
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
DeepOmicsAE: تمثيل وحدات الإشارات في مرض الزهايمر مع تحليل التعلم العميق للبروتينات والأيض والبيانات السريرية
In This Article
Summary
DeepOmicsAE هو سير عمل يركز على تطبيق طريقة التعلم العميق (أي التشفير التلقائي) لتقليل أبعاد بيانات الأوميكس المتعددة ، مما يوفر أساسا للنماذج التنبؤية ووحدات الإشارات التي تمثل طبقات متعددة من بيانات omics.
Abstract
أصبحت مجموعات بيانات omics الكبيرة متاحة بشكل متزايد للبحث في صحة الإنسان. تقدم هذه الورقة DeepOmicsAE ، وهو سير عمل محسن لتحليل مجموعات البيانات متعددة الأوميكس ، بما في ذلك البروتينات والأيض والبيانات السريرية. يستخدم سير العمل هذا نوعا من الشبكات العصبية يسمى autoencoder ، لاستخراج مجموعة موجزة من الميزات من بيانات الإدخال متعددة الأبعاد عالية الأبعاد. علاوة على ذلك ، يوفر سير العمل طريقة لتحسين المعلمات الرئيسية اللازمة لتنفيذ برنامج التشفير التلقائي. لعرض سير العمل هذا ، تم تحليل البيانات السريرية من مجموعة من 142 فردا كانوا إما أصحاء أو تم تشخيصهم بمرض الزهايمر ، إلى جانب البروتين والتمثيل الغذائي لعينات الدماغ بعد الوفاة. تحتفظ الميزات المستخرجة من الطبقة الكامنة للمشفر التلقائي بالمعلومات البيولوجية التي تفصل بين المرضى الأصحاء والمرضى. بالإضافة إلى ذلك ، تمثل الميزات المستخرجة الفردية وحدات إشارات جزيئية متميزة ، يتفاعل كل منها بشكل فريد مع السمات السريرية للأفراد ، مما يوفر وسيلة لدمج البروتينات والأيض والبيانات السريرية.
Introduction
نسبة كبيرة بشكل متزايد من السكان يشيخون ومن المتوقع أن يزداد عبء الأمراض المرتبطة بالعمر ، مثل التنكس العصبي ، بشكل حاد في العقود القادمة1. مرض الزهايمر هو النوع الأكثر شيوعا من الأمراض التنكسية العصبية2. كان التقدم في العثور على علاج بطيئا نظرا لفهمنا الضعيف للآليات الجزيئية الأساسية التي تقود ظهور المرض وتقدمه. يتم الحصول على غالبية المعلومات حول مرض الزهايمر بعد الوفاة من فحص أنسجة المخ ، مما جعل التمييز بين الأسباب والعواقب مهمة صعبة3. مشروع دراسة الأوامر الدينية / الذاكرة والشيخوخة (ROSMAP) هو جهد طموح لاكتساب فهم أوسع للتنكس العصبي ، والذي يتضمن دراسة آلاف الأفراد الذين التزموا بالخضوع لفحوصات طبية ونفسية سنويا والمساهمة بأدمغتهم للبحث بعد وفاتهم4. تركز الدراسة على الانتقال من الأداء الطبيعي للدماغ إلى مرض الزهايمر2. ضمن المشروع ، تم تحليل عينات الدماغ بعد الوفاة مع عدد كبير من مناهج omics ، بما في ذلك علم الجينوم ، وعلم الجينوم ، وعلم النسخ ، والبروتينات5 ، وعلم الأيض.
تقنيات أوميكس التي تقدم قراءات وظيفية للحالات الخلوية (أي البروتينات والأيض)6,7 هي المفتاح لتفسير المرض8،9،10،11،12 ، بسبب العلاقة المباشرة بين وفرة البروتين والمستقلبات والأنشطة الخلوية. البروتينات هي المنفذين الأساسيين للعمليات الخلوية ، في حين أن المستقلبات هي ركائز ومنتجات التفاعلات الكيميائية الحيوية. يوفر تحليل بيانات Multi-omics إمكانية فهم العلاقات المعقدة بين بيانات البروتينات والأيض بدلا من تقديرها بمعزل عن غيرها. Multi-omics هو تخصص يدرس طبقات متعددة من البيانات البيولوجية عالية الأبعاد ، بما في ذلك البيانات الجزيئية (تسلسل الجينوم والطفرات ، والنسخ ، والبروتين ، والأيض) ، وبيانات التصوير السريري ، والميزات السريرية. على وجه الخصوص ، يهدف تحليل البيانات متعددة الأوميكس إلى دمج هذه الطبقات من البيانات البيولوجية ، وفهم تنظيمها المتبادل وديناميكيات التفاعل ، وتقديم فهم شامل لبداية المرض وتطوره. ومع ذلك ، لا تزال طرق دمج البيانات متعددة الأوميكس في المراحل الأولى من التطوير13.
تعد أجهزة التشفير التلقائي ، وهي نوع من الشبكات العصبية غير الخاضعة للإشراف14 ، أداة قوية لتكامل البيانات متعددة الأوميكس. على عكس الشبكات العصبية الخاضعة للإشراف ، لا تقوم أجهزة التشفير الذاتي بتعيين عينات إلى قيم مستهدفة محددة (مثل صحية أو مريضة) ، ولا يتم استخدامها للتنبؤ بالنتائج. يكمن أحد تطبيقاتها الأساسية في تقليل الأبعاد. ومع ذلك ، توفر أجهزة التشفير التلقائي العديد من المزايا مقارنة بطرق تقليل الأبعاد الأبسط مثل تحليل المكون الرئيسي (PCA) ، أو تضمين الجار العشوائي الموزع على t (tSNE) ، أو التقريب والإسقاط المشعب الموحد (UMAP). على عكس PCA ، يمكن لأجهزة التشفير التلقائي التقاط العلاقات غير الخطية داخل البيانات. على عكس tSNE و UMAP ، يمكنهم اكتشاف العلاقات الهرمية ومتعددة الوسائط داخل البيانات لأنها تعتمد على طبقات متعددة من الوحدات الحسابية تحتوي كل منها على وظائف تنشيط غير خطية. لذلك ، فهي تمثل نماذج جذابة لالتقاط تعقيد بيانات الأوميكس المتعددة. أخيرا ، في حين أن التطبيق الأساسي ل PCA و tSNE و UMAP هو تجميع البيانات ، تقوم أجهزة التشفير التلقائي بضغط بيانات الإدخال إلى ميزات مستخرجة مناسبة تماما للمهام التنبؤية النهائية15,16.
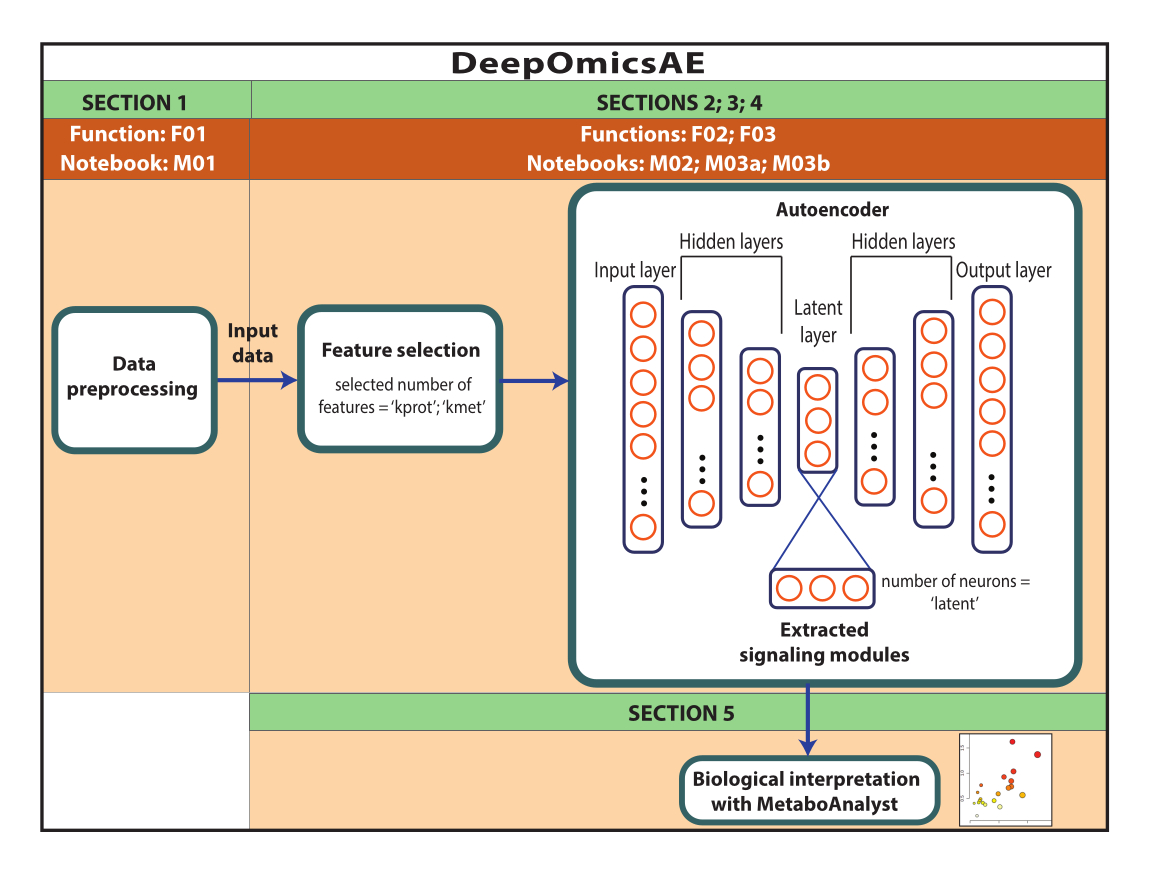
باختصار ، تتكون الشبكات العصبية من عدة طبقات ، تحتوي كل منها على وحدات حسابية متعددة أو "خلايا عصبية". يشار إلى الطبقات الأولى والأخيرة باسم طبقات الإدخال والإخراج ، على التوالي. التشفير الذاتي عبارة عن شبكات عصبية ذات بنية الساعة الرملية ، تتكون من طبقة إدخال ، تليها طبقة واحدة إلى ثلاث طبقات مخفية وطبقة صغيرة "كامنة" تحتوي عادة على ما بين اثنين وستة خلايا عصبية. يعرف النصف الأول من هذا الهيكل باسم التشفير ويتم دمجه مع وحدة فك ترميز تعكس برنامج التشفير. ينتهي جهاز فك التشفير بطبقة خرج تحتوي على نفس عدد الخلايا العصبية مثل طبقة الإدخال. تأخذ أجهزة التشفير التلقائي المدخلات من خلال عنق الزجاجة وتعيد بنائها في طبقة الإخراج ، بهدف إنشاء مخرجات تعكس المعلومات الأصلية بأكبر قدر ممكن. يتم تحقيق ذلك عن طريق التقليل رياضيا من معلمة تسمى "خسارة إعادة الإعمار". يتكون المدخل من مجموعة من الميزات ، والتي في التطبيق المعروض هنا ستكون وفرة البروتين والأيض ، والخصائص السريرية (أي الجنس والتعليم والعمر عند الوفاة). تحتوي الطبقة الكامنة على تمثيل مضغوط وغني بالمعلومات للمدخلات ، والذي يمكن استخدامه للتطبيقات اللاحقة مثل النماذج التنبؤية17,18.
يقدم هذا البروتوكول سير عمل ، DeepOmicsAE ، والذي يتضمن: 1) المعالجة المسبقة للبروتينات والأيض والبيانات السريرية (أي التطبيع والقياس والإزالة الخارجية) للحصول على بيانات بمقياس ثابت لتحليل التعلم الآلي ؛ 2) اختيار ميزات إدخال التشفير التلقائي المناسبة ، لأن الحمل الزائد للميزة قد يحجب أنماط المرض ذات الصلة ؛ 3) تحسين وتدريب التشفير الذاتي ، بما في ذلك تحديد العدد الأمثل للبروتينات والمستقلبات للاختيار ، والخلايا العصبية للطبقة الكامنة ؛ 4) استخراج الميزات من الطبقة الكامنة ؛ و 5) استخدام الميزات المستخرجة للتفسير البيولوجي من خلال تحديد وحدات الإشارات الجزيئية وعلاقتها بالسمات السريرية.
يهدف هذا البروتوكول إلى أن يكون بسيطا وقابلا للتطبيق من قبل علماء الأحياء ذوي الخبرة الحسابية المحدودة الذين لديهم فهم أساسي للبرمجة باستخدام بايثون. يركز البروتوكول على تحليل بيانات الأوميكس المتعددة ، بما في ذلك البروتينات والأيض والسمات السريرية ، ولكن يمكن توسيع استخدامه ليشمل أنواعا أخرى من بيانات التعبير الجزيئي ، بما في ذلك النسخ. أحد التطبيقات الجديدة المهمة التي قدمها هذا البروتوكول هو تعيين درجات أهمية الميزات الأصلية على الخلايا العصبية الفردية في الطبقة الكامنة. نتيجة لذلك ، تمثل كل خلية عصبية في الطبقة الكامنة وحدة إشارات ، توضح بالتفصيل التفاعلات بين التغيرات الجزيئية المحددة والخصائص السريرية للمرضى. يتم الحصول على التفسير البيولوجي لوحدات الإشارات الجزيئية باستخدام MetaboAnalyst ، وهي أداة متاحة للجمهور تدمج بيانات الجينات / البروتين والأيض لاشتقاق مسارات إشارات التمثيل الغذائي والخليةالمخصب 17.
Protocol
ملاحظة: البيانات المستخدمة هنا هي بيانات ROSMAP التي تم تنزيلها من بوابة AD المعرفية. ليست هناك حاجة إلى موافقة مستنيرة لتنزيل البيانات وإعادة استخدامها. يستخدم البروتوكول المقدم هنا التعلم العميق لتحليل بيانات الأوميكس المتعددة وتحديد وحدات الإشارات التي تميز مجموعات معينة من المرضى أو العينات بناء على تشخيصهم على سبيل المثال. يقدم البروتوكول أيضا مجموعة صغيرة من الميزات المستخرجة التي تلخص البيانات الأصلية واسعة النطاق ويمكن استخدامها لمزيد من التحليل مثل تدريب نموذج تنبؤي باستخدام خوارزميات التعلم الآلي (الشكل 1). ارجع إلى الملف التكميلي 1 وجدول المواد للحصول على معلومات حول الوصول إلى الكود وإعداد البيئة الحسابية قبل تنفيذ البروتوكول. يجب تنفيذ الطرق باتباع الترتيب المحدد أدناه.

الشكل 1: رسم تخطيطي لسير عمل DeepOmicsAE. تمثيل تخطيطي لسير العمل لتحليل بيانات متعددة الأوميكس باستخدام سير العمل. في تصوير المشفر التلقائي ، تمثل المستطيلات طبقات من الشبكة العصبية وتمثل الدوائر الخلايا العصبية داخل الطبقات. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
1. المعالجة المسبقة للبيانات
ملاحظة: الهدف من هذا القسم هو المعالجة المسبقة للبيانات ، بما في ذلك معالجة البيانات المفقودة ؛ تطبيع وتوسيع نطاق التعبير البروتيني والأيضي والبيانات السريرية ؛ وإزالة القيم المتطرفة. تم تصميم البروتوكول لمجموعة بيانات تتضمن بيانات البروتينات المعبر عنها بالسجل2 (النسبة) ؛ بيانات الأيض معبرا عنها كتغيير أضعاف ؛ والسمات السريرية بما في ذلك السمات المستمرة والفئوية. يجب تجميع المرضى أو العينات بناء على التشخيص أو معايير أخرى مماثلة. يجب أن تكون العينات أو المرضى عبر الصفوف والميزات عبر الأعمدة.
- لبدء مثيل جديد من Jupyter Notebook في المستعرض ، افتح نافذة طرفية جديدة ، واكتب ما يلي واضغط على Enter.
دفتر جوبيتر - في صفحة Jupyter الرئيسية على المتصفح ، انقر فوق دفتر الملاحظات M01 - بيانات التعبير pre-processing.ipynb لفتحه في علامة تبويب جديدة (الملف التكميلي 2 ، الخطوة 1.1).
- في الخلية الثانية من دفتر الملاحظات، اكتب اسم ملف مجموعة البيانات بدلا من your_dataset_name.csv.
- في الخلية الأخيرة من دفتر الملاحظات ، اكتب الاسم المطلوب لملف بيانات الإخراج بدلا من M01_output_data.csv.
- في الخلية الخامسة من دفتر الملاحظات ، حدد موضع الأعمدة لكل نوع بيانات كما يلي: بيانات البروتينات (cols_prot) ، بيانات الأيض (cols_met) ، البيانات السريرية المستمرة (على سبيل المثال ، العمر) (cols_clin_con) ، البيانات السريرية الثنائية (مثل الجنس) (cols_clin_bin). أدخل فهرس العمود الأول لكل نوع بيانات بدلا من col_start وفهرس الأعمدة الأخير بدلا من col_end ؛ على سبيل المثال: cols_prot = شريحة (0 ، 8817). تأكد من أن القيم المحددة في كائنات الشريحة تتوافق مع فهارس العمودين الأول والأخير المقابلة لكل نوع بيانات. استخدم الأمر الموجود في الخلية الرابعة من دفتر الملاحظات نفسه (df.iloc[:, :]) لتحديد موضع البداية والنهاية لكل نوع بيانات (الملف التكميلي 2، الخطوة 1.2).
- حدد الخلية | قم بتشغيل الكل من شريط القائمة في Jupyter لإنشاء ملف بيانات الإخراج في المجلد المحدد (الملف التكميلي 2 ، الخطوة 1.3).
ملاحظة: سيتم استخدام هذه البيانات كمدخلات للبروتوكولات الموضحة في الأقسام 2 أو 3 أو 4.
2. التحسين المخصص لسير العمل (اختياري)
ملاحظة: القسم 2 اختياري لأنه يتطلب استخدام الكمبيوتر بشكل مكثف. يجب على المستخدمين التخطي مباشرة إلى القسم 4 إذا قرروا عدم تنفيذ القسم 2. سيوجه هذا البروتوكول المستخدم من خلال تحسين سير العمل بطريقة آلية. على وجه التحديد ، تحدد الطريقة المعلمات التي تقدم أفضل أداء للمشفر التلقائي من حيث إنشاء الميزات المستخرجة التي تفصل مجموعات العينات جيدا. تتضمن المعلمات المحسنة التي تم إنشاؤها كمخرجات عدد الميزات التي يجب استخدامها لاختيار الميزة (k_prot و k_met) وعدد الخلايا العصبية في طبقة التشفير الذاتي الكامنة (الكامنة). يمكن بعد ذلك استخدام هذه المعلمات في البروتوكول الموضح في القسم 3 لإنشاء النموذج.
- في الصفحة الرئيسية ل Jupyter على المتصفح ، انقر فوق دفتر الملاحظات M02 - DeepOmicsAE model optimization.ipynb لفتحه في علامة تبويب جديدة (الملف التكميلي 2 ، الخطوة 2.1).
- في الخلية الثانية من دفتر الملاحظات، اكتب اسم ملف الإدخال بدلا من M01_output_data.csv. الإدخال في هذه الوظيفة هو بيانات الإخراج من القسم 1.
- في الخلية الخامسة من دفتر الملاحظات ، حدد موضع الأعمدة لكل نوع بيانات كما يلي: بيانات البروتينات (cols_X_prot) ، بيانات الأيض (cols_X_met) ، البيانات السريرية (cols_clin ؛ تتضمن جميع البيانات السريرية) ، جميع بيانات التعبير الجزيئي ، بما في ذلك بيانات البروتينات والأيض (cols_X_expr). أدخل فهرس العمود الأول لكل نوع بيانات بدلا من col_start وفهرس الأعمدة الأخير بدلا من col_end ؛ على سبيل المثال ، cols_prot = شريحة (0 ، 8817). تأكد من أن القيم المحددة في كائنات الشريحة تتوافق مع فهرس العمودين الأول والأخير المقابل لكل نوع بيانات، واستخدم الأوامر الموجودة في الخليتين الثالثة والرابعة من دفتر الملاحظات لاستكشاف البيانات وتحديد مواضع البداية والنهاية لكل نوع بيانات. حدد اسم العمود الذي يحتوي على المتغير الهدف بدلا من y_column_name ك y_label (الملف التكميلي 2، الخطوة 2.2).
ملاحظة: ستختلف قيم الفهارس المحددة في cols_X_prot و cols_X_met و cols_clin و cols_X_expr عن تلك المستخدمة في القسم 1 بسبب إعادة تشكيل إطار البيانات الذي يحدث أثناء المعالجة المسبقة للبيانات. - في الخلية السادسة من دفتر الملاحظات، حدد عدد جولات التحسين التي يجب تنفيذها عن طريق تعيين قيمة إلى n_comb. أوقات المعالجة حوالي 4-5 دقائق لمدة 10 جولات ؛ 20 دقيقة ل 50 طلقة ، و 40 دقيقة ل 100 طلقة (الملف التكميلي 2 ، الخطوة 2.3).
- حدد الخلية | قم بتشغيل الكل من شريط القائمة في Jupyter.
ملاحظة: سيتم تخزين متغيرات الإخراج kprot و kmet و cament ويمكن الوصول إليها من دفاتر الملاحظات الأخرى ، والتي سيتم استخدامها لمتابعة سير العمل التحليلي. سيتم إنشاء AE_optimization_plot.pdf المؤامرة وحفظها في المجلد المحلي (الشكل 2).
3. تنفيذ سير العمل مع معلمات محسنة مخصصة
ملاحظة: قم بإجراء هذا البروتوكول فقط بعد تحسين الأسلوب (القسم 2). إذا اختار المستخدمون عدم إجراء تحسين الطريقة، فانتقل مباشرة إلى القسم 4. سيوجه هذا البروتوكول المستخدم من خلال إنشاء نموذج باستخدام المعلمات المحسنة المخصصة المستمدة من القسم 2. سيقوم المشفر التلقائي 1) بإنشاء مجموعة من الميزات المستخرجة التي تلخص البيانات الأصلية و 2) تحديد الميزات المهمة التي تقود كل خلية عصبية في الطبقة الكامنة ، والتي تمثل بشكل فعال وحدات إشارات فريدة. سيتم تفسير وحدات التشوير باستخدام البروتوكول المنصوص عليه في القسم 5.
- في الصفحة الرئيسية ل Jupyter على المتصفح ، انقر فوق تطبيق دفتر الملاحظات M03a - DeepOmicsAE مع parameters.ipynb المحسن المخصص لفتحه في علامة تبويب جديدة (الملف التكميلي 2 ، الخطوة 3.1).
- في الخلية الثانية من دفتر الملاحظات، اكتب اسم ملف الإدخال بدلا من M01_output_data.csv. الإدخال في هذه الوظيفة هو بيانات الإخراج من القسم 1.
- في الخلية الخامسة من دفتر الملاحظات ، حدد موضع الأعمدة لكل نوع بيانات كما يلي: بيانات البروتينات (cols_prot) ، بيانات الأيض (cols_met) ، البيانات السريرية (cols_clin ؛ تتضمن جميع البيانات السريرية). أدخل فهرس العمود الأول لكل نوع بيانات بدلا من col_start وفهرس الأعمدة الأخير بدلا من col_end ؛ على سبيل المثال: cols_prot = شريحة (0 ، 8817). تأكد من أن القيم المحددة في كائنات الشريحة تتوافق مع فهارس العمودين الأول والأخير المقابلة لكل نوع بيانات، واستخدم الأوامر الموجودة في الخليتين الثالثة والرابعة من دفتر الملاحظات لاستكشاف البيانات وتحديد مواضع البداية والنهاية لكل نوع بيانات. حدد اسم العمود الذي يحتوي على المتغير الهدف (على سبيل المثال ، 0 أو 1 ، المقابل لسليم أو مريض) بدلا من y_column_name ك y_label.
ملاحظة: ستختلف قيمة الفهارس المحددة في cols_X_prot و cols_X_met و cols_clin و cols_X_expr عن تلك المستخدمة في القسم 1 بسبب إعادة تشكيل إطار البيانات الذي يحدث أثناء المعالجة المسبقة للبيانات. - حدد الخلية | قم بتشغيل الكل من شريط القائمة في Jupyter لإنشاء وحفظ المؤامرات PCA_initial_data.pdf و PCA_extracted_features.pdf و distribution_important_feature_scores.pdf في المجلد المحلي (الشكل 3 والشكل التكميلي S1). بالإضافة إلى ذلك ، سيتم تخزين قوائم الميزات المهمة لكل وحدة تشوير محددة في ملفات نصية في المجلد المحلي ، المسمى module_n.txt ، حيث سيتم استبدال n برقم الوحدة.
4. تنفيذ سير العمل مع معلمات محددة مسبقا
- راجع القسم 3 للحصول على إرشادات مفصلة حول كيفية تشغيل هذه الطريقة (الملف التكميلي 2، الخطوة 4.1). الفرق الوحيد بين هذين البروتوكولين هو أن المعلمات kprot و kmet والكامنة (في الخلية السابعة من دفتر الملاحظات) مشتقة رياضيا بناء على نتائج التحسين الذي تم إجراؤه كما هو موضح في الشكل 2.
ملاحظة: إذا كان القسم 4 يوفر فصلا ضعيفا لمجموعات العينات، مما يشير إلى أداء النموذج دون المستوى الأمثل، فمن المستحسن تنفيذ تحسين النموذج (القسم 2) باستخدام 15 تكرارا على الأقل، وإذا أمكن، حتى 50.
5. التفسير البيولوجي باستخدام MetaboAnalyst
- افتح المتصفح وانتقل إلى الرابط أدناه للوصول إلى وظيفة تحليل المسار المشترك على موقع MetaboAnalyst : https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml.
- قم بالوصول إلى المجلد حيث تم حفظ ملفات الإخراج من الطريقة 3 أو الطريقة 4 وافتح الملفات النصية module_n.txt لكل وحدة إشارة n تم إنشاؤها بواسطة الطريقة 3 أو الطريقة 4.
- حدد موقع البروتينات في الملفات النصية وانسخها.
- الصق قائمة البروتينات في النافذة الجينات / البروتينات مع تغييرات الطي الاختيارية في صفحة ويب MetaboAnalyst.
- كرر الخطوة أعلاه للمستقلبات والصقها في النافذة قائمة المركبات مع تغييرات الطي الاختيارية على نفس صفحة الويب.
- حدد الكائن الحي المناسب ونوع المعرف ، ثم انقر فوق إرسال في أسفل الصفحة (الملف التكميلي 2 ، الخطوة 5.1).
ملاحظة: تأكد من التعرف على المعرفات بواسطة MetaboAnalyst. تشمل المعرفات المعترف بها معرف Entrez ورموز الجينات الرسمية ومعرف Uniprot للبروتينات. الاسم المركب ومعرف HMDB ومعرف KEGG للمستقلبات. إذا كانت المعرفات غير هذه الأنواع ، فإن التحويل المناسب ضروري قبل التحليل. - في الصفحة التالية، تحقق من تعيين المعرف قبل النقر على متابعة للتحقق من التعرف على المعرفات.
- في صفحة إعداد المعلمات ، حدد المسارات الأيضية (متكاملة) أو كل المسارات (متكاملة) لتصور مساهمة المدخلات في المسارات الأيضية فقط أو في جميع مسارات الإشارات على التوالي (الملف التكميلي 2، الخطوة 5.2). في لوحة تحديد الخوارزمية ، اختر تحليل الإثراء: اختبار الهندسة الفائقة، وقياس الطوبولوجيا: مركزية الدرجة، وطريقة التكامل: دمج قيم p (مستوى المسار). انقر فوق إرسال في أسفل الصفحة.
- الصفحة الأخيرة هي عرض النتائج ، والتي تعرض نتائج تحليل الإثراء. يتم رسم المسارات المخصبة بناء على تأثيرها وأهميتها ، كما يتم توفير قائمة المسارات في شكل جدول.
النتائج
لعرض البروتوكول ، قمنا بتحليل مجموعة بيانات تضم البروتينات والأيض والمعلومات السريرية المستمدة من أدمغة ما بعد الوفاة ل 142 فردا كانوا إما أصحاء أو تم تشخيصهم بمرض الزهايمر.
بعد إجراء قسم البروتوكول 1 للمعالجة المسبقة للبيانات ، تضمنت مجموعة البيانات 6,497 بروتينا و 443 مستقلبا...
Discussion
هيكل مجموعة البيانات أمر بالغ الأهمية لنجاح البروتوكول ويجب التحقق منه بعناية. يجب تنسيق البيانات كما هو موضح في قسم البروتوكول 1. يعد التعيين الصحيح لمواضع الأعمدة أمرا بالغ الأهمية أيضا لنجاح الطريقة. تتم معالجة بيانات البروتيوميات والأيض بشكل مختلف ويتم اختيار الميزة بشكل منفصل بسبب ا...
Disclosures
يعلن صاحب البلاغ أنه ليس لديهم تضارب في المصالح.
Acknowledgements
تم دعم هذا العمل من قبل CA201402 منحة المعاهد الوطنية للصحة وجائزة الباحث المتميز لمركز كورنيل لعلم جينوم الفقاريات (CVG). تستند النتائج المنشورة هنا كليا أو جزئيا إلى البيانات التي تم الحصول عليها من بوابة المعرفة الخاصة بأبوظبي (https://adknowledgeportal.org). تم توفير بيانات الدراسة من خلال شراكة الطب المسرع لمرض الزهايمر (U01AG046161 و U01AG061357) بناء على العينات المقدمة من مركز مرض الزهايمر راش ، المركز الطبي بجامعة راش ، شيكاغو. تم دعم جمع البيانات من خلال التمويل من قبل منح NIA P30AG10161 و R01AG15819 و R01AG17917 و R01AG30146 و R01AG36836 و U01AG32984 و U01AG46152 وإدارة الصحة العامة في إلينوي ومعهد أبحاث الجينوم الانتقالي. تم إنشاء مجموعة بيانات الأيض في Metabolon ومعالجتها مسبقا بواسطة ADMC.
Materials
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
References
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved