
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
DeepOmicsAE: Representación de módulos de señalización en la enfermedad de Alzheimer con análisis de aprendizaje profundo de proteómica, metabolómica y datos clínicos
En este artículo
Resumen
DeepOmicsAE es un flujo de trabajo centrado en la aplicación de un método de aprendizaje profundo (es decir, un codificador automático) para reducir la dimensionalidad de los datos multiómicos, proporcionando una base para modelos predictivos y módulos de señalización que representan múltiples capas de datos ómicos.
Resumen
Los grandes conjuntos de datos ómicos están cada vez más disponibles para la investigación de la salud humana. Este artículo presenta DeepOmicsAE, un flujo de trabajo optimizado para el análisis de conjuntos de datos multiómicos, incluidos proteómicos, metabolómicos y datos clínicos. Este flujo de trabajo emplea un tipo de red neuronal denominada autocodificador para extraer un conjunto conciso de características de los datos de entrada multiómicos de alta dimensión. Además, el flujo de trabajo proporciona un método para optimizar los parámetros clave necesarios para implementar el autocodificador. Para mostrar este flujo de trabajo, se analizaron los datos clínicos de una cohorte de 142 individuos sanos o diagnosticados con la enfermedad de Alzheimer, junto con el proteoma y el metaboloma de sus muestras cerebrales postmortem. Las características extraídas de la capa latente del autocodificador retienen la información biológica que separa a los pacientes sanos de los enfermos. Además, las características individuales extraídas representan distintos módulos de señalización molecular, cada uno de los cuales interactúa de manera única con las características clínicas de los individuos, proporcionando un medio para integrar la proteómica, la metabolómica y los datos clínicos.
Introducción
Una proporción cada vez mayor de la población está envejeciendo y se espera que la carga de las enfermedades relacionadas con la edad, como la neurodegeneración, aumente drásticamente en las próximas décadas1. La enfermedad de Alzheimer es el tipo más común de enfermedad neurodegenerativa2. El progreso en la búsqueda de un tratamiento ha sido lento debido a nuestra escasa comprensión de los mecanismos moleculares fundamentales que impulsan la aparición y el progreso de la enfermedad. La mayor parte de la información sobre la enfermedad de Alzheimer se obtiene post mortem a partir del examen del tejido cerebral, lo que ha dificultado la distinción de causas y consecuencias3. El Proyecto de Estudio de las Órdenes Religiosas/Memoria y Envejecimiento (ROSMAP, por sus siglas en inglés) es un ambicioso esfuerzo para obtener una comprensión más amplia de la neurodegeneración, que implica el estudio de miles de personas que se han comprometido a someterse a exámenes médicos y psicológicos anualmente y a contribuir con sus cerebros para la investigación después desu fallecimiento. El estudio se centra en la transición del funcionamiento normal del cerebro a la enfermedad de Alzheimer2. Dentro del proyecto, se analizaron muestras cerebrales postmortem con una gran cantidad de enfoques ómicos, que incluyen genómica, epigenómica, transcriptómica, proteómica5 y metabolómica.
Las tecnologías ómicas que ofrecen lecturas funcionales de los estados celulares (es decir, proteómica y metabolómica)6,7 son clave para interpretar la enfermedad 8,9,10,11,12, debido a la relación directa entre la abundancia de proteínas y metabolitos y las actividades celulares. Las proteínas son los principales ejecutores de los procesos celulares, mientras que los metabolitos son los sustratos y productos de las reacciones bioquímicas. El análisis de datos multiómicos ofrece la posibilidad de comprender las complejas relaciones entre los datos proteómicos y metabolómicos en lugar de apreciarlos de forma aislada. La multiómica es una disciplina que estudia múltiples capas de datos biológicos de alta dimensión, incluidos datos moleculares (secuencia y mutaciones del genoma, transcriptoma, proteoma, metaboloma), datos de imágenes clínicas y características clínicas. En particular, el análisis de datos multiómicos tiene como objetivo integrar dichas capas de datos biológicos, comprender su regulación recíproca y su dinámica de interacción, y ofrecer una comprensión holística de la aparición y progresión de la enfermedad. Sin embargo, los métodos para integrar datos multiómicos aún se encuentran en las primeras etapas de desarrollo13.
Los autocodificadores, un tipo de red neuronal no supervisada14, son una poderosa herramienta para la integración de datos multiómicos. A diferencia de las redes neuronales supervisadas, los autocodificadores no asignan muestras a valores objetivo específicos (como sanos o enfermos), ni se utilizan para predecir resultados. Una de sus principales aplicaciones radica en la reducción de la dimensionalidad. Sin embargo, los autocodificadores ofrecen varias ventajas sobre los métodos de reducción de dimensionalidad más simples, como el análisis de componentes principales (PCA), la incrustación de vecinos estocásticos distribuidos en t (tSNE) o la aproximación y proyección de variedades uniformes (UMAP). A diferencia de PCA, los autocodificadores pueden capturar relaciones no lineales dentro de los datos. A diferencia de tSNE y UMAP, pueden detectar relaciones jerárquicas y multimodales dentro de los datos, ya que se basan en múltiples capas de unidades computacionales, cada una de las cuales contiene funciones de activación no lineales. Por lo tanto, representan modelos atractivos para capturar la complejidad de los datos multiómicos. Por último, mientras que la aplicación principal de PCA, tSNE y UMAP es la agrupación de los datos, los autocodificadores comprimen los datos de entrada en características extraídas que son adecuadas para tareas predictivas posteriores15,16.
En resumen, las redes neuronales comprenden varias capas, cada una de las cuales contiene múltiples unidades computacionales o "neuronas". La primera y la última capa se denominan capas de entrada y salida, respectivamente. Los autocodificadores son redes neuronales con una estructura de reloj de arena, que consta de una capa de entrada, seguida de una a tres capas ocultas y una pequeña capa "latente" que normalmente contiene entre dos y seis neuronas. La primera mitad de esta estructura se conoce como codificador y se combina con un decodificador que refleja el codificador. El decodificador termina con una capa de salida que contiene el mismo número de neuronas que la capa de entrada. Los autocodificadores toman la entrada a través del cuello de botella y la reconstruyen en la capa de salida, con el objetivo de generar una salida que refleje la información original lo más fielmente posible. Esto se logra minimizando matemáticamente un parámetro denominado "pérdida de reconstrucción". La entrada consiste en un conjunto de características, que en la aplicación que se muestra en este documento serán abundancias de proteínas y metabolitos, y características clínicas (es decir, sexo, educación y edad de muerte). La capa latente contiene una representación comprimida y rica en información de la entrada, que puede ser utilizada para aplicaciones posteriores como modelos predictivos17,18.
Este protocolo presenta un flujo de trabajo, DeepOmicsAE, que implica: 1) preprocesamiento de proteómica, metabolómica y datos clínicos (es decir, normalización, escalado, eliminación de valores atípicos) para obtener datos con una escala consistente para el análisis de aprendizaje automático; 2) seleccionar las características de entrada apropiadas del autocodificador, ya que la sobrecarga de características puede ocultar los patrones de enfermedades relevantes; 3) optimizar y entrenar el autocodificador, incluida la determinación del número óptimo de proteínas y metabolitos a seleccionar, y de neuronas para la capa latente; 4) extracción de características de la capa latente; y 5) utilizar las características extraídas para la interpretación biológica mediante la identificación de módulos de señalización molecular y su relación con las características clínicas.
Este protocolo pretende ser simple y aplicable por biólogos con experiencia computacional limitada que tengan un conocimiento básico de programación con Python. El protocolo se centra en el análisis de datos multiómicos, incluidos los proteómicos, los metabolómicos y las características clínicas, pero su uso puede extenderse a otros tipos de datos de expresión molecular, incluida la transcriptómica. Una nueva aplicación importante introducida por este protocolo es el mapeo de las puntuaciones de importancia de las características originales en neuronas individuales en la capa latente. Como resultado, cada neurona en la capa latente representa un módulo de señalización, detallando las interacciones entre alteraciones moleculares específicas y las características clínicas de los pacientes. La interpretación biológica de los módulos de señalización molecular se obtiene mediante el uso de MetaboAnalyst, una herramienta disponible públicamente que integra datos de genes/proteínas y metabolitos para derivar vías de señalización metabólica y celular enriquecidas17.
Protocolo
NOTA: Los datos utilizados aquí fueron datos de ROSMAP descargados del portal de conocimiento de AD. No se necesita el consentimiento informado para descargar y reutilizar los datos. El protocolo presentado en este documento utiliza el aprendizaje profundo para analizar datos multiómicos e identificar módulos de señalización que distinguen a pacientes específicos o grupos de muestras basándose, por ejemplo, en su diagnóstico. El protocolo también ofrece un pequeño conjunto de características extraídas que resumen los datos originales a gran escala y se pueden utilizar para análisis posteriores, como el entrenamiento de un modelo predictivo mediante algoritmos de aprendizaje automático (Figura 1). Consulte el Archivo Suplementario 1 y la Tabla de Materiales para obtener información sobre el acceso al código y la configuración del entorno computacional antes de realizar el protocolo. Los métodos deben realizarse siguiendo el orden que se especifica a continuación.

Figura 1: Esquema del flujo de trabajo de DeepOmicsAE. Representación esquemática del flujo de trabajo para analizar datos multiómicos mediante el flujo de trabajo. En la representación del autocodificador, los rectángulos representan capas de la red neuronal y los círculos representan neuronas dentro de capas. Haga clic aquí para ver una versión más grande de esta figura.
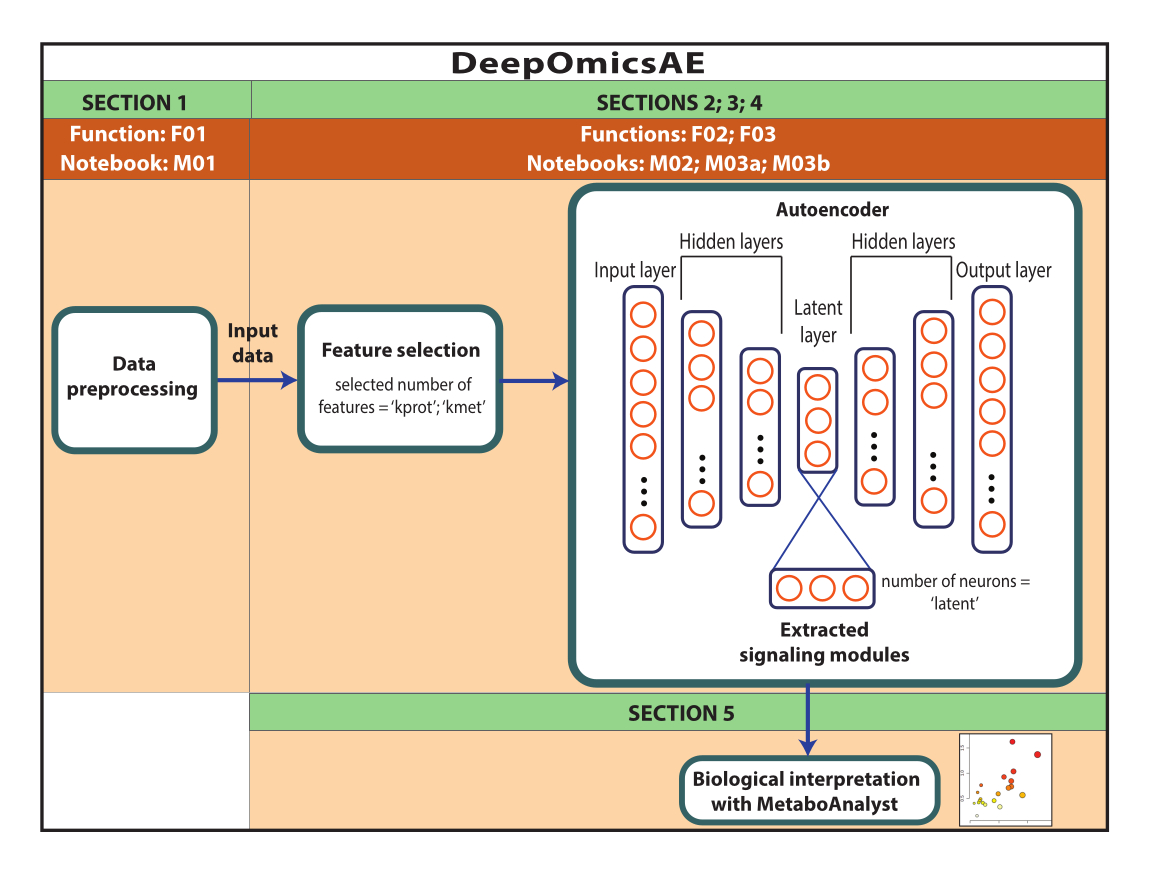{kind=link}
1. Preprocesamiento de datos
NOTA: El objetivo de esta sección es preprocesar los datos, incluido el manejo de los datos faltantes; normalización y escalado de la expresión proteómica, metabolómica y de datos clínicos; y la eliminación de valores atípicos. El protocolo está diseñado para un conjunto de datos que incluye datos proteómicos expresados como log2 (ratio); datos metabolómicos expresados como cambio de pliegue; y características clínicas, incluidas las características continuas y categóricas. Los pacientes o las muestras deben agruparse en función del diagnóstico u otros parámetros similares. Las muestras o los pacientes deben estar en las filas y las entidades en las columnas.
- Para iniciar una nueva instancia de Jupyter Notebook en el explorador, abra una nueva ventana de terminal, escriba lo siguiente y presione Entrar.
Jupyter Notebook - En la página de inicio de Jupyter en el navegador, haga clic en el cuaderno M01 - expression data pre-processing.ipynb para abrirlo en una nueva pestaña (Archivo complementario 2, paso 1.1).
- En la segunda celda del bloc de notas, escriba el nombre del archivo de conjunto de datos en lugar de your_dataset_name.csv.
- En la última celda del bloc de notas, escriba el nombre deseado del archivo de datos de salida en lugar de M01_output_data.csv.
- En la quinta celda del cuaderno, especifique la posición de las columnas para cada tipo de datos de la siguiente manera: datos proteómicos (cols_prot), datos metabolómicos (cols_met), datos clínicos continuos (p. ej., edad) (cols_clin_con), datos clínicos binarios (p. ej., sexo) (cols_clin_bin). Introduzca el índice de la primera columna para cada tipo de datos en lugar de col_start y el índice de la última columna en lugar de col_end; Por ejemplo: cols_prot = slice(0, 8817). Asegúrese de que los valores especificados en los objetos de sector correspondan a los índices de la primera y la última columna correspondientes a cada tipo de datos. Utilice el comando de la cuarta celda del mismo cuaderno (df.iloc[:, :]) para determinar la posición inicial y final de cada tipo de datos (Archivo complementario 2, paso 1.2).
- Seleccionar celda | Ejecute todo desde la barra de menús de Jupyter para crear el archivo de datos de salida en la carpeta especificada (Archivo complementario 2, paso 1.3).
NOTA: Estos datos se utilizarán como entrada para los protocolos descritos en las secciones 2, 3 o 4.
2. Optimización personalizada del flujo de trabajo (opcional)
NOTA: La sección 2 es opcional porque requiere un uso intensivo de computadoras. Los usuarios deben saltar directamente a la sección 4 si deciden no realizar la sección 2. Este protocolo guiará al usuario a través de la optimización del flujo de trabajo de manera automatizada. En concreto, el método identifica los parámetros que ofrecen el mejor rendimiento del autocodificador en términos de generación de características extraídas que separan bien los grupos de muestras. Los parámetros optimizados generados como salida incluyen el número de entidades que se van a utilizar para la selección de características (k_prot y k_met) y el número de neuronas en la capa latente del autocodificador (latente). Estos parámetros se pueden utilizar en el protocolo descrito en la sección 3 para generar el modelo.
- En la página de inicio de Jupyter en el navegador, haga clic en el bloc de notas M02 - DeepOmicsAE model optimization.ipynb para abrirlo en una nueva pestaña (Archivo complementario 2, paso 2.1).
- En la segunda celda del bloc de notas, escriba el nombre del archivo de entrada en lugar de M01_output_data.csv. La entrada de esta función son los datos de salida de la sección 1.
- En la quinta celda del cuaderno, especifique la posición de las columnas para cada tipo de datos de la siguiente manera: datos proteómicos (cols_X_prot), datos metabolómicos (cols_X_met), datos clínicos (cols_clin; incluye todos los datos clínicos), todos los datos de expresión molecular, incluidos los datos proteómicos y metabolómicos (cols_X_expr). Introduzca el índice de la primera columna para cada tipo de datos en lugar de col_start y el índice de la última columna en lugar de col_end; Por ejemplo, cols_prot = slice(0, 8817). Asegúrese de que los valores especificados en los objetos de sector correspondan al índice de la primera y la última columna correspondientes a cada tipo de datos, y utilice los comandos de la tercera y cuarta celda del cuaderno para explorar los datos y determinar las posiciones inicial y final de cada tipo de datos. Especifique el nombre de la columna que contiene la variable de destino en lugar de y_column_name como y_label (Archivo complementario 2, paso 2.2).
NOTA: Los valores de los índices especificados en cols_X_prot, cols_X_met, cols_clin y cols_X_expr serán diferentes de los utilizados en la sección 1 debido a la remodelación de la trama de datos que se produce durante el preprocesamiento de datos. - En la sexta celda del bloc de notas, especifique el número de rondas de optimización que se van a realizar asignando un valor a n_comb. Los tiempos de procesamiento son de aproximadamente 4-5 minutos para 10 rondas; 20 minutos para 50 rondas y 40 minutos para 100 rondas (Archivo Suplementario 2, Paso 2.3).
- Seleccionar celda | Ejecute todo desde la barra de menús de Jupyter.
NOTA: Las variables de salida kprot, kmet y latent se almacenarán y se podrá acceder a ellas desde los otros cuadernos, que se utilizarán para continuar con el flujo de trabajo analítico. El AE_optimization_plot.pdf de la gráfica se generará y guardará en la carpeta local (Figura 2).
3. Implementación del flujo de trabajo con parámetros optimizados a medida
NOTA: Realice este protocolo solo después de la optimización del método (sección 2). Si los usuarios deciden no realizar la optimización del método, vaya directamente a la sección 4. Este protocolo guiará al usuario a través de la generación de un modelo utilizando los parámetros optimizados a medida derivados de la sección 2. El autocodificador 1) generará un conjunto de características extraídas que recapitulan los datos originales y 2) identificará las características importantes que impulsan cada neurona en la capa latente, representando efectivamente módulos de señalización únicos. Los módulos de señalización se interpretarán utilizando el protocolo proporcionado en la sección 5.
- En la página de inicio de Jupyter en el navegador, haga clic en el cuaderno M03a - Implementación de DeepOmicsAE con parámetros optimizados personalizados.ipynb para abrirlo en una nueva pestaña (Archivo complementario 2, paso 3.1).
- En la segunda celda del bloc de notas, escriba el nombre del archivo de entrada en lugar de M01_output_data.csv. La entrada de esta función son los datos de salida de la sección 1.
- En la quinta celda del cuaderno, especifique la posición de las columnas para cada tipo de datos de la siguiente manera: datos proteómicos (cols_prot), datos metabolómicos (cols_met), datos clínicos (cols_clin; incluye todos los datos clínicos). Introduzca el índice de la primera columna para cada tipo de datos en lugar de col_start y el índice de la última columna en lugar de col_end; Por ejemplo: cols_prot = slice(0, 8817). Asegúrese de que los valores especificados en los objetos de sector corresponden a los índices de la primera y la última columna correspondientes a cada tipo de datos, y utilice los comandos de la tercera y cuarta celda del cuaderno para explorar los datos y determinar las posiciones inicial y final de cada tipo de datos. Especifique el nombre de la columna que contiene la variable de destino (por ejemplo, 0 o 1, correspondiente a sano o enfermo) en lugar de y_column_name como y_label.
NOTA: El valor de los índices especificados en cols_X_prot, cols_X_met, cols_clin y cols_X_expr será diferente de los utilizados en la sección 1 debido a la remodelación de la trama de datos que se produce durante el preprocesamiento de datos. - Seleccionar celda | Ejecute todo desde la barra de menús de Jupyter para generar y guardar los gráficos PCA_initial_data.pdf, PCA_extracted_features.pdf y distribution_important_feature_scores.pdf en la carpeta local (Figura 3 y Figura complementaria S1). Además, las listas de características importantes para cada módulo de señalización identificado se almacenarán en archivos de texto en la carpeta local, llamada module_n.txt, donde n se sustituirá por el número de módulo.
4. Implementación del flujo de trabajo con parámetros preestablecidos
- Consulte la sección 3 para obtener instrucciones detalladas sobre cómo ejecutar este método (Archivo complementario 2, Paso 4.1). La única diferencia entre estos dos protocolos es que los parámetros kprot, kmet y latent (en la séptima celda del cuaderno) se derivan matemáticamente en función de los resultados de la optimización realizada, como se muestra en la Figura 2.
NOTA: Si la sección 4 ofrece una separación deficiente de los grupos de muestra, lo que indica un rendimiento subóptimo del modelo, se recomienda ejecutar la optimización del modelo (sección 2) utilizando al menos 15 iteraciones y, si es posible, hasta 50.
5. Interpretación biológica con MetaboAnalyst
- Abra el navegador y navegue hasta el siguiente enlace para acceder a la funcionalidad de Análisis de Vías Conjuntas en el sitio web de MetaboAnalyst : https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml.
- Acceda a la carpeta donde se guardaron los archivos de salida del Método 3 o del Método 4 y abra los archivos de texto module_n.txt para cada módulo de señalización n generado por el Método 3 o por el Método 4.
- Localiza las proteínas en los archivos de texto y cópialas.
- Pegue la lista de proteínas en la ventana Genes/proteínas con cambios de plegado opcionales en la página web de MetaboAnalyst.
- Repita el paso anterior para los metabolitos y péguelos en la ventana Lista de compuestos con cambios de plegado opcionales en la misma página web.
- Seleccione el organismo y el tipo de ID apropiados, luego haga clic en Enviar en la parte inferior de la página (Archivo complementario 2, Paso 5.1).
NOTA: Asegúrese de que MetaboAnalyst reconozca los identificadores. Los identificadores reconocidos incluyen Entrez ID, símbolos genéticos oficiales y Uniprot ID para proteínas; nombre del compuesto, ID de HMDB e ID de KEGG para los metabolitos. Si los identificadores son distintos de estos tipos, es necesaria una conversión adecuada antes del análisis. - En la página siguiente, compruebe la asignación de ID antes de hacer clic en Continuar para comprobar que se reconocen los identificadores.
- En la página Configuración de parámetros , seleccione Rutas metabólicas (integradas) o Todas las vías (integradas) para visualizar, respectivamente, la contribución de la entrada solo a las vías metabólicas o a todas las vías de señalización (Archivo complementario 2, paso 5.2). En el panel de selección Algoritmo , elija Análisis de enriquecimiento: Prueba hipergeométrica, Medida de topología: Centralidad de grado y Método de integración: Combinar valores p (nivel de ruta). Haga clic en Enviar en la parte inferior de la página.
- La última página es la vista de resultados, que presenta los resultados del análisis de enriquecimiento. Las vías enriquecidas se trazan en función de su impacto e importancia, y la lista de vías también se proporciona en formato tabular.
Resultados
Para mostrar el protocolo, analizamos un conjunto de datos que comprendía el proteoma, el metaboloma y la información clínica derivada de los cerebros postmortem de 142 individuos sanos o diagnosticados con la enfermedad de Alzheimer.
Después de realizar la sección 1 del protocolo para preprocesar los datos, el conjunto de datos incluyó 6.497 proteínas, 443 metabolitos y tres características clínicas (sexo, edad de muerte y educación). La característica objetivo es el diagn?...
Discusión
La estructura del conjunto de datos es fundamental para el éxito del protocolo y debe comprobarse cuidadosamente. Los datos deben formatearse como se indica en la sección 1 del protocolo. La asignación correcta de las posiciones de las columnas también es fundamental para el éxito del método. Los datos proteómicos y metabolómicos se preprocesan de manera diferente y la selección de características se realiza por separado debido a la diferente naturaleza de los datos. Por lo tanto, es fundamental asignar correct...
Divulgaciones
El autor declara que no tiene conflictos de intereses.
Agradecimientos
Este trabajo fue financiado por el CA201402 de subvenciones de los NIH y el Premio al Académico Distinguido del Centro de Genómica de Vertebrados de Cornell (CVG). Los resultados publicados aquí se basan total o parcialmente en datos obtenidos del Portal de Conocimiento de AD (https://adknowledgeportal.org). Los datos del estudio se proporcionaron a través de la Asociación de Medicina Acelerada para la EA (U01AG046161 y U01AG061357) en base a muestras proporcionadas por el Centro de la Enfermedad de Alzheimer Rush, Centro Médico de la Universidad Rush, Chicago. La recopilación de datos fue financiada por subvenciones del NIA P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, el Departamento de Salud Pública de Illinois y el Instituto de Investigación Genómica Traslacional. El conjunto de datos metabolómicos se generó en Metabolon y fue preprocesado por el ADMC.
Materiales
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
Referencias
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados