Method Article
Cultivating Ex Vivo Patient-Derived Glioma Organoids Using a Tissue Chopper
In This Article
Summary
This study introduces an automated method to generate patient-derived glioblastoma 3-dimensional organoids utilizing a tissue chopper. The method provides a suitable and effective approach to obtain such organoids for therapeutical testing.
Abstract
Glioblastoma, IDH-wild type, CNS WHO grade 4 (GBM) is a primary brain tumor associated with poor patient survival despite aggressive treatment. Developing realistic ex vivo models remain challenging. Patient-derived 3-dimensional organoid (PDO) models offer innovative platforms that capture the phenotypic and molecular heterogeneity of GBM, while preserving key characteristics of the original tumors. However, manual dissection for PDO generation is time-consuming, expensive and can result in a number of irregular and unevenly sized PDOs. This study presents an innovative method for PDO production using an automated tissue chopper. Tumor samples from four GBM and one astrocytoma, IDH-mutant, CNS WHO grade 2 patients were processed manually as well as using the tissue chopper. In the manual approach, the tumor material was dissected using scalpels under microscopic control, while the tissue chopper was employed at three different angles. Following culture on an orbital shaker at 37 °C, morphological changes were evaluated using bright field microscopy, while proliferation (Ki67) and apoptosis (CC3) were assessed by immunofluorescence after 6 weeks. The tissue chopper method reduced almost 70% of the manufacturing time and resulted in a significantly higher PDOs mean count compared to the manually processed tissue from the second week onwards (week 2: 801 vs. 601, P = 0.018; week 3: 1105 vs. 771, P = 0.032; and week 4:1195 vs. 784, P < 0.01). Quality assessment revealed similar rates of tumor-cell apoptosis and proliferation for both manufacturing methods. Therefore, the automated tissue chopper method offers a more efficient approach in terms of time and PDO yield. This method holds promise for drug- or immunotherapy-screening of GBM patients.
Introduction
Low-grade gliomas (LGGs) are a group of relatively rare brain tumors that typically present as slow-growing and less aggressive compared to high-grade gliomas like glioblastoma. They can occur in both adults and children, with a slightly higher prevalence in adults. The exact prevalence varies by region and population, but LGGs account for approximately 15%-20% of all primary brain tumors1. Treatment strategies for LGGs often involve a combination of surgery, radiation therapy, and chemotherapy, aiming to maximize tumor resection while preserving neurological function. The management of LGGs can be complex, and the choice of therapy may depend on factors such as tumor location and molecular characteristics2. Advances in understanding the genetic and molecular underpinnings of LGGs have led to more targeted therapies, and ongoing research continues to refine treatment approaches.
Glioblastoma, IDH-wild type, CNS WHO grade 4 (GBM), on the other hand, is the most prevalent primary brain tumor found in adults, with an incidence rate between 3.19-4.17 cases per 100,000 person-years3. GBM causes symptoms such as headaches, seizures, focal neurological deficits, changes in personality, and increased intracranial pressure. The standard treatment for GBM involves debulking of the tumor, if feasible, followed by radiation therapy combined with Temozolomide4. Furthermore, combining Temozolomide and Lomustine may enhance the median overall survival rate in patients with O6-methylguanine-methyltransferase (MGMT)-promoter methylation5. However, despite these recent therapeutic approaches, GBM remains an incurable disease with poor prognosis, characterized by a patients' median overall survival rate of 16 months up to 20.9 months when Tumor Treating Fields (TTFields) is added3,6. Several immunotherapeutic approaches have been investigated in GBM but demonstrated limited efficacy in vivo. Moreover, clinical and preclinical limitations hinder therapeutic breakthroughs7. The establishment of a suitable and realistic ex vivo model has been challenging due to the inter-8 and intratumoral9 heterogeneity of GBM.
Conventional 2-dimensional (2D) patient cell lines represent homogeneous cell populations and are suitable for high-throughput drug screening. However, patient-derived and immortalized cell lines fail to mimic GBM adequately due to differences in growth conditions and deviations in genotypic and phenotypic features after multiple passages10,11,12.
On the other hand, 3D organoid models have recently emerged as promising systems that replicate the phenotypic and molecular heterogeneity of organ and various cancer types13,14,15,16,17,18. In the context of GBM, cerebral organoids have been genetically modified to simulate tumor-like characteristics16,17 or co-cultured with GSCs or spheroids to induce tumor cell infiltration18,19. While patient-derived GBM organoids cultured with Matrigel and EGF/bFGF exhibit GBM hallmarks such as stem cell heterogeneity and hypoxia20, it remains uncertain to what extent this model can represent the key molecular properties of patients' neoplasms.
Patient-derived GBM organoids (PDOs) are promising models that can maintain the predominant features of their analogous parental tumors, including histological characteristics, cellular diversity, gene expression, and mutational profiles. Additionally, they are rapidly infiltrated upon implantation into adult rodent brains, providing a realistic model for drug testing and personalized therapy21. However, manually dissecting tumor tissue to generate PDOs is time-consuming and costly. Therefore, an urgent need exists for a rapid method that can produce large numbers of PDOs, enabling comprehensive assessment of different therapeutic approaches holding promise for individualized drug testing. This study describes a new method for manufacturing PDOs directly from freshly dissected tumor tissue using an automatic tissue chopper. Furthermore, PDOs generated by this method were compared with manually dissected PDOs from the same patients in terms of PDO count, morphological features, apoptosis and proliferation of tumor cells.
Protocol
All patients were treated at the Department of Neurosurgery, University Hospital Würzburg, Germany, after giving written informed consent in accordance with the declaration of Helsinki and as approved by the Institutional Review Board of the University of Würzburg (#22/20-me). Tumor tissue material from four GBM patients and one astrocytoma, IDH-mutant, CNS WHO grade 2 patients (low grade glioma, LGG) (Table 1) was obtained from surgery and processed using the following protocol. The automated process of generating PDOs utilizing a tissue chopper is referred to as chopper (C) and the process of manually cutting the tissue with two scalpels under microscopic control as manual (M). Six equally sized sections (1-2 cm3) were dissected from the tumor sample, then each one was cut in half and processed homogenously using the two methods. Due to the aforementioned intratumoral heterogeneity, one 6-well plate for each approach was generated from each patient with each well representing PDOs from a different site within the original tumor. Both procedures took place under a laminar airflow cabinet and all used instruments were sterilized prior to use. The overview of the approach is illustrated in Figure 1.
1. Preparing agarose blocks (for the C-approach only, optional)
- Fill 50 mL of phosphate buffered saline (PBS) into a beaker, add one tablet of agarose (see Table of Materials), and mix well until suspended.
- Heat the mixture in the microwave for 30-40 s, while avoiding boiling. Then cool the mixture down until it reaches 47 °C.
- Pour the agarose mixture into a sealed cylinder-shaped casting mold and avoid any bubble-formation. Immediately cool the cast using a frozen (-20 °C) clamp or by placing it on dry ice for 30 min.
2. Processing the tumor material
- Prepare a box of ice to keep the tumor material cooled on the way from the operating room to the laboratory.
- Transfer the tumor tissue (4-5 cm³) into a sterile 50 mL tube containing 25 mL of Hibernate A (see Table of Materials) covering the tumor and place the tube into the ice box.
- Under a laminar airflow cabinet, transfer the tumor material together with the Hibernate A into a sterilized glass Petri dish.
- Eliminate the necrotic tissue and dissect the blood vessels carefully using a scalpel and tissue forceps under microscopic control. Identify necrotic tissue by hemorrhagic areas exhibiting a brownish hue resulting from bleeding, or tissue exhibiting a paler or whiter appearance relative to the adjacent viable tissue. Pay attention not to squeeze or disrupt the tissue.
- Cut the tumor material into six pieces with an approximate size of 1-2 cm3. Distribute the pieces to plastic Petri dishes (n = 6) pre-filled with 3 mL of H-GPSA medium (Table 2), each22. Place the Petri dishes on ice.
3. Setting up the tissue chopper
- Position the blade as described in the manufacturers' manual23.
- Adjust the slice thickness to 0.45-0.50 mm. Set the blade force to medium. Fix the table release knob to "start" mode.
4. Processing the tumor tissue pieces
- Processing tumor tissue with the chopper (C method)
- Cut the agarose blocks into cylinders of 2 cm length and glue one of these cylinders onto the chopper circular plastic dish using the histoacryl glue (see Table of Materials).
- Create a deepening in the agarose cylinder using a scalpel, and then fit the tumor tissue from the first well (step 2.5) within this pit.
NOTE: The tumor material should be handled carefully and not be squeezed or pushed into the gap. The gap should be big enough to fit the tumor easily, but small enough to keep the tumor material stable during the cutting process. Steps 4.1.1. and 4.1.2. are optional. - Position the plastic disc onto the mounting disc of the cutting table (see Table of Materials).
- Switch the chopper on and press the reset button. The chopper now starts cutting (first round). The machine stops automatically after the table reaches the end and both agarose and tumor tissue are cut into the desired diameter.
- Rotate the mounting disc by 90°, and then adjust the releasing table knob to the starting mode.
- Press the reset button and let the machine cut the tissue again creating rectangular shaped tissue (second round).
- Remove the plastic disc with the processed material and carefully rotate only the tumor tissue by 90° using tissue spatula.
- Place the plastic disc onto the cutting table, then adjust the releasing table knob to the starting mode and press the reset button for a final cutting round (third round).
- Switch the chopper off and remove the plastic disc. Clean the chopper and the blades.
- Using a 5 mL single channel pipette, aspirate the processed material along with the medium into the pipette and flush the suspension back into the dish.
- Repeat the previous step 2-3 times to separate the tissue properly.
- Place the Petri dish back onto ice and repeat steps (4.1.1-4.1.12.) with the other 5 dishes of each tumor.
- Processing tumor tissue manually (M method)
- Transfer the tumor tissue from the first plastic Petri dish (step 2.5) together with 3 mL of H-GPSA medium (Table 2) into a glass Petri dish. Dissect the segment manually under the microscope into sections of 0.5 mm using two scalpels.
- Transfer the dissected tissue back to its plastic Petri dish using a 2 mL pipette.
- Repeat steps (4.2.1.-4.2.3.) for the tumor sections in the other five Petri dishes (step 2.5).
5. Washing the tumor tissue
- Tilt each Petri dish upwards to 45° and wait for 30 s until the tumor chunks sink to the bottom of the dish.
- Aspirate 2.5 mL of the H-GPSA medium (Table 2) carefully using a 1 mL pipette and be aware not to take up any tumor tissue.
- Add 2 mL of RBCs lysis buffer (see Table of Materials) to each sample. The processed tumor pieces must be covered completely by lysis buffer.
- Place the 6 dishes on a laboratory orbital shaking machine on slow speed for 10 min.
- Aspirate 2 mL of the lysis buffer carefully to not take up any tumor tissue.
- Repeat the previous washing steps (step 5.1-5.5) twice using 2 mL of H-GPSA medium (Table 2) instead of lysis buffer each time.
6. Culturing the tumor tissue
- Aspirate the H-GPSA medium (Table 2) from each dish and replace it with 4 mL of PDO medium (Table 2).
- Transfer the tissue chunks of each dish to one respective well of an ultra-low attachment 6-well plate (see Table of Materials).
- Place the plate onto an orbital shaker inside an incubator and incubate at 37 °C, 5% CO2 and 150 rpm for 2-4 weeks.
- Perform a half medium change every two days by aspirating 2 mL of medium from each well and replacing it with 2 mL of fresh PDO medium (Table 2) pre-warmed to 37 °C.
- Observe the tissue under the microscope (morphology, growth, medium color) and cut growing PDOs (>0.7 mm) or adhesive tissue to prevent tissue hypoxia.
- To do so, transfer the PDOs from the ultra-low attachment well to a sterilized glass Petri dish and use a scalpel for cutting. Alternatively, adhesive PDOs can be resolved by aspirating them with a 1 mL pipette. Be careful not to squeeze the PDOs and handle them gently.
- Evaluate PDO formation by counting the PDO every two days and thoroughly check for the desired round morphology (Figure 2).
7. Fixing and embedding the PDOs
- Fix two PDOs from each well of each patient with 4% formalin for 24 h after 6 weeks of culture.
- Immerse the fixed PDOs in neutrally buffered (sodium phosphate) formalin until embedding.
- Place each PDO into a cassette (see Table of Materials) for further processing.
- Initiate a dehydration process by immersing the cassette in the following solutions as mentioned in the NOTE below:
NOTE: 50% ethanol for 20 min, 70% ethanol for 20 min, 80% ethanol for 20 min, 96% ethanol for 20 min, 100% ethanol for 20 min, 100% ethanol for 30 min, 100% ethanol + chloroform (1:1 ratio) for 30 min, 100% ethanol + chloroform (1:1 ratio) for 30 minutes, Absolute chloroform for 30 min, Absolute chloroform for 30 min, Paraffin for 30 min, Paraffin for 30 min using the STP 120. - Embed the dehydrated PDOs in paraffin wax at 58-60 °C.
- Slice the embedded PDOs at 2.5 µm thickness and mount them on slides for staining.
8. Immunofluorescence staining
- Mount the PDOs after 6 weeks of culture.
- Subsequently, perform double staining against glial fibrillary acidic protein (GFAP, dilution: 1:100) and the proliferation marker Ki67 (dilution: 1:1000) (see Table of Materials), as previously reported22.
- Similarly, evaluate apoptosis by double staining PDOs against GFAP and anti-caspase-3 (CC3, dilution: 1:400) (see Table of Materials).
- Capture images of the PDOs using a fluorescence microscope at 40x magnification.
- Analyze the images for GFAP-, Ki67-, and CC3-positive cells, as well as GFAP/Ki67 and GFAP/CC3 double-positive cells.
- Utilize the open-source program Fiji (ImageJ-win 32) for image analysis.
9. Evaluation and data analysis
- Capture daily microscopic bright field pictures during the first week of culture at standard settings and 5x magnification.
- Observe the morphological changes and track the maturing process in both manual and automated processing approaches.
- Conduct morphological analysis using the microscope in standard brightfield mode settings.
- Evaluate the PDO number and morphology during the first 4 weeks of culture in PDOs from all five patients.
- Obtain three readings of each PDO count from each patient to calculate the mean and standard error of the mean (SEM).
- Investigate proliferation and apoptosis in PDOs from three patients.
- Analyze the data using a commercially available statistical software package (see Table of Materials).
- Apply Students-T and Mann-Whitney U Tests to determine differences between the manual and automated PDO generation in terms of PDO count, proliferation, and apoptosis.
Results
Four patients with GBM and one with LGG were included after pathological confirmation by an experienced neuropathologist (CMM). The majority of patients had an unmethylated MGMT promoter, and all GBM patients were IDH1 and IDH2 wild type (Table 1). On average, the manufacturing process lasted 88.8 min (+/- 6.3 min) in the C approach and 322 min (+/- 17.2 min) in the M approach. The overall success rate was 87% in the manual and 93% in the chopper approach after 4 weeks of culture (n = 5). Moreover, PDOs derived from the C group reached the desired rounded shape within 1 week and were mature enough to be used in in vitro experiments, while the PDOs of the M group mostly remained sharply edged and undefined (Figure 2). The tumor tissue processed with the C approach resulted in overall 281 PDOs (Mean per patient = 56 +/- 43) after the first week of culture, while 250 PDOs (Mean per patient= 50 +/- 41) developed with the M approach. During the second week of culture, the tissue of all five patients yielded higher PDO numbers when they were generated with the C approach (801; Mean per patient= 130 +/- 38) compared to the M approach (601; Mean per patient= 76 +/- 44; P = 0.018). During the third week of culture, the C approach accumulated overall 1105 PDOs from all patients (Mean per patient = 221 +/- 32) compared to 771 PDOs (Mean per patient= 155 +/- 34) in the M approach (P = 0.032). Furthermore, a total of 1195 PDOs (Mean per patient= 239 +/- 50) formed after four weeks of culture when generated with the C approach compared to 784 (Mean per patient= 157 +/- 36) utilizing the M approach (P < 0.01). Therefore, the C method showed a significantly higher PDOs count starting from the second week (Figure 3). Furthermore, the relative fluctuations in PDO counts were evaluated to explore the dynamic trends between successive weeks. The analysis unveiled an impressive surge in PDO counts during the initial transition from the first to the second week in the C approach (265%), which was indicative of rapid progress. Subsequently, there was a lower rise in counts during the third week (75%), reflecting a temporary adjustment. In contrast, the M approach demonstrated a consistent and steady increase in PDO counts (92% in second week, respectively 67% in the third week), which contributed to a remarkable stability in counts during the fourth week. This consistent upward trend in PDO counts underscores the reliability and resilience of the C approach throughout the observation period.
Two GBM patients and one LGG patient were included for the analysis of astrocyte-numbers (GFAP) within PDOs, PDO cell proliferation (Ki67), and apoptosis (CC3). The determined astrocyte count revealed no significant differences between the two processing methods with an average of 43% in the C approach and 45% in the M approach (Figure 4 and Figure 5, Supplementary Figure 1 and Supplementary Figure 2). Similarly, the proliferation rates within the PDOs were comparable between the C (3%) and M approaches (1%). Only PDOs generated with the C approach from patient 5 displayed a proliferation rate of 26% compared to 1% with the M approach (P = 0.001; Figure 4C). Overall low apoptosis rates were detected in PDOs processed with the C approach (3%) compared to 2% from the M approach for all patients, which were not significantly different (Figure 5C). Furthermore, there was no significant difference between the two methods regarding the number of astrocytes undergoing apoptosis (Figure 5D).

Figure 1: Graphical overview of the patient-derived organoid (PDO) manufacturing process using an automated chopper versus a manual approach. The illustration depicts the various steps involved, including (A) sample collection, (B) tumor material dissection, (C) washing, and (D) incubation. Please click here to view a larger version of this figure.
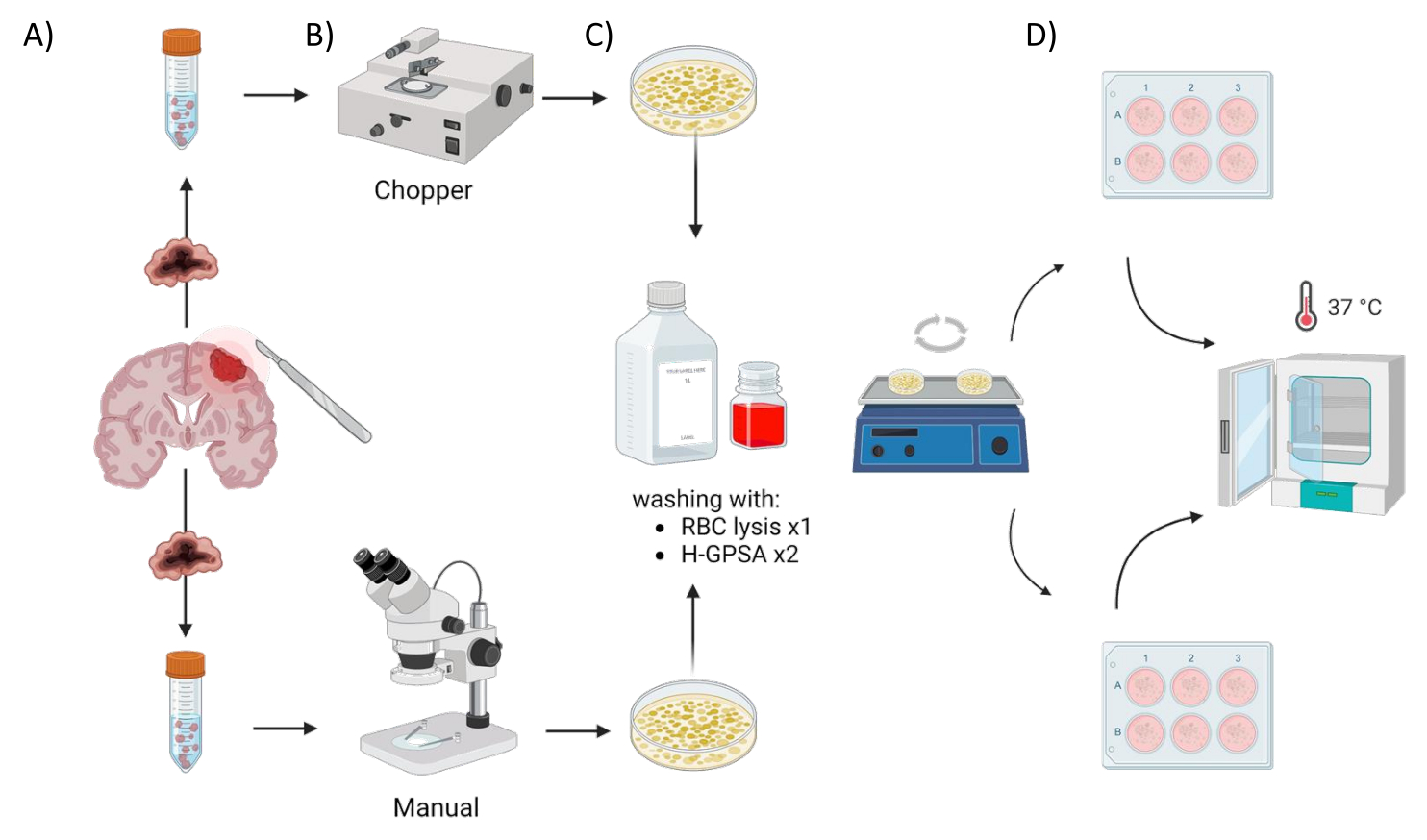{kind=link}

Figure 2: PDO morphology during the first week of culture. Comparison of PDOs formation post-dissection using both the automated chopper and manual method. Scale bars = 1000 µm. Please click here to view a larger version of this figure.
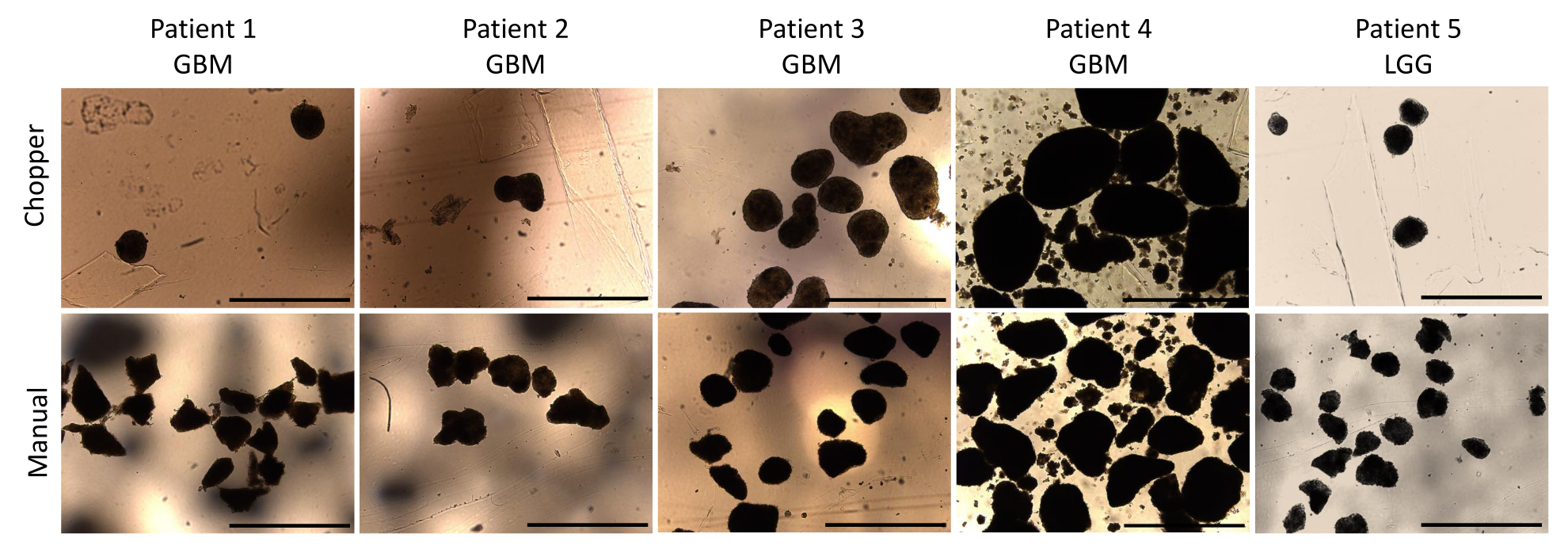{kind=link}

Figure 3: PDO count during the first four weeks of culture. The x-axis displays the time in weeks (W), and the y-axis the number of PDOs of the C (blue) and M (red) approach (n = 5). Each data point represents the mean count, with error bars indicating the standard error of the mean. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Cell proliferation rates within PDOs. (A) Representative immunofluorescence images (n = 3) of GFAP-positive cells (green), Ki67-positive cells (red), and DAPI (blue) in PDOs from patients 3, 4 and 5 (Table 1). All PDOs were processed using the chopper (C) and manual (M) methods. Scale bars = 100 µm. (B) Comparison of the two methods regarding the relative number of GFAP-positive cells, (C) Ki67-positive cells, and (D) Ki67/GFAP-double positive cells. Not significant results are indicated by "ns". Please click here to view a larger version of this figure.
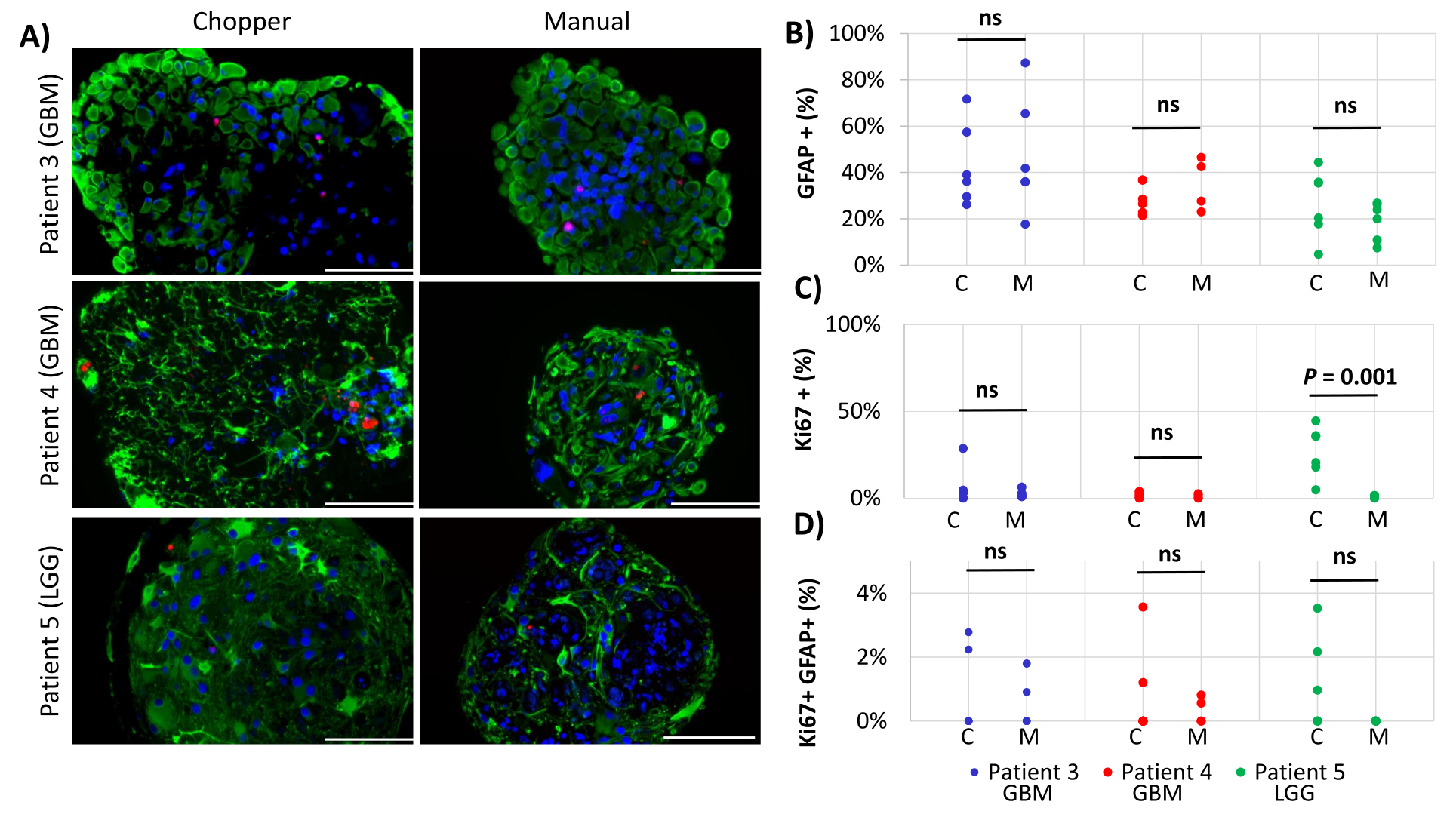{kind=link}

Figure 5: Apoptosis rates within PDOs. (A) Representative immunofluorescence images (n = 3) of GFAP-positive cells (green), CC3-positive cells (red), and DAPI (blue) in PDOs from patients 3, 4 and 5 (Table 1). All PDOs were processed using the chopper (C) and manual (M) methods. Scale bars = 100 µm. (B) Comparison of the two methods regarding the relative number of GFAP-positive cells, (C) CC3-positive cells, and (D) CC3/GFAP-double positive cells. Not significant results are indicated by "ns". Please click here to view a larger version of this figure.
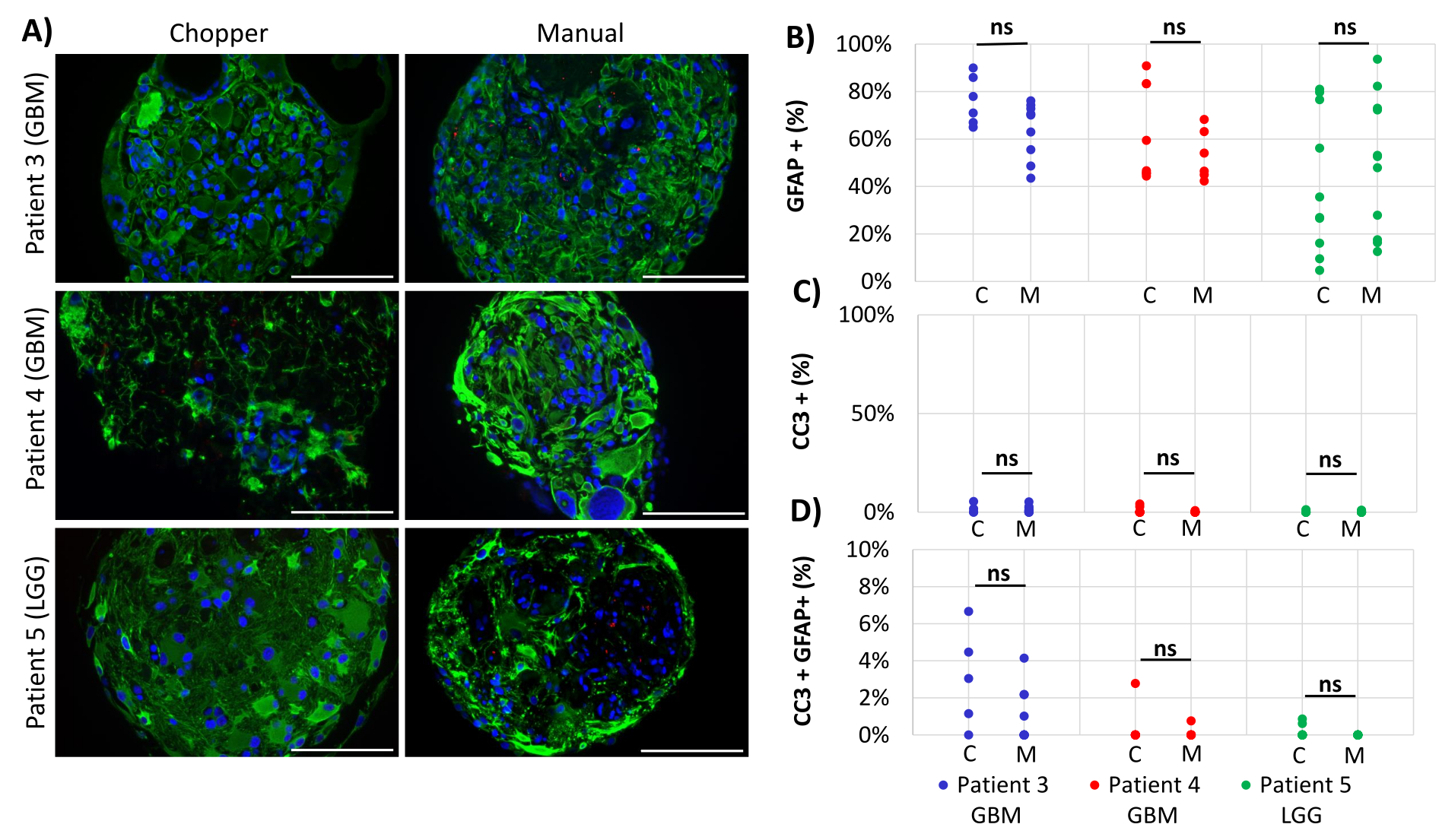{kind=link}
Table 1: Patients' characteristics and clinical parameters. GBM = glioblastoma, IDH-wild type, CNS WHO grade 4; LGG = low grade glioma; KPS = Karnofsky performance score; MGMT = O6-methylguanine-DNA methyltransferase; IDH1 = isocitrate dehydrogenase 1, IDH2 = isocitrate dehydrogenase 2, ATRX = α-thalassemia/mental retardation, X-linked gene; M = morphology; CC = cell count; p = proliferation; A = apoptosis. Please click here to download this Table.
Table 2: Medium compositions. H-GPSA = Hibernate A-Glutamax pencillin streptomycin amphotericin B. PDO = Patient-derived organoids DMEM: Dulbecco’s Modified Eagle’s Medium. NEAA: non-essential amino acids. Pen Strep: Penicillin/ Streptomycin Please click here to download this Table.
Table 3: Overview of the used techniques to generate cell culture models. Please click here to download this Table.
Supplementary Figure 1: Individual channels of proliferation staining of PDOs. (A) DAPI (blue), (B) GFAP-positive cells (green), (C) Ki67-positive cells (red) and (D) overlay channel in PDOs from patients 3, 4 and 5. Scale bars = 100 µm. Please click here to download this File.
Supplementary Figure 2: Individual channels of apoptosis staining of PDOs. (A) DAPI (blue), (B) GFAP-positive cells (green), (C) CC3-positive cells (red) and (D) overlay channel in PDOs from patients 3, 4 and 5. Scale bars = 100 µm. Please click here to download this File.
Discussion
This study presents a quick and efficient method for generating PDOs. GBM remains a challenging tumor to treat, often characterized by relapse and a high disease burden3,6. Innovative therapeutic approaches are urgently needed, as promising results observed in vitro often fail to demonstrate efficacy in vivo during phase-I trials. One of the reasons for this discrepancy could be the limited ability of patient-derived immortalized cell lines, grown in monolayer cultures, to reflect the complex cell-cell interactions and genetic properties of the parental tumor. Given the high inter- and intratumoral heterogeneity of GBM8,9, personalized targeted therapies are preferred and may hold promise for future applications. In contrast to 2D adherent cell lines, organoids have the ability to retain the properties of the parental tissue21, yet complex cell-cell interactions between the tumor and the normal brain are of paramount importance and could potentially be overlooked by this model. However, manual generation of PDOs is a time-consuming process, and tissue damage caused by squeezing with scalpels during cutting can hinder successful PDO growth. Therefore, an automated method was optimized using a tissue chopper to generate higher numbers of PDOs with reduced time and effort. Additionally, we demonstrated that overall proliferation and apoptosis rates did not differ between the two approaches.
The C approach is straightforward, easy to implement, and enables the generation of a larger number of PDOs (Figure 3). The rotation of the tissue between the second and third rounds of chopping was identified as a critical step in the protocol. At this stage, the tissue has already lost its integrity and can easily fall apart, resulting in larger pieces that require additional cutting or manual dissection under the microscope. While the automated chopper approach allows for a preset cutting size with greater accuracy, the manual approach lacks precision in determining the size of PDOs, leading to unevenly shaped and sized PDOs, which is a disadvantage for comparative drug screening (Figure 2). Nonetheless, with the proposed method, the standardization of cell numbers per PDO is not achieved, potentially posing a drawback for standardized drug screening protocols. The advantages and disadvantages of different organoid generation techniques18,19,20,24,25,26,27,28,29,30,31,32,33,34,35,36,
37,38,39,40,41,42 and their applications are summarized in Table 3.
GBM tissue can vary in consistency, ranging from tough (infiltration zone) to soft (necrotic core), which can pose challenges for the automated chopper approach. If the tissue is too tough, the chopper may squeeze and damage it, whereas too soft tissue might be squashed. The chosen tissue displayed distinctive attributes, including an intermediate level of firmness, sporadically featuring a pinkish-grayish coloration rather than manifesting brown or yellow discoloration. Tissue possessing a spongy and readily crumbly texture demonstrated superior preservation within the agarose blocks, whereas exceedingly delicate and liquified tumor tissue was omitted from the sampling procedure. However, the chopper approach enabled a successful generation of a higher number of PDOs compared to the manual approach, even with tissue of suboptimal consistency. The key solution is to maintain close interaction with the surgeon performing the tumor resection to process tissue from different areas of the tumor. In cases of suboptimal tissue consistency, manually reworking the tissue under the microscope was a helpful addition after chopping. To account for heterogeneity, the tumor tissue was initially divided into six segments, each subsequently halved for either the C or M approach. Within these six distinct sections, a substantial degree of heterogeneity is anticipated. Furthermore, even within the PDOs from the same section or well, the presence of distinct subpopulations is plausible.
As proof of concept, the proliferation and apoptosis data were reported from two patients with GBM and one patient with LGG, which show no significant differences between the two methods. The generation of PDOs is not limited to highly malignant brain tumors but can also be applied to LGGs. This study highlights that LGG seldom exhibit growth in 2D culture, making the development of an accurate model for their study highly valuable. This protocol aims to demonstrate the versatility of this approach in generating PDOs from GBM as well as LGG quickly and effectively.
Overall, PDOs could be utilized in the future for patient-oriented pre-therapeutic testing of targeted therapies in malignant brain tumors. Providing a quick and efficient method for individualized drug screening is crucial, as tumor progression occurs rapidly, and salvage treatment options are desperately needed. As a next step, the PDO model could be evaluated with various immunotherapeutic approaches to better mimic real treatment responses. In the future, PDOs could be utilized to draw sophisticated conclusions regarding the need for further exploration and evaluation of therapies in a clinical setting.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This research was funded by the Interdisciplinary Center of Clinical Research (IZKF, B-450) Würzburg, Bavarian Center of Cancer Research (BZKF) and the publication supported by Open Access Publishing Fund of the University of Würzburg. We would like to thank Dagmar Hemmerich and Siglinde Kühnel, both Section Experimental Neurosurgery, Department of Neurosurgery, University Hospital Würzburg, for technical support. Figure 1 was created using www.biorender.com.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (1000x) | Gibco | 21985023 | |
| 30% formaldehyde methanol-free | Carl Roth | 4235.1 | Used in 4% concentration |
| 70% ethanol solution | For sterilisation | ||
| Agarose tablets 0.5 g | Carl Roth | HP67.7 | |
| Amphotericin B 250 µg/mL | Gibco | 15290018 | |
| Anatomical forceps | Hartstein | N/A | |
| Anatomical spatula | Hartstein | N/A | |
| B-27 Supplement without vitamin A (50x) | Gibco | 12587010 | |
| Biopsy cassette with cover | Resolab | 37001-b | |
| Blades for McIlwain Tissue Chopper | Campden instruments | Model TC752-1 | |
| CC3 antibody (Asp 175) | Cells signaling technology | 9661 | |
| Disposable scalpel | Feather | 0200130015 | |
| Distilled water | Gibco | 15230089 | To dilute the formaldehyd |
| Dulbecco's Modified Eagle Serum Nutrient Mixture (DMEM) F-12 (1:1) (1x) | Gibco | 11330032 | Includes L-Glutamine and 15 mM HEPES |
| Dulbecco's Phosphate Buffered Saline (PBS) | Sigma Life Sciences | D8537-500ML | Modified, without calcium, chloride and magnesium chloride, liquid, sterile-filtered, suitable for cell culture |
| eBioscience 1x RBC Lysis Buffer | Invitrogen | 433357 | |
| Falcon tube 50 ml Cellstar | Greiner Bio-One | 227261 | |
| GFAP antibody | Santa Cruz Biotechnology | sc33673 | |
| Glass beaker | N/A | N/A | |
| Glass petri dish | N/A | N/A | |
| GlutaMAX (100x) | Gibco | 35050061 | |
| Heracell 240i CO2 Incubator | Thermo scientific | 51032875 | |
| Herasafe 2025 Biological Safety Cabinet | Thermo scientific | 5016643 | |
| Hibernate-A | Gibco | A1247501 | |
| Histoacryl glue | B. Braun surgical | 1050052 | |
| Human Insulin, Solution | Santa Cruz Biotechnology | sc-360248 | |
| Ice box | N/A | N/A | |
| Ki67 antibody | Abcam | ab16667 | |
| McIlwain Tissue Chopper | Cavey Laboratory Engineering | 51350V | |
| Microscope Leica DMI 3000B, DMI 4000B, DMI 6000B | Leica | DMI6000B | For brightfield and immunofluorescence pictures |
| Microscope stereozoom S9D | Leica | W841832 | For manual cutting and to organoids monitoring |
| Microwave | Bosch | N/A | To heat the agarose solution |
| Mounting plastic discs | Cavey Laboratory Engineering | 51354 | |
| N-2 Supplement (100x) | Gibco | 17502048 | |
| NEM Non-Essential Amino Acids (NEAA) (100x) | Gibco | 11140050 | |
| Neurobasal (1x) | Gibco | 21103049 | |
| Orbital shaking machine Rotamax120 | Heidolph | 10304491 | |
| Penicilin Streptomycin | Gibco | 15140122 | |
| Plastic petri dishes Cellstar | greiner bio-one | 628160 | n = 12 |
| Single channel pipette 1000 µm | Eppendorf | 4924000010 | |
| Single channel pipette 5000 µm | Eppendorf | EP3123000276 | |
| Statistical Package for the Social Sciences (SPSS) version 23.0 | IBM | ||
| Surgipath Paraplast | Leica | 39601006 | Embedding medium |
| Ultra-low attachment Nucleon Sphera 6-well plate | Thermo Scientific | 174932 |
References
- Gatto, L., et al. IDH inhibitors and beyond: the cornerstone targeted glioma treatment. Mol Diagn Ther. 25 (4), 457-473 (2021).
- Buckner, J. C., et al. Radiation plus Procarbazine, CCNU, and Vincristine in low-grade glioma. N Engl J Med. 374 (14), 1344-1355 (2016).
- Grochans, S., et al. Epidemiology of glioblastoma multiforme-literature review. Cancers (Basel). 14 (10), 2412 (2022).
- Stupp, R., et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma). N Engl J Med. 352 (10), 987-996 (2005).
- Herrlinger, U., et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): a randomised, open-label, phase 3 trial. Lancet. 393 (10172), 678-688 (2019).
- Stupp, R., et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. Jama. 318 (23), 2306-2316 (2017).
- Desbaillets, N., Hottinger, A. F. Immunotherapy in glioblastoma: a clinical perspective. Cancers (Basel). 13 (15), 3721 (2021).
- Gularyan, S. K., et al. Investigation of inter- and intratumoral heterogeneity of glioblastoma using TOF-SIMS). Mol Cell Proteomics. 19 (6), 960-970 (2020).
- Brennan, C. W., et al. The somatic genomic landscape of glioblastoma. Cell. 155 (2), 462-477 (2013).
- Lathia, J. D., Mack, S. C., Mulkearns-Hubert, E. E., Valentim, C. L., Rich, J. N. Cancer stem cells in glioblastoma. Genes Dev. 29 (12), 1203-1217 (2015).
- Pollard, S. M., et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 4 (6), 568-580 (2009).
- Timerman, D., Yeung, C. M. Identity confusion of glioma cell lines. Gene. 536 (1), 221-222 (2014).
- Boj, S. F., et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 160 (1-2), 324-338 (2015).
- Kopper, O., et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat Med. 25 (5), 838-849 (2019).
- Lee, S. H., et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell. 173 (2), 515-528 (2018).
- Bian, S., et al. Genetically engineered cerebral organoids model brain tumor formation. Nat Methods. 15 (8), 631-639 (2018).
- Ogawa, J., Pao, G. M., Shokhirev, M. N., Verma, I. M. Glioblastoma model using human cerebral organoids. Cell Rep. 23 (4), 1220-1229 (2018).
- Linkous, A., et al. Modeling patient-derived glioblastoma with cerebral organoids. Cell Rep. 26 (12), 3203-3211 (2019).
- da Silva, B., Mathew, R. K., Polson, E. S., Williams, J., Wurdak, H. Spontaneous glioblastoma spheroid infiltration of early-stage cerebral organoids models brain tumor invasion. SLAS Discov. 23 (8), 862-868 (2018).
- Hubert, C. G., et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 76 (8), 2465-2477 (2016).
- Jacob, F., et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell. 180 (1), 188-204 (2020).
- Nickl, V., et al. Glioblastoma-derived three-dimensional ex vivo models to evaluate effects and efficacy of tumor treating fields (TTFields). Cancers (Basel). 14 (21), 5177 (2022).
- Klein, E., Hau, A. C., Oudin, A., Golebiewska, A., Niclou, S. P. Glioblastoma organoids: pre-clinical applications and challenges in the context of immunotherapy. Front Oncol. 10, 604121 (2020).
- Golebiewska, A., et al. Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol. 140 (6), 919-949 (2020).
- Bougnaud, S., et al. Molecular crosstalk between tumour and brain parenchyma instructs histopathological features in glioblastoma. Oncotarget. 7 (22), 31955-31971 (2016).
- Chua, C. W., et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat Cell Biol. 16 (10), 951-954 (2014).
- Collingridge, G. L. The brain slice preparation: a tribute to the pioneer Henry McIlwain. J Neurosci Methods. 59 (1), 5-9 (1995).
- Kato, H., Ogawa, T. A technique for preparing in vitro slices of cat's visual cortex for electrophysiological experiments. J Neurosci Methods. 4 (1), 33-38 (1981).
- Teyler, T. J. Brain slice preparation: hippocampus. Brain Res Bull. 5 (4), 391-403 (1980).
- Schulz, E., et al. Preparation and culture of organotypic hippocampal slices for the analysis of brain metastasis and primary brain tumor growth. Methods Mol Biol. 2294, 59-77 (2021).
- Driehuis, E., Gracanin, A., Vries, R. G. J., Clevers, H., Boj, S. F. Establishment of pancreatic organoids from normal tissue and tumors. STAR Protoc. 1 (3), 100192 (2020).
- Driehuis, E., et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc Natl Acad Sci U S A. 116 (52), 26580-26590 (2019).
- Neal, J. T., et al. Organoid modeling of the tumor immune microenvironment. Cell. 175 (7), 1972-1988 (2018).
- Li, X., et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med. 20 (7), 769-777 (2014).
- Ootani, A., et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 15 (6), 701-706 (2009).
- Zhao, Z., et al. Organoids. Nat Rev Methods Primers. 2, 94 (2022).
- Toh, Y. C., et al. A novel 3D mammalian cell perfusion-culture system in microfluidic channels. Lab Chip. 7 (3), 302-309 (2007).
- Zhang, C., Zhao, Z., Abdul Rahim, N. A., van Noort, D., Yu, H. Towards a human-on-chip: culturing multiple cell types on a chip with compartmentalized microenvironments. Lab Chip. 9 (22), 3185-3192 (2009).
- Homan, K. A., et al. Flow-enhanced vascularization and maturation of kidney organoids in vitro. Nat Methods. 16 (3), 255-262 (2019).
- Huh, D., et al. Reconstituting organ-level lung functions on a chip. Science. 328 (5986), 1662-1668 (2010).
- Kengla, C., Atala, A., Sang Jin, L. Bioprinting of organoids. Essentials of 3D Biofabrication and Translation. , 271-282 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved