
Method Article
Deep Fluorescence Observation in Rice Shoots via Clearing Technology
In This Article
Summary
The present protocol describes a clearing technique for rice shoots, which are difficult to prepare for internal structural observations owing to the hard, thick, or layered nature of the tissues. This method facilitates continuous and deep fluorescence observations, even in adult rice plants.
Abstract
The recently developed clearing technology that eliminates refractive index mismatches and diminishes auto-fluorescent material has made it possible to observe plant tissues in three dimensions (3D) while preserving their internal structures. In rice (Oryza sativa L.), a monocot model plant and a globally important crop, clearing technology has been reported in organs that are relatively easy to observe, such as the roots and leaves. Applications of clearing technology in shoot apical meristem (SAM) and stems have also been reported, but only to a limited degree because of the poor penetration of the clearing solution (CS) in these tissues. The limited efficiency of the clearing solutions in these tissues has been attributed to auto-fluorescence, thickening, and hardening of the tissues in the stem as the vascular bundles and epidermis develop and layering of the SAM with water-repellent leaves. The present protocol reports the optimization of a clearing approach for continuous and 3D observation of gene expression from the SAM/young panicle to the base of the shoots during development. Fixed tissue samples expressing a fluorescent protein reporter were trimmed into sections using a vibrating micro-slicer. When an appropriate thickness was achieved, the CS was applied. By specifically targeting the central tissue, the penetration rate and uniformity of the CS increased, and the time required to make the tissue transparent decreased. Additionally, clearing of the trimmed sections enabled the observation of the internal structure of the whole shoot from a macro perspective. This method has potential applications in deep imaging of tissues of other plant species that are difficult to clear.
Introduction
Recently developed clearing technology has made it possible to observe the deep tissues of plants while preserving their internal structure1,2,3. In the dicot model plant Arabidopsis, many studies on fluorescent protein imaging have been conducted using clearing technology to eliminate refractive index mismatches and remove auto-fluorescent materials4,5,6. Although the use of clearing technology7,8 and 3D imaging at cellular resolution9 have been reported in rice (Oryza sativa L.), a monocot model plant and a globally important crop, these are limited to relatively thin and soft organs, such as roots, leaves, and shoot apical meristem (SAM), which are easy to observe.
The shoot is the main organ constituting the aboveground parts of vascular plants. In rice, shoots are composed of a series of vertically stacked "phytomers," comprising axillary buds, leaves, and the stem10. At the tip of the shoot, the SAM is composed of undifferentiated stem cells in the center. Phytomers are formed by the differentiation of cells derived from the SAM. After the plants shift from the vegetative to the reproductive phase, rice stems elongate and the SAM differentiates into young panicles10. This developmental change is accompanied by fluctuations in the expression of various genes in the stems and SAM/young panicles. To understand the mechanisms underlying cell differentiation into various tissues, it is important to structurally observe the cell morphology and gene expression in internal shoot tissues. However, the deep imaging of stems (nodes and internodes) in the shoot presents a challenge because of the inefficiency of clearing solutions to penetrate the tissue. Stems immediately undergo a rapid volume increase from lateral growth after differentiation from the SAM. The hardening of the rice node tissues due to the thickening of the vascular bundles and the horizontal complex link of the nodal vascular anastomosis, in addition to the high repellency of rice shoots, all contribute to limiting the penetration of the CS in stems10.
This study aimed to observe changes in gene expression in rice shoot tissues using a structural deep fluorescence technique. This work optimizes a clearing protocol for rice to continuously observe gene expression from the SAM/young panicle to the base in a 3D structure, rather than on a flat surface, using a confocal laser microscope.
Protocol
1. Preparation of the fixative solution
- Transfer 70 mL of sterile water into a glass bottle and add 10 g of paraformaldehyde (PFA).
CAUTION: PFA is toxic; hence, wearing gloves is recommended. - Add 2 mL of 1 N NaOH to dissolve the PFA solution.
- Dissolve the PFA solution with continuous stirring (300-500 rpm) at 60 °C for about 1 h until the PFA solution is transparent.
- Add sterilized water to increase the volume of the PFA solution to 100 mL.
NOTE: The PFA solution was prepared freshly before use. Moreover, a PFA solution stored at 4 °C can be used for approximately 2 months. - Immediately before sampling, add 25 mL of 60 mM HEPES (pH 7.4), 3 mL of 1 M sucrose, and 2 mL of sterile water into 20 mL of 10 % PFA to prepare a 50 mL fixative solution.
2. Preparation of the clearing solution (CS)
- Weigh and mix 7.5 g of sodium deoxycholate, 5 g of xylitol, and 12.5 g of urea (see Table of Materials) in 50 mL of sterile water.
NOTE: Sodium deoxycholate powder was weighed in a general laboratory draft chamber to prevent airborne dispersion. - Dissolve the ingredients (step 2.1.) using a magnetic stirrer to prepare the CS.
NOTE: The present study used in-house prepared clearing solution (CS), but a commercially available clearing solution, ClearSee (see Table of Materials), can also be used. - Transfer the CS into a 50 mL conical tube and store it at room temperature (15-30 °C) in the dark.
NOTE: The CS can be stored for more than 1 year.
3. Sampling
- Cut the roots carefully without damaging the plants. Wash the plants with water to remove the dirt.
NOTE: This method studied the observation of tissues from the three-leaf stage (LS) to flag LS. The present study used the rice cultivars Nipponbare (Nip) and Taichung 65 (T65) at the 8-10 LS. - Peel off the old outer leaves with hands and tweezers. There were about 2-3 leaves left.
- Cut the tissue from the SAM/young panicle to the base with a single-edged razor using a sliding motion to avoid crushing the cells (Figure 1A-C). If the position of the SAM/young panicle is not visible, cut it longer.
- Since the cross-section of the rice shoot has an oval shape (Figure 1D), shave off one side of the oval thinly in the direction of its long axis with a double-edged razor (Figure 1E-F).
NOTE: This step made it easier for the fixative solution to penetrate the sample. A horizontal cutting surface makes the sample easier to stick to a vibrating micro-slicer (see Table of Materials) tray. - Confirm the position of the SAM/young panicle by the appearance of a whitish color in the sample (Figure 1G). Trim off excess tissue.
NOTE: The length of the sample should be adjusted to the maximum length that can be trimmed using a vibrating micro-slicer. If the sample is too long, divide it into several pieces.
4. Fixation of the samples
- Pipette 1 mL of the fixative solution (prepared in step 1.) to a 1.5 mL microcentrifuge tube and immediately put in the samples.
NOTE: Ensure that the amount of sample does not exceed 20% of the volume of the fixative solution. - Cut a piece of paraffin film into 2.5 cm2. Shape the paraffin film into balls and place them on top of the samples to prevent them from floating out of the fixative solution.
- Place the tube containing the sample in a desiccator. Then, close the lid of the desiccator and start the vacuum pump to depressurize the inside of the desiccator until −0.095 MPa slowly.
- After closing the desiccator at −0.095 MPa, turn off the vacuum pump and allow it to stand for 30 min at room temperature.
NOTE: The vacuum level was such that bubbles appeared gradually in the samples. - Open the desiccator slightly and slowly return it to atmospheric pressure. Reduce the pressure of the desiccator to −0.095 MPa, turn off the vacuum pump, and allow the mixture to stand for 30 min at room temperature.
- Open the desiccator slightly and slowly return it to atmospheric pressure. If the sample is correctly fixed, it sinks into the fixative solution. If it did not sink, reduce the pressure again.
- Remove the paraffin film, close the lid, and keep the tubes overnight at 4 °C.
NOTE: Samples were fixed in fixative solution for at least 2 h. It can be stored for ~1 week.
5. Trimming the fixed sample
- Remove the excess fixative solution from the sample using lint-free wipes.
NOTE: Excess fixative solution prevents the sample from sticking properly onto the tray. - Fix the sample on a vibrating micro-slicer tray with instant glue. Place it on a tray with the flat side down, which was shaved off during sampling.
NOTE: Due to the structure of the rice shoot, it must be trimmed from the base toward the SAM/young panicle. - Set the trimming conditions according to the fixed sample.
NOTE: In this study, trimming was set to THICK, 130 µm; WIDE, 15 mm; FREQUENCY, 15%; and CUTTING SPEED, 32 mm. - Adjust the sample position with the RVS or FWD button and the UP or DOWN button. Fill the tray with deionized water until all the samples are immersed. After filling the tray, press the START button. Place the trimmed samples on a glass slide with a drop of 1x PBS.
NOTE: Shift the position of the sample to the other side after each trimming since the hard sample wears out the razor blade. - After checking the sample containing the target site under a stereomicroscope, transfer the trimmed sample to a multiwell plate with 1 mL of 1x PBS.
NOTE: Choose the number of wells according to the size of the sample. In this study, a 12-multiwell plate was used.
6. Treatment with the clearing solution
- Remove the 1x PBS from the multiwell plate and add fresh 1x PBS. Let it stand for 1 min.
- Remove 1x PBS and add the CS.
NOTE: The amount of CS must be five times the sample volume. - Place the multiwell plate containing the sample in a desiccator. Then, close the lid of the desiccator and start the vacuum pump to depressurize the inside of the desiccator until −0.09 MPa slowly.
- After closing the desiccator at −0.09 MPa, turn off the vacuum pump and let it stand for 1 h at room temperature.
- Open the desiccator slightly and slowly return it to atmospheric pressure.
- Pour deionized water into the gap between the wells to prevent the evaporation of the CS. Store the multiwell plates at room temperature in the dark for clearing.
NOTE: Gently shake the multiwell plate every 1-2 days to diffuse the chlorophyll and other components. - When the CS turns green, replace it with fresh solution.
7. Cell wall staining with chemical dye
- Pipet the calcofluor white solution (1 mL, final concentration: 1 mg/mL in the clearing solution, see Table of Materials) into a multiwell plate. Place the samples in the dye solution and incubate for 1 h.
NOTE: The volume of the dye must not exceed 10% of the volume of the CS. - Wash the stained samples in another multiwell plate with 1 mL of the CS for at least 1 h.
8. Observation with a confocal laser microscope
- Place a drop of CS onto a microscope slide and transfer the treated (with CS) samples onto the slide.
NOTE: As the treated samples are soft, they must be carefully handled with tweezers. - Cover the sample with a glass coverslip to prevent the evaporation of the CS.
NOTE: The CS precipitates easily. - Observe the treated samples under a confocal laser microscope.
NOTE: After observation, the treated samples can be returned to the CS for further observations later.
Results
First, the extent to which the hard and thick tissues of adult rice plants could be made transparent by the clearing treatment was validated. Nip at the 9-10 LS, which is the panicle formation stage, and 12-16 mm long tissues cut out from the young panicle to the base of the shoot were used. These samples, including the young panicle, were fixed in the fixative solution and observed before and after 1-4 weeks of the clearing treatment (Figure 2). The untrimmed samples remained green and did not become transparent after 4 weeks or even after 3 months of treatment with the clearing solution (Figure 2A). In samples that were peeled off all leaves, the internodes turned white after 1 week of treatment with the CS, although the nodes remained green. This sample did not become transparent after 4 weeks (Figure 2B). After 3 months of the treatment with the CS, only the young panicle became transparent. The hand-trimmed samples changed to white, except for the outer leaves and vascular bundles, after 1 week of treatment with the CS. Some parts of the leaves, young panicle, and internodes became translucent but did not become transparent after 4 weeks (Figure 2C). After 3 months, only the young panicle and inner leaves became transparent. The samples that were trimmed to 130 µm thickness with the vibrating micro-slicer became translucent after 1 week of treatment with the CS, with the leaves turning transparent. After 2 weeks, the sample was almost transparent and remained unchanged after 4 weeks (Figure 2D). After 3 months, the samples became completely transparent.
Next, the depth by which the clearing treatment allowed gene expression observations in the shoot of T65 at the 8 LS was validated. The UBQpro::NLS-sGFP-nClover3-mNeonGreen (UBQpro::NGCN, see Table of Materials) expressed shoots were used. Tissue samples were fixed in the fixative solution before trimming them to 130 µm thickness using a vibrating micro-slicer. The samples were then observed under a confocal laser microscope (laser: 488 nm/13%, objective: 20x, pinhole: 0.93 AU, averaging: 16, detector gain: 800) at 10 µm intervals in the z-axis direction every week from 0-4 weeks of treatment with the CS. The representative results are shown in Figure 3. All figures were adjusted with the same processing to compare the fluorescence intensity. In the node without the treatment, faint fluorescent signals were observed at a depth of −40 µm (Figure 3A). The observable depth was −60 µm after 1 week of the CS treatment and −90 µm after 2 weeks, but auto-fluorescence of the cytoplasm was noticeable at both weeks at a depth of −20 µm. The autofluorescence became somewhat weaker after 3 weeks and was no longer visible after 4 weeks. Fluorescent signals were observed at a depth of −80 µm at 3 weeks and at a depth of −100 µm at 4 weeks. In the internode, fluorescent signals were observed at a depth of −60 µm without the CS treatment (Figure 3B). The observable depth was −90 µm after 1 week to 3 weeks of the CS treatment and −100 µm after 4 weeks, but auto-fluorescence in the cytoplasm was noticeable at a depth of −20 µm after 1 week. Auto-fluorescence was no longer visible after 2 weeks. In the leaves, fluorescent signals were observed at a depth of −90 µm without the CS treatment (Figure 3C). The fluorescent signals were observed deeper in the leaves than in the nodes and internodes. However, the fluorescent signals were weaker. The fluorescent signals became clear after 1 week of CS treatment and were observed at a depth of −120 µm. After 2 weeks and 3 weeks, fluorescent signals were observed at a depth of −110 µm and, after 4 weeks, at a depth of −120 µm.
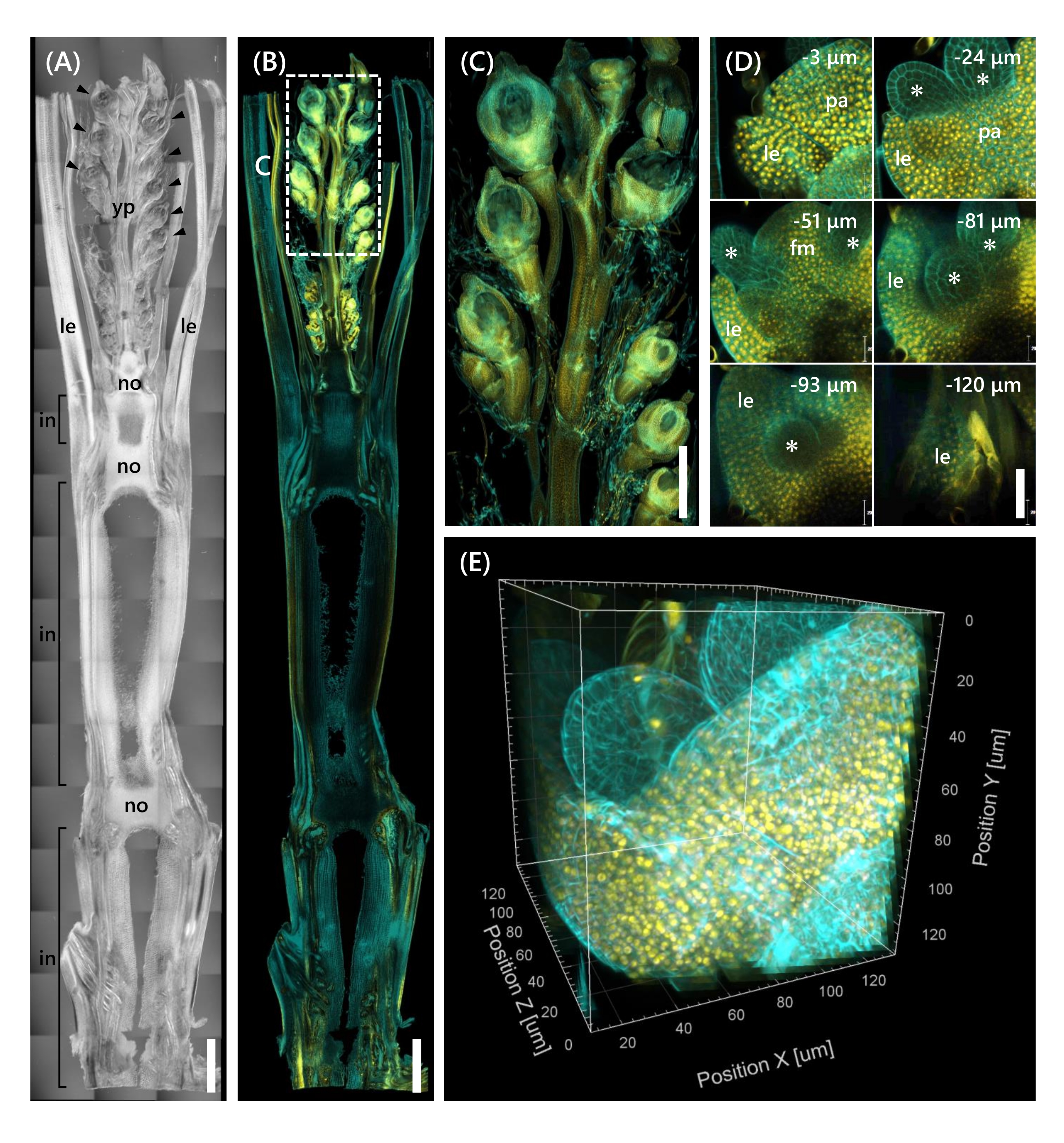
Finally, the expression of the transcription factor OsMADS1511 fused with mOrange12 was observed, which regulates the transition of SAM from the vegetative to the reproductive phase13. Tissue samples from Nip at the 10 LS with OsMADS15-mOrange were fixed, trimmed to 130 µm thickness, and treated with the CS for 2 weeks. Figure 4 shows that the clearing solution could be used simultaneously to observe the fluorescent proteins (OsMADS15-mOrange) of natural gene expression and fluorescent dye staining (calcofluor white). The whole floret at depths from 0 to −130 µm was observed, and the 3D image was reconstructed from 43 z-stack images at 3 µm intervals (Figure 4E).

Figure 1: Sampling position and processing. (A) Nip seedling at the 8 LS under the short-day condition. (B) Enlarged view of the base. (C) A sample cut-out from the SAM/young panicle to the base. (D) An oval cross-section from C (inset). (E) Thinly shaving off one side of the shoot with a blade. (F) Thinly shaved surface. (G) Sample showing internode elongation. The whitish section indicated by the arrowheads is nodes. The upper side of the upper node has the young panicle. Scale bars: (A) 10 cm, (B-C,E-G) 1 cm, (D) 0.5 cm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Tissue clearing depends on the sample thickness and CS treatment period. (A) Untrimmed samples with partial shoots including the young panicle. Maximum thickness: 4 mm. (B) All leaves peeled off from the samples to expose the young panicle. Maximum thickness: 2.5 mm. yp: young panicle, in: internode; arrowheads indicate node. (C) Samples that were hand-trimmed to 1 mm thickness. (D) Samples trimmed with the vibrating micro-slicer to 130 µm thickness. Scale bar: 5 mm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: The depth of fluorescence observation depends on the tissue type and CS treatment period. Deep imaging of UBQpro::NGCN in the (A) node, (B) internode, and (C) leaf. "Merge" means the images were fused with the differential interference contrast (DIC) image and all z-stack images. Images in which no fluorescence was observed were marked as "n.d." (not detected). Settings of the confocal laser microscopy are as follows: laser: 488 nm/13%, objective: 20x, pinhole: 0.93 AU, averaging: 16, detector gain: 800, interval: 10 µm. Scale bars: 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Deep fluorescence observation of OsMADS15-mOrange. (A) The DIC image of Nip at the 10 LS. yp: young panicle, le: leaf, no: node, in: internode; arrowheads indicate florets. (B) Fluorescence image of OsMADS15-mOrange. Yellow dots indicate the fluorescent proteins of OsMADS15-mOrange at the nucleus. The cell wall was stained cyan with the calcofluor white solution. (C) Enlarged view of the young panicle. (D) Deep imaging of the floret. pa: palea, le: lemma, fm: floral meristem; asterisks indicate stamen primordia. The confocal laser microscope was set as follows: OsMADS15-mOrange; laser: 555 nm/13%, objective: 20x, pinhole: 0.96 AU, averaging: 16, detector gain: 800, interval: 3 µm. Calcofluor white; laser: 405 nm/3%, objective: 20x, pinhole: 0.93 AU, averaging: 16, detector gain: 470, interval: 3 µm. (E) The 3D image of the figure (D) constructed from the z-stack images. Scale bars: (A-B) 1 mm, (C) 500 µm, (D) 40 µm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Critical steps of the protocol
The critical steps in this protocol are fixation and trimming. Rice shoots have hard, thick, or layered tissues that limit penetration of the fixative solution. To improve the permeability of the fixative solution, one side of the tissue was thinly shaved at sampling, as shown in Figure 1E-F. In addition, the vacuum treatments were repeated twice using higher pressure. Furthermore, the samples were fixed overnight at 4 °C instead of the usual 2 h fixation at 4 °C.
The key point in the trimming step is to determine the thickness of the tissues to be prepared to observe the fluorescent proteins while preserving their internal structure following a short period of CS treatment. As shown in Figure 2C, the 1 mm thick samples, hand-trimmed as thin as possible, became transparent in only a limited number of tissues even after 3 months of CS treatment. Therefore, the trimming step is essential for deep fluorescence observation of adult rice shoots. In this study, the samples were trimmed to a thickness of 130 µm, as shown in Figure 2D. The 130 µm thickness allowed for clearing the leaves after 1 week of CS treatment and the whole sample after 2 weeks. Adult rice shoots at the 9-10 LS were used in this study. Thick but softer tissues from younger rice shoots may be cleared faster with the CS treatment. The thickness of the samples and the duration of the CS treatment should be adjusted according to the tissue type, condition, and thickness of the 3D structure to be observed.
Modifications and troubleshooting methods
The CS precipitated easily at low temperatures. The precipitated CS cannot preserve the fluorescent proteins; hence, care must be taken when storing the samples at the proper temperature. In addition, both the CS and fixative solution have no antiseptic effect; therefore, fluorescent proteins will be degraded if contaminated. Soil-grown rice is prone to fungal growth; hence, the sampling and handling of samples must be performed with care to avoid contamination.
Excess fluorescent dyes in the buffer may give off background fluorescence and interfere with microscopic observations. For example, a calcofluor white solution containing Evans blue dye was previously used. After staining for 1 h and washing for 1 h, the fluorescent proteins of OsMADS15-mOrange were observed using a 555 nm laser. However, fluorescent proteins could not be observed because of the background fluorescence derived from Evans blue dye. This background fluorescence was almost eliminated by washing the samples for 2 h. Moreover, the fluorescent proteins were clearer if the samples were left overnight. Therefore, a pure calcofluor white solution was used in this study. Background fluorescence derived from the fluorescent dye should be checked using different laser wavelengths before observations.
Limitations of the method
As shown in Figure 3, deep fluorescent proteins were observed in the samples that were 130 µm thick after 2 weeks of CS treatment. This is consistent with the results shown in Figure 2D, where the 130 µm thick sample became transparent after 2 weeks of CS treatment. However, as shown in Figure 3A, auto-fluorescence of the cytoplasm was still noticeable in the nodes after 2 weeks and was only completely removed after 4 weeks of CS treatment. Nodes have a high cell density and, therefore, require a longer time to remove auto-fluorescent materials.
As shown in Figure 3C, deep fluorescent proteins were observed in the leaves without CS treatment, but the brightness was weaker than that in the nodes and internodes at the same depth of 20 µm. After 1 week of CS treatment, the fluorescent proteins were brighter. Chlorophyll is abundant in leaves and absorbs 488 nm of excitation light. They also have orange/red auto-fluorescence, which can interfere with the observation of fluorescent proteins using a 555 nm laser. After 1 week of CS treatment, chlorophyll and other auto-fluorescent materials were removed, resulting in high signal-to-noise ratio images.
The depths that could be observed in tissues after 2 weeks and 4 weeks of CS treatment were not significantly different, although the fluorescent proteins appeared weaker after 4 weeks (Figure 3). Normally, the brightness of fluorescent proteins and auto-fluorescence weakens with time, resulting in a higher signal-to-noise ratio. Therefore, fluorescent proteins can be observed more clearly by adjusting the microscopic conditions and image processing. Based on these results, it was concluded that 2 weeks of CS treatment could facilitate the observation of deep fluorescent proteins, given our sample conditions. However, 4 weeks are needed to observe clearer images that completely exclude the auto-fluorescent materials.
Structures with strong autofluorescence, such as vascular bundles and multi-arm cells, cannot be cleared in the CS. To observe these structures without autofluorescence, it is necessary to use a time-gating method12 or to obtain images by spectroscopy of the fluorescence spectrum. A two-photon microscope may be more suitable for observing deeper tissues if thicker tissues are observed.
Significance of the method with respect to existing and alternative methods
Generally, the internal structures of rice plants have been observed using either cryostat or vibratome sectioning. A cryostat is suitable for preparing thin sections, which allow easier observation, but the preparation of the samples and the operation of the equipment are time-consuming. Reconstructing the original 3D structure from thin sections is also difficult. The vibratome is relatively easy to operate and suitable for producing thick sections. However, thick sections of target tissues only allow observations of the cut surface and not deep tissues that light cannot reach. For these reasons, neither method is suitable for deep fluorescence observations.
This study addressed challenges in deep fluorescence observation in rice shoots, such as the limited tissue penetration of the CS and the poor object resolution under a confocal microscope, by combining existing methods. As shown in Figure 4, we observed fluorescent proteins (OsMADS15-mOrange) expressed in the deep tissues of adult rice shoots from the young panicle to the base. Figure 4D focuses on the floret and shows deep fluorescent proteins at 3 µm intervals. Tissues over −130 µm depth were observed after 2 weeks of CS treatment, but only the tissues within −27 µm depth were observed (data not shown) in the floret at the same size and growth stage without CS treatment. The current improved protocol allowed observation not only of overexpression of genes but also natural gene expression in the deep tissues of adult rice shoots.
Importance and potential applications of the method in specific research areas
This protocol, which optimizes the deep fluorescence observation of adult rice shoots, enables the efficient clearing of hard, thick, or layered tissues by trimming off unnecessary tissues and increasing the permeability of the CS. In addition, the thickness of the samples for analysis was optimized to allow continuous and structural deep fluorescence observation using a confocal laser microscope, which normally cannot resolve thick or opaque tissues.
It is difficult to compare rice samples at different growth stages because the fluorescent proteins degrade over time in the fixative and PBS solutions. However, the fluorescent proteins in the CS can be stored for more than 5 months1. The long shelf life of the CS is a major advantage for deep fluorescence observation in rice.
Recently, many clearing technologies have been developed, making it possible to observe deep tissues in 3D while preserving their internal structures. These technologies have continued to evolve, and new clearing solutions have been developed. A good example is iTOMEI14, which enables efficient chlorophyll removal and brighter fluorescence detection. Another example is ClearSeeAlpha15, which prevents the browning of tissues during clearing treatment and makes them appear transparent. Combining these clearing solutions with the present method may allow more efficient and effective clearing.
It is expected that the current method will help gain new insights through deep imaging of not only rice but also other plants.
Disclosures
The authors have no conflicts of interest to declare.
Acknowledgements
We thank Dr. R. Terada, Dr. Z. Shimatani, and Dr. H. Tsuji for providing us with the OsMADS15-mOrange seeds; Dr. D. Kurihara for providing us with the NGCN construction; and Dr. R. Shim for editing our manuscript. This work was funded by JSPS KAKENHI (grant numbers JP20H05912, 20H05778, 20H05779) and by the SATREPS program (no. JPMJSA1706) of the JST and JICA.
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mL microcentrifuge tube | BIO-BIK | ST-0150F | |
| 12-multiwell plate | Corning | 353043 | |
| 50 mL conical tube | Corning | 352070 | |
| Calcofluor white solution | Sigma-Aldrich | 910090 | |
| ClearSee | FUJIFILM Wako Pure Chemical | 031-25151 | This can be made or purchased. |
| Confocal laser microscope | Carl Zeiss | LSM700 | |
| Desiccator | SANPLATEC | Custom made of acrylic. 30 cm (L), 30 cm (W), 14.5 cm (H) | |
| Glass coverslip (18 × 18 No.1) | MATSUNAMI | C018181 | |
| HEPES | FUJIFILM Wako Pure Chemical | 342-01375 | |
| Microscope slide (76 × 26) | MATSUNAMI | S2441 | |
| Paraffin film | Bemis | PM-996 | |
| Paraformaldehyde | FUJIFILM Wako Pure Chemical | 162-16065 | |
| Sodium deoxycholate | Tokyo Chemical Industry | C0316 | |
| Sucrose | FUJIFILM Wako Pure Chemical | 190-00013 | |
| UBQpro::NLS-sGFP-nClover3-mNeonGreen (UBQpro::NGCN) | provided by Dr. Kurihara | ||
| Urea | FUJIFILM Wako Pure Chemical | 211-01213 | |
| Vacuum pump | AS One | AS-01 | |
| Vibrating micro-slicer | DOSAKA | DTK-3000 | |
| Xylitol | FUJIFILM Wako Pure Chemical | 248-00545 |
References
- Kurihara, D., Mizuta, Y., Sato, Y., Higashiyama, T. ClearSee: A rapid optical clearing reagent for whole-plant fluorescence imaging. Development. 142 (23), 4168-4179 (2015).
- Hasegawa, J., et al. Three-dimensional imaging of plant organs using a simple and rapid transparency technique. Plant and Cell Physiology. 57 (3), 462-472 (2016).
- Musielak, T. J., Slane, D., Liebig, C., Bayer, M. A versatile optical clearing protocol for deep tissue imaging of fluorescent proteins in Arabidopsis thaliana. PLoS One. 11 (8), 0161107 (2016).
- Pandey, K. B., et al. Plant roots sense soil compaction through restricted ethylene diffusion. Science. 371 (6526), 276-280 (2021).
- Ejaza, M., Bencivenga, S., Tavaresa, R., Busha, M., Sablowski, R. ARABIDOPSIS THALIANA HOMEOBOX GENE 1 controls plant architecture by locally restricting environmental responses. Proceedings of the National Academy of Sciences of the United States of America. 118 (17), (2021).
- Smetana, O., et al. High levels of auxin signalling define the stem-cell organizer of the vascular cambium. Nature. 565 (7740), 485-489 (2019).
- Chu, T. T. H., et al. Sub-cellular markers highlight intracellular dynamics of membrane proteins in response to abiotic treatments in rice. Rice. 11 (1), 23 (2018).
- Nagaki, K., Yamaji, N., Murata, M. ePro-ClearSee: A simple immunohistochemical method that does not require sectioning of plant samples. Scientific Reports. 7, 42203 (2017).
- Sato, M., Akashi, H., Sakamoto, Y., Matsunaga, S., Tsuji, H. Whole-tissue three-dimensional imaging of rice at single-cell resolution. International Journal of Molecular Sciences. 23 (1), 40 (2022).
- Matsuo, T., Hoshikawa, K. Science of the Rice Plant: Morphology. Food and Agriculture Policy Research Center. , (1993).
- Kobayashi, K., et al. Inflorescence meristem identity in rice is specified by overlapping functions of three AP1/FUL-Like MADS box genes and PAP2, a SEPALLATA MADS box gene. Plant Cell. 24 (5), 1848-1859 (2012).
- Tamaki, S., et al. FT-like proteins induce transposon silencing in the shoot apex during floral induction in rice. Proceedings of the National Academy of Sciences of the United States of America. 112 (8), 901-910 (2015).
- Kodama, Y. Time gating of chloroplast autofluorescence allows clearer fluorescence imaging In Planta. PLoS One. 11 (3), 0152484 (2016).
- Sakamoto, Y., et al. Improved clearing method contributes to deep imaging of plant organs. Communications Biology. 5 (1), 12 (2022).
- Kurihara, D., Mizuta, Y., Nagahara, S., Higashiyama, T. ClearSeeAlpha: Advanced optical clearing for whole-plant imaging. Plant and Cell Physiology. 62 (8), 1302-1310 (2021).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved