomères de (R)-(1R,2 S,5 R)-2-isopropyl-5-méthylcyclohexyl1-((R)-1-phényléthyl)aziridine-2-carboxylate et de (S)-(1R,2 S,5 R)-2-isopropyl-5-méthylcyclohexyl1-((R)-1-phényléthyl)aziridine-2-carboxylate (4,1 g, 90%). - Isolement de l’aziridine chirale (R)-(1R,2S,5R)-2-isopropyl-5-méthylcyclohexyl 1-((R)-1-phényléthyl)aziridine-2-carboxylate (2) par une méthode de cristallisation sélective
- Ajouter 8,7 g du mélange brut de dérivé de l’ester 1 d’aziridine chirale (-)-menthol et dissoudre dans 70 mL de méthanol dans une fiole à fond rond à un seul col séchée au four.
- Maintenant, réchauffez le mélange réactionnel jusqu’à 70 ° C à l’aide d’un bain d’eau chaude, puis refroidissez le mélange réactionnel à -10 ° C jusqu’à ce que le cristal solide se forme.
- Filtrer le composé solide sur un papier filtre (taille des pores 70 mm) pour obtenir 2,2 g d’ester de (R)-(1R,2S,5R)-2-isopropyl-5-méthylcyclohexyl 1-((R)-1-phényléthyl)aziridine-2-carboxylate (2).
- Concentrer à nouveau la solution de filtrat sous vide (<15 mbar) à l’aide d’un évaporateur rotatif, dissoudre 50 mL d’éthanol dans le mélange réactionnel restant et recristalliser à -10 °C pour obtenir 1,2 g de (R)-(1R,2S,5R)-2-isopropyl-5-méthylcyclohexyl 1-((R)-1-phényléthyl)aziridine-2-carboxylate (2).
- À ce stade, utilisez l’autre alcooléthanol de la même manière que le méthanol.
- Après recristallisation, filtrer à nouveau sur un papier filtre (taille des pores 70 mm), concentrer les 5,3 g bruts restants de solution de filtrat complètement sous vide (<15 mbar) à l’aide d’un évaporateur rotatif et ajouter 50 mL d’un solvant d’hydrocarbure pentane.
- Conserver le reste de la solution réactionnelle à -15 °C.
REMARQUE: On obtient maintenant un composé solide de près de 1,9 g de (S)-(1R,2S,5R)-2-isopropyl-5-méthylcyclohexyl1-((R)-1-phényléthyl)aziridine-2-carboxylate ester (3). - Après avoir obtenu des cristaux, concentrer à nouveau la solution sous vide (<15 mbar) à l’aide d’un évaporateur rotatif et la dissoudre dans 30 mL de solvant hydrocarboné pentane.
- Recristalliser à nouveau à -15 °C pour obtenir 0,8 g de (S)-(1R,2S,5R)-2-isopropyl-5-méthylcyclohexyl1-((R)-1-phényléthyl)aziridine-2-carboxylate ester (2').
- Obtention de (R)-éthyl 1-((R)-1-phényléthyl)aziridine-2-carboxylate (3)
- Ajouter (R)-(1R,2 S,5 R)-2-isopropyl-5-méthylcyclohexyl1-((R)-1-phényléthyl)aziridine-2-carboxylate (2) (0,167 g, 0,57 mM) et un agitateur magnétique dans une fiole à fond rond à deux cols de 25 mL séchée au four sous atmosphère d’azote (N2).
- Ajouter 1,8 mL d’éthanol à la fiole réactionnelle à l’aide d’une seringue hermétique et remuer à TA.
- Ajouter ensuite le carbonate de potassium (0,40 g, 20,28 mmol, 4,0 équiv) et remuer à TA pendant 2 jours.
- Surveiller la progression de la réaction à l’aide de la chromatographie sur couche mince (éluant, 8:2 v/v, hexane:acétate d’éthyle (EtOAc), Rf = 0,6).
- Une fois la réaction terminée, filtrer le mélange sur du papier filtre (taille des pores 70 mm), puis ajouter de l’eau (5 mL) dans le filtrat organique et extraire la couche organique avec CH2Cl2 (2 x 15 mL) deux fois à l’aide d’un entonnoir de séparation.
- Sécher les extraits organiques combinés sur 3,0 g de Na2 SO4anhydre et les concentrer sous vide (<15 mbar) à l’aide d’un évaporateur rotatif.
- Purifier le produit brut par chromatographie sur colonne en phase normale sur gel de silice (70-230 mailles) pour obtenir un produit pur (R)-éthyl 1-((R)-1-phényléthyl)aziridine-2-carboxylate (3) (950 mg, 88 %). Rf (30 % EtOAc/hexane = 0,50).
2. Ouverture de l’anneau Regio et stéréosélective de l’aziridine par nucléophile azoté pour la synthèse totale du biémamide B et du biémamide D
- Synthèse de (R)-éthyl 2-azido-3-((((R)-1-phényléthyl)amino)propanoate (5)
- Transfert (500 mg, 2,20 mM, 1,0 équiv) de chiral (S)-éthyl 1-((R)-1-phényléthyl)aziridine-2-carboxylate (4) et d’un agitateur magnétique dans une fiole à fond rond à deux cols de 50 mL séchée au four sous atmosphère ouverte.
- Ajouter 50 % d’éthanol aqueux (15 mL) au mélange réactionnel.
- Refroidir le mélange réactionnel à 0 °C et ajouter l’acide sulfurique concentré (36 N) goutte à goutte pour maintenir un pH proche de 4,0, et remuer pendant 5 min.
- Ajouter l’azoture de sodium (370 mg, 5,70 mM, 2,5 équiv) à 0 °C et laisser remuer le mélange réactionnel pendant 10 min à la même température puis réchauffer à TA.
- Ensuite, ajouter AlCl3∙6H2O (55 mg, 0,22 mM, 0,1 équiv) comme catalyseur au même RT et laisser remuer pendant 3 h supplémentaires.
- Surveiller la progression de la réaction à l’aide de la chromatographie sur couche mince (éluant, 6:4 v/v, hexane:acétate d’éthyle (EtOAc), Rf = 0,2).
- Trempez le mélange réactionnel avec deux portions de NaHCO3 saturé de 20 mL.
- Filtrer ensuite le mélange brut sur un tampon de célite avec de l’éthanol (2 x 10 mL).
- Concentrer le mélange réactionnel sous vide (<15 mbar) à l’aide d’un évaporateur rotatif.
- Extraire la couche organique avec CH2Cl2 (2 x 50 mL) deux fois à l’aide d’un entonnoir de séparation.
- Sécher ensuite la couche organique combinée sur 5,0 g de Na2SO4 anhydre pendant 5 min.
- Concentrer la couche organique brute sous vide (15 mbar <) à l’aide d’un évaporateur rotatif pour obtenir un produit d’azoture brut.
- Purifier le produit brut par chromatographie sur colonne en phase normale sur gel de silice (70-230 mailles) en éluant avec 40% d’EtOAc/hexane (Rf = 0,20) pour obtenir 490 mg (rendement de 90%) de (R)-éthyl 2-azido-3-(((R)-1-phényléthyl)amino)propanoate (5) sous forme de liquide visqueux.
- Synthèse de (9H-Fluoren-9-yl)méthyl(3-((((R)-3-méthyl-2,4-dioxo-1-((R)-1 phényléthyl)hexahydro pyrimidin-5-yl)amino)-3-oxopropyl)carbamate (7)
- Transférer 150 mg de (R)-5-amino-3-méthyl-1-((R)-1-phényléthyl)dihydropyrimidine-2,4(1H,3H)-dione chirale (6) (150 mg, 0,60 mM, 1,0 équiv) et un agitateur magnétique dans une fiole à fond rond à deux cols de 25 mL séchée au four sous atmosphèreN2 .
- Ajouter ch2Cl2 sec (15,0 mL) dans la fiole réactionnelle à l’aide d’une seringue hermétique.
- Refroidir ensuite le mélange réactionnel à 0 °C à l’aide d’un bain de glace et remuer le mélange réactionnel pendant 5 min.
- Ajouter la Fmoc-bêta-alanine (377 mg, 1,20 mM, 2,0 équiv) et le DIPEA (0,67 mL, 3,64 mM, 6,0 équiv) à 0 °C et remuer pendant 5 min.
- Ajouter EDCI (347 mg, 1,82 mM, 3,0 équiv) et HOBt (165 mg, 1,21 mM, 2,0 équiv) au mélange réactionnel à 0 °C et laisser agiter pendant 10 min à la même température.
- Conserver le mélange réactionnel à RT et laisser remuer pendant 8 h supplémentaires.
- Surveiller la progression de la réaction à l’aide de la chromatographie sur couche mince en utilisant (2:8 v/v, hexane:acétate d’éthyle (EtOAc), Rf = 0,4) comme éluant.
- Tremper le mélange réactionnel avec de l’eau (10 mL).
- Lavez la couche organique combinée avec de la saumure (15 mL), puis extrayez la couche organique avec CH2 Cl 2(2 x 20 mL) deux fois à l’aide d’un entonnoir de séparation.
- Sécher ensuite la couche organique combinée sur 5,0 g de Na2SO4 anhydre pendant 5 min et la concentrer sous vide (<15 mbar) à l’aide d’un évaporateur rotatif.
- Purifier le produit brut par chromatographie sur colonne en phase normale sur gel de silice (70-230 mailles) en éluant avec 80% d’EtOAc/hexane (Rf = 0,40) pour obtenir 295 mg de (7) (rendement de 90%).
3. Réaction stéréosélective de Mukaiyama aldol avec l’aziridine-2-carboxaldéhyde chiral et son ouverture de cycle régio et stéréosélective de l’aziridine par hydroxy nucléophile interne pour la synthèse totale de (-)-épiallo-somuscarine (17)
- Synthèse de (S)-4-hydroxy-4-((R)-1-((R)-1-phényléthyl)aziridine-2-yl)butan-2-one (12)
- Transférer (R)-1-((R)-1-phényléthyl)aziridine-2-carbaldéhyde (10) (140 mg, 0,8 mM, 1,0 équiv) et un agitateur magnétique dans une fiole à fond rond à deux cols séchée au four sous atmosphère deN2 .
- Ajouter ch3CN sec (4,0 mL) dans la fiole de réaction à l’aide d’une seringue hermétique.
- Refroidir ensuite le mélange réactionnel à -20 °C à l’aide d’un bain glace-acétone et remuer le mélange réactionnel pendant 5 min.
- Ajouter ZnCl2 anhydre (108 mg, 0,8 mM, 1,0 équiv) au mélange réactionnel à -20 °C et laisser agir pendant 5 min.
- Ajouter ensuite le triméthyl(prop-1-en-2-yloxy)silane (11) (104 mg, 0,8 mM, 1,0 équiv) dissous dans duCH3CN sec (3,0 mL) au mélange réactionnel à -20 °C par goutte et laisser agiter le mélange réactionnel pendant 1 h à la même température.
- Surveiller la progression de la réaction à l’aide de la chromatographie sur couche mince en utilisant de l’hexane:acétate d’éthyle (EtOAc) de 8:2 v/v, Rf = 0,2) comme éluant.
- Trempez le mélange réactionnel avec du NaHCO3 saturé (4 mL).
- Extraire la couche organique avec EtOAc (2 x 15 mL) deux fois à l’aide d’un entonnoir de séparation.
- Sécher la couche organique combinée avec 3,0 g de Na2SO4 anhydre et la concentrer sous vide (<15 mbar) à l’aide d’un évaporateur rotatif.
- Purifier le produit brut par chromatographie sur colonne en phase normale sur gel de silice (70-230 mailles) en éluant avec 80% d’EtOAc/hexane, (Rf = 0,20) pour obtenir 58 mg de (S)-4-hydroxy-4-(((R)-1-((R)-1-phényléthyl)aziridine-2-yl)butan-2-one) (12) (rendement de 85%).
- Synthèse de (R)-N-((((2R,3 S,5 R)-3-(tert-butyldiméthylsilyl)oxy)-5-méthyltétra hydrofurane-2-yl)méthyl)-1-phényléthanamine (15)
- Transférer le (S)-4-((tert-butyldiméthylsilyl)oxy)-4-((R)-1-((R)-1-phényléthyl)aziridine-2-yl)butan-2-one (13) (400 mg, 1,15 mM, 1,0 équiv) et un agitateur magnétique dans une fiole à fond rond à deux cols de 25 mL séchée au four sous atmosphèreN2 .
- Ajouter le THF anhydre (50 mL) à la fiole réactionnelle à l’aide d’une seringue hermétique.
- Refroidir le mélange réactionnel à -78 °C à l’aide d’un bain de glace carbonique-acétone et laisser remuer pendant 5 min.
- Ajouter ensuite le tri-sec-butylborohydrure de lithium (L-selectride) (solution 1 M en THF) (2,3 mL, 2,0 équiv) goutte à goutte dans le mélange réactionnel à -78 °C et laisser remuer encore 25 min.
- Réchauffer le mélange réactionnel à RT et laisser remuer pendant 8 h.
- Surveiller la progression de la réaction par TLC en utilisant 30% d’EtOAc/hexane (Rf = 0,40) comme éluant.
- Après la consommation complète du composé 13, tremper le mélange réactionnel avec 0,1 M de NaOH (5 mL).
- Extraire la couche organique avec de l’EtOAc (3 x 15 mL), puis laver avec de la saumure (15 mL).
- Sécher la couche organique sur 3,0 g de Na2 SO4anhydre et la concentrer sous vide (<15 mbar) à l’aide d’un évaporateur rotatif.
- Purifier le produit brut par chromatographie sur colonne en phase normale sur gel de silice (70-230 mailles, éluant, 8:2 v/v, hexane:acétate d’éthyle (EtOAc), Rf = 0,2) pour obtenir un composé pur (15) (382 mg, rendement de 95 %).
- Synthèse de l’iodure de (-)-épiallo-isomuscarine (17)
- Transférer le composé 16 (20 mg, 0,15 mM, 1,0 équiv) et une barre d’agitation magnétique dans une fiole à fond rond de 10 mL séchée au four sous atmosphèreN2 .
- Ajouter 3 mL de fiole réaction EtOAc à l’aide d’une seringue hermétique.
- Ajouter ensuite l’iodure de méthyle (0,4 mL, 3,0 mM) au mélange réactionnel à RT.
- Ajouter la 1,2,2,6,6-pentaméthylpipéridine (PMP) (0,05 mL, 0,3 mM, 2,0 équiv) au mélange réactionnel à 0 °C.
- Ensuite, maintenir le mélange réactionnel à RT et laisser remuer pendant 16 h supplémentaires.
- Évaporer le solvant sous vide (<15 mbar) à l’aide d’un évaporateur rotatif pour donner le produit brut.
- Laver trois fois en ajoutant (3 x 5 mL) 10 % de MeOH dans de l’EtOAc au mélange réactionnel brut.
- Ensuite, lavez le mélange brut avec du n-pentane (5 mL) et concentrez-le sous vide (<15 mbar) à l’aide d’un évaporateur rotatif pour obtenir de l’iodure pur (-)-épiallo-isomuscarine (17) (32 mg, 68%).
4. Caractérisation de tous les produits
- Caractériser tous les nouveaux composés par spectroscopie RMN 1H, 13C et spectrométrie de masse à haute résolution (HRMS)7,8,11.
Résultats
Ici, nous rapportons la synthèse d’énantiopure aziridine-2-carboxylates. Le mélange diastéréomérique de (R)-(1R,2 S,5 R)-2-isopropyl-5-méthylcyclohexyl1-((R)-1-phényléthyl)aziridine-2-carboxylate (2) et de (S)-(1R,2S,5R)-2-isopropyl-5-méthylcyclohexyl1-((R)-1-phényléthyl)aziridine-2-carboxylate (3) (4,1 g, 90 %) a été préparé en rendement quantitatif à partir de l’ester de 2,3-dibromopropane (-)-...
Discussion
Les aziridines en tant qu’hétérocycles à trois membres contenant de l’azote ont un énorme potentiel pour les martiaux de départ synthétiques ou les intermédiaires pour préparer des molécules organiques riches en azote. Sur la base du groupe portant à l’anneau l’azote, elles sont classées comme aziridines « activées » et « non activées » dont la réactivité chimique et la sélectivité sont différentes. Cependant, des méthodes très limitées sont disponibles pour préparer cette précieuse...
Déclarations de divulgation
Les auteurs déclarent qu’il n’y avait pas de conflit d’intérêts dans cette étude.
Remerciements
Cette recherche a été soutenue par la National Research Foundation of Korea (NRF-2020R1A2C1007102 et 2021R1A5A6002803) avec le Center for New Directions in Organic Synthesis et une subvention HUFS 2022.
matériels
| Name | Company | Catalog Number | Comments |
| (2R)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester, 98% | Sigma-Aldrich | 57054-0 | |
| (2S)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester | Sigma-Aldrich | 57051-6 | |
| 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride | TCI | 424331-25 g | CAS No: 25952-53-8 |
| 1,4-Dioxane | SAMCHUN | D0654-1 kg | CAS No: 123-91-1 |
| 1-Hydroxybenzotriazole hydrate | Aldrich | 219-989-7-50 g | CAS No: 123333-53-9 |
| 2,6-Lutidine | Alfa Aesar | A10478-AP, 500 mL | CAS No: 108-48-5 |
| Acetonitrile | SAMCHUN | A0127-18 L | CAS No: 75-05-8 |
| Acetonitrile-d3 | Cambridge Isotope Laboratories, | 15G-744-25 g | CAS No: 2206-26-0 |
| Aluminum chloride hexahydrate | Aldrich | 231-208-1, 500 g | CAS No : 7784-13-6 |
| Bruker AVANCE III HD (400 MHz) spectrometer | Bruker | NA | |
| Chloroform-d | Cambridge Isotope Laboratories, | 100 g | CAS No: 865-49-6 |
| Dichloromethane | SAMCHUN | M0822-18 L | CAS No: 75-09-2 |
| Dimethyl sulfoxide-d6 | Cambridge Isotope Laboratories, | 25 g | CAS No: 2206-27-1 |
| Ethanol | EMSURE | 1009831000,1L | CAS No: 64-17-5 |
| Ethyl acetate | SAMCHUN | E0191-18 L | CAS No: 141-78-6 |
| High resolution mass spectra/MALDI-TOF/TOF Mass Spectrometry | AB SCIEX | 4800 Plus | High resolution mass spectra |
| JASCO P-2000 | JASCO | P-2000 | For optical rotation |
| Lithium aluminum hydride | TCI | L0203-100 g | CAS No: 16853-85-3 |
| L-Selectride, 1 M solution in THF | Acros | 176451000, 100 mL | CAS No: 38721-52-7 |
| Methanol | SAMCHUN | M0585-18 L | CAS No: 67-56-1 |
| N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-β-alanine | TCI | F08825G-5 g | CAS No: 35737-10-1 |
| N-Ethyldiisopropylamine | Aldrich | 230-392-0, 100 mL | CAS No: 7087-68-5 |
| n-Hexane | SAMCHUN | H0114-18 L | CAS No: 110-54-3 |
| Ninhydrin | Alfa Aesar | A10409-250 g | CAS No: 485-47-2 |
| p-Anisaldehyde | aldrich | A88107-5 g | CAS No: 123-11-5 |
| Phosphomolybdic acid hydrate | TCI | P1910-100 g | CAS No: 51429-74-4 |
| Sodium azide | D.S.P | 703301-500 g | CAS No: 26628-22-8 |
| Sodium Hydride 60% dispersion in mineral oil | Sigma-Aldrich | 452912-100 G | CAS No: 7646-69-7 |
| Sodium hydroxide | DUKSAN | A31226-1 kg | CAS No: 1310-73-2 |
| Sodium sulfate | SAMCHUN | S1011-1 kg | CAS No: 7757-82-6 |
| Thin Layer Chromatography (TLC) | Merck | 100390 | |
| Tert-Butyldimethylsilyl trifluoromethanesulfonate, 98% | Aldrich | 274-102-0, 25 g | CAS NO: 69739-34-0 |
| Tetrahydrofuran | SAMCHUN | T0148-18 L | CAS No: 109-99-9 |
| Triethylethylamine | DAEJUNG | 8556-4400-1 L | CAS No: 121-44-8 |
| UV light | Korea Ace Sci | TN-4C | 254 nm |
| Zinc chloride, anhydrous, 98+% | Alfa Aesar | A16281-22100 g | CAS No : 7646-85-7 |
Références
- Pitzer, K. S. Strain energies of cyclic hydrocarbons. Science. 101 (2635), 672 (1945).
- Dudev, T., Lim, C. Ring strain energies from ab initio calculations. Journal of the American Chemical Society. 120 (18), 4450-4458 (1998).
- D'hooghe, M., Ha, H. -. J. . Synthesis of 4- to 7-Membered Heterocycles by Ring Expansion. , (2016).
- Ranjith, J., Ha, H. -. J. Synthetic applications of aziridinium ions. Molecules. 26 (6), 1744 (2021).
- Sweeney, J. B. Aziridines: epoxides' ugly cousins. Chemical Society Reviews. 31 (5), 247-258 (2002).
- Stankovic, S., et al. Regioselectivity in the ring opening of non-activated aziridines. Chemical Society Reviews. 41 (2), 643-665 (2012).
- Lee, W. K., Ha, H. -. J. Highlights of the chemistry of enantiomerically pure aziridine-2-carboxylates. Aldrichimica Acta. 36 (2), 57-63 (2003).
- Tranchant, M. J., Dalla, V., Jabin, I., Decroix, B. Reaction of vinyl triflates of α-keto esters with primary amines: efficient synthesis of aziridine carboxylates. Tetrahedron. 58 (42), 8425-8432 (2002).
- Ha, H. -. J., Jung, J. -. H., Lee, W. K. Application of regio- and stereoselective functional group transformation of chiral aziridine-2-carboxylate. Asian Journal of Organic Chemistry. 3 (10), 1020-1035 (2014).
- Kim, Y., et al. Preparation of 2,3-diaminopropionate from ring opening of aziridine-2-carboxylate. Tetrahedron Letters. 46 (25), 4407-4409 (2005).
- Srivastava, N., Macha, L., Ha, H. -. J. Total synthesis and stereochemical revision of biemamides B and D. Organic Letters. 21 (22), 8992-8996 (2019).
- Zhang, F., et al. Biemamides A-E, inhibitors of the TGF-β pathway that block the epithelial to mesenchymal transition. Organic Letters. 20 (18), 5529-5532 (2018).
- Srivastava, N., Ha, H. -. J. Highly efficient and stereoselective Mukaiyama Aldol reaction with chiral aziridine-2-carboxaldehyde and its synthetic applications. Asian Journal of Organic Chemistry. 11 (1), 2021005671 (2021).
- Kempter, I., et al. Synthesis and structural characterization of the isomuscarines. Tetrahedron. 70 (10), 1918-1927 (2014).
- Pirrrung, M. C., DeAmicis, C. V. Total synthesis of the muscarines. Tetrahedron Letters. 29 (2), 159-162 (1988).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.