
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
DeepOmicsAE : Représentation de modules de signalisation dans la maladie d’Alzheimer avec l’analyse de l’apprentissage profond de la protéomique, de la métabolomique et des données cliniques
Dans cet article
Résumé
DeepOmicsAE est un flux de travail centré sur l’application d’une méthode d’apprentissage profond (c’est-à-dire un auto-encodeur) pour réduire la dimensionnalité des données multi-omiques, fournissant une base pour les modèles prédictifs et les modules de signalisation représentant plusieurs couches de données omiques.
Résumé
De grands ensembles de données omiques sont de plus en plus disponibles pour la recherche sur la santé humaine. Cet article présente DeepOmicsAE, un flux de travail optimisé pour l’analyse d’ensembles de données multi-omiques, y compris la protéomique, la métabolomique et les données cliniques. Ce flux de travail utilise un type de réseau neuronal appelé auto-encodeur, pour extraire un ensemble concis de caractéristiques à partir des données d’entrée multi-omiques de grande dimension. De plus, le flux de travail fournit une méthode pour optimiser les paramètres clés nécessaires à la mise en œuvre de l’auto-encodeur. Pour présenter ce flux de travail, les données cliniques d’une cohorte de 142 personnes en bonne santé ou diagnostiquées avec la maladie d’Alzheimer ont été analysées, ainsi que le protéome et le métabolome de leurs échantillons de cerveau post-mortem. Les caractéristiques extraites de la couche latente de l’auto-encodeur retiennent les informations biologiques qui séparent les patients sains et malades. De plus, les caractéristiques individuelles extraites représentent des modules de signalisation moléculaire distincts, chacun interagissant de manière unique avec les caractéristiques cliniques des individus, fournissant un moyen d’intégrer la protéomique, la métabolomique et les données cliniques.
Introduction
Une proportion de plus en plus importante de la population vieillit et le fardeau des maladies liées à l’âge, telles que la neurodégénérescence, devrait fortement augmenter au cours des prochaines décennies1. La maladie d’Alzheimer est le type le plus courant de maladie neurodégénérative2. Les progrès dans la recherche d’un traitement ont été lents étant donné notre mauvaise compréhension des mécanismes moléculaires fondamentaux à l’origine de l’apparition et de la progression de la maladie. La majorité des informations sur la maladie d’Alzheimer sont obtenues post-mortem à partir de l’examen du tissu cérébral, ce qui a rendu difficile la distinction des causes et des conséquences3. Le projet ROSMAP (Religious Orders Study/Memory and Aging) est un effort ambitieux visant à mieux comprendre la neurodégénérescence, ce qui implique l’étude de milliers de personnes qui se sont engagées à subir des examens médicaux et psychologiques chaque année et à contribuer à la recherche après leur décès4. L’étude se concentre sur la transition du fonctionnement normal du cerveau à la maladie d’Alzheimer2. Dans le cadre du projet, des échantillons de cerveau post-mortem ont été analysés avec une pléthore d’approches omiques, notamment la génomique, l’épigénomique, la transcriptomique, la protéomique5 et la métabolomique.
Les technologies omiques qui offrent des lectures fonctionnelles des états cellulaires (c.-à-d. protéomique et métabolomique)6,7 sont essentielles à l’interprétation de la maladie 8,9,10,11,12, en raison de la relation directe entre l’abondance des protéines et des métabolites et les activités cellulaires. Les protéines sont les principaux exécuteurs des processus cellulaires, tandis que les métabolites sont les substrats et les produits des réactions biochimiques. L’analyse de données multi-omiques offre la possibilité de comprendre les relations complexes entre les données protéomiques et métabolomiques au lieu de les apprécier isolément. La multi-omique est une discipline qui étudie plusieurs couches de données biologiques de grande dimension, y compris des données moléculaires (séquence et mutations du génome, transcriptome, protéome, métabolome), des données d’imagerie clinique et des caractéristiques cliniques. En particulier, l’analyse de données multi-omiques vise à intégrer ces couches de données biologiques, à comprendre leur régulation réciproque et leur dynamique d’interaction, et à fournir une compréhension holistique de l’apparition et de la progression de la maladie. Cependant, les méthodes d’intégration des données multi-omiques en sont encore aux premiers stades de développement13.
Les auto-encodeurs, un type de réseau neuronal non supervisé14, sont un outil puissant pour l’intégration de données multi-omiques. Contrairement aux réseaux neuronaux supervisés, les auto-encodeurs ne font pas correspondre les échantillons à des valeurs cibles spécifiques (telles que sain ou malade), et ne sont pas utilisés pour prédire les résultats. L’une de leurs principales applications réside dans la réduction de la dimensionnalité. Cependant, les auto-encodeurs offrent plusieurs avantages par rapport aux méthodes de réduction de dimensionnalité plus simples telles que l’analyse en composantes principales (PCA), l’intégration de voisins stochastiques distribués en t (tSNE) ou l’approximation et la projection de variétés uniformes (UMAP). Contrairement à l’ACP, les auto-encodeurs peuvent capturer des relations non linéaires dans les données. Contrairement au tSNE et à l’UMAP, ils peuvent détecter des relations hiérarchiques et multimodales dans les données car ils reposent sur plusieurs couches d’unités de calcul contenant chacune des fonctions d’activation non linéaires. Par conséquent, ils représentent des modèles attrayants pour capturer la complexité des données multi-omiques. Enfin, alors que l’application principale de l’ACP, du tSNE et de l’UMAP est le clustering des données, les auto-encodeurs compressent les données d’entrée en caractéristiques extraites qui sont bien adaptées aux tâches prédictives en aval15,16.
En bref, les réseaux neuronaux comprennent plusieurs couches, chacune contenant plusieurs unités de calcul ou « neurones ». La première et la dernière couche sont appelées respectivement couches d’entrée et de sortie. Les auto-encodeurs sont des réseaux neuronaux avec une structure en sablier, composée d’une couche d’entrée, suivie d’une à trois couches cachées et d’une petite couche « latente » contenant généralement entre deux et six neurones. La première moitié de cette structure est connue sous le nom d’encodeur et est combinée à un décodeur reflétant l’encodeur. Le décodeur se termine par une couche de sortie contenant le même nombre de neurones que la couche d’entrée. Les auto-encodeurs font passer l’entrée à travers le goulot d’étranglement et la reconstruisent dans la couche de sortie, dans le but de générer une sortie qui reflète le plus fidèlement possible les informations d’origine. Ceci est réalisé en minimisant mathématiquement un paramètre appelé « perte de reconstruction ». L’entrée consiste en un ensemble de caractéristiques qui, dans l’application présentée ici, seront l’abondance des protéines et des métabolites et les caractéristiques cliniques (c’est-à-dire le sexe, l’éducation et l’âge au décès). La couche latente contient une représentation compressée et riche en informations de l’entrée, qui peut être utilisée pour des applications ultérieures telles que les modèles prédictifs17,18.
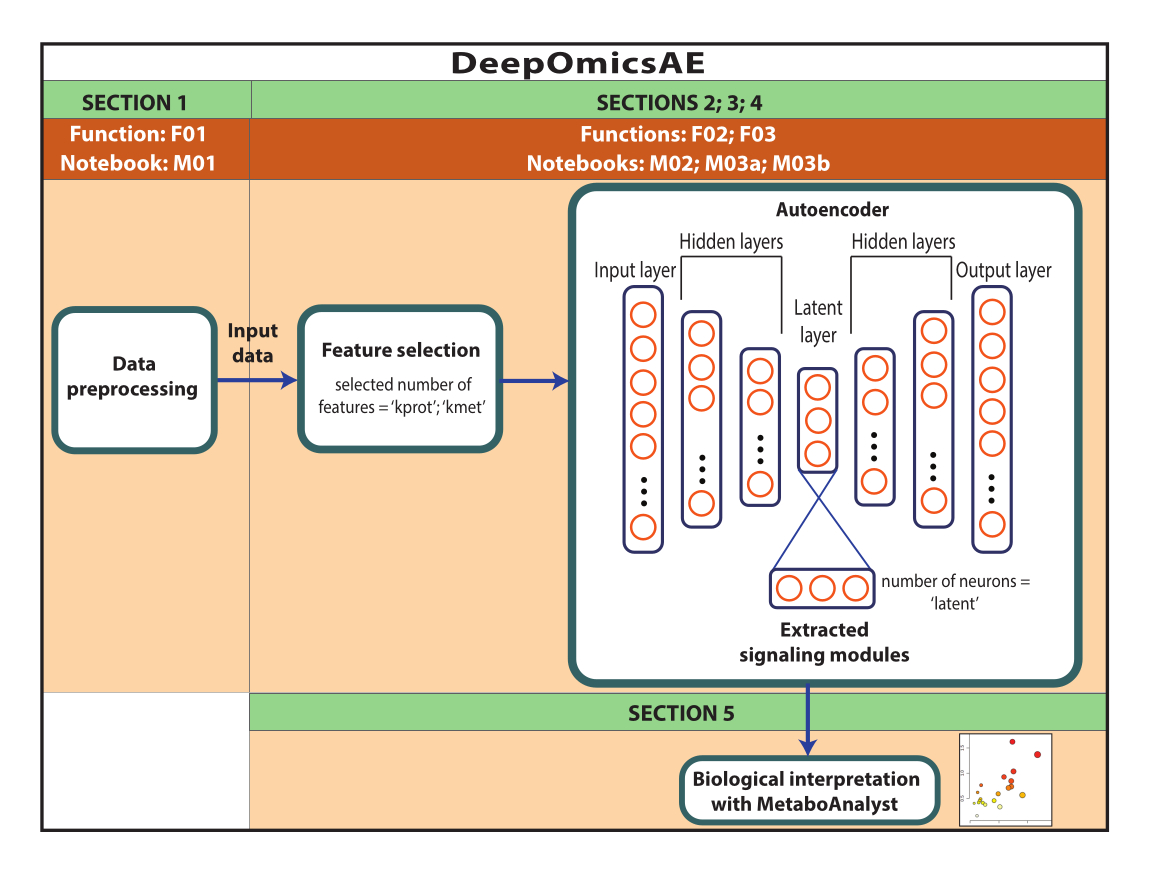
Ce protocole présente un flux de travail, DeepOmicsAE, qui implique : 1) le prétraitement des données protéomiques, métabolomiques et cliniques (c’est-à-dire la normalisation, la mise à l’échelle, la suppression des valeurs aberrantes) pour obtenir des données avec une échelle cohérente pour l’analyse de l’apprentissage automatique ; 2) sélectionner les caractéristiques d’entrée de l’auto-encodeur appropriées, car la surcharge de caractéristiques peut masquer les modèles de maladie pertinents ; 3) l’optimisation et l’entraînement de l’auto-encodeur, y compris la détermination du nombre optimal de protéines et de métabolites à sélectionner, et de neurones pour la couche latente ; 4) extraire les caractéristiques de la couche latente ; et 5) l’utilisation des caractéristiques extraites pour l’interprétation biologique en identifiant les modules de signalisation moléculaire et leur relation avec les caractéristiques cliniques.
Ce protocole se veut simple et applicable par des biologistes ayant une expérience informatique limitée qui ont une compréhension de base de la programmation avec Python. Le protocole se concentre sur l’analyse des données multi-omiques, y compris la protéomique, la métabolomique et les caractéristiques cliniques, mais son utilisation peut être étendue à d’autres types de données d’expression moléculaire, y compris la transcriptomique. Une nouvelle application importante introduite par ce protocole est la cartographie des scores d’importance des caractéristiques originales sur les neurones individuels de la couche latente. En conséquence, chaque neurone de la couche latente représente un module de signalisation, détaillant les interactions entre des altérations moléculaires spécifiques et les caractéristiques cliniques des patients. L’interprétation biologique des modules de signalisation moléculaire est obtenue à l’aide de MetaboAnalyst, un outil accessible au public qui intègre des données sur les gènes/protéines et les métabolites pour en déduire des voies de signalisation métaboliques et cellulaires enrichies17.
Protocole
REMARQUE : Les données utilisées ici sont des données ROSMAP téléchargées à partir du portail de connaissances AD. Le consentement éclairé n’est pas nécessaire pour télécharger et réutiliser les données. Le protocole présenté ici utilise l’apprentissage profond pour analyser les données multi-omiques et identifier les modules de signalisation qui distinguent des patients ou des groupes d’échantillons spécifiques en fonction, par exemple, de leur diagnostic. Le protocole fournit également un petit ensemble de caractéristiques extraites qui résument les données originales à grande échelle et peuvent être utilisées pour une analyse plus approfondie, comme l’entraînement d’un modèle prédictif à l’aide d’algorithmes d’apprentissage automatique (Figure 1). Reportez-vous au fichier supplémentaire 1 et à la table des matériaux pour plus d’informations sur l’accès au code et la configuration de l’environnement de calcul avant d’exécuter le protocole. Les méthodes doivent être effectuées dans l’ordre spécifié ci-dessous.

Figure 1 : Schéma du flux de travail DeepOmicsAE. Représentation schématique du flux de travail pour l’analyse des données multi-omiques à l’aide du flux de travail. Dans la représentation de l’auto-encodeur, les rectangles représentent les couches du réseau neuronal et les cercles représentent les neurones à l’intérieur des couches. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
1. Prétraitement des données
REMARQUE : L’objectif de cette section est de prétraiter les données, y compris le traitement des données manquantes ; normalisation et mise à l’échelle de l’expression protéomique, métabolomique et des données cliniques ; et supprimer les valeurs aberrantes. Le protocole est conçu pour un ensemble de données qui comprend des données protéomiques exprimées sous forme de log2(ratio) ; données métabolomiques exprimées en changement de pli ; et les caractéristiques cliniques, y compris les caractéristiques continues et catégorielles. Les patients ou les échantillons doivent être regroupés en fonction du diagnostic ou d’autres paramètres similaires. Les échantillons ou les patients doivent être répartis sur les lignes et les caractéristiques sur les colonnes.
- Pour démarrer une nouvelle instance de Jupyter Notebook dans le navigateur, ouvrez une nouvelle fenêtre de terminal, tapez ce qui suit et appuyez sur Entrée.
Carnet Jupyter - Dans la page d’accueil Jupyter du navigateur, cliquez sur le notebook M01 - expression data pre-processing.ipynb pour l’ouvrir dans un nouvel onglet (Fichier supplémentaire 2, étape 1.1).
- Dans la deuxième cellule du bloc-notes, tapez le nom du fichier de jeu de données à la place de your_dataset_name.csv.
- Dans la dernière cellule du bloc-notes, tapez le nom souhaité du fichier de données de sortie à la place de M01_output_data.csv.
- Dans la cinquième cellule du bloc-notes, spécifiez la position des colonnes pour chaque type de données comme suit : données protéomiques (cols_prot), données métabolomiques (cols_met), données cliniques continues (p. ex., âge) (cols_clin_con), données cliniques binaires (p. ex., sexe) (cols_clin_bin). Entrez le premier index de colonne pour chaque type de données à la place de col_start et l’index des dernières colonnes à la place de col_end ; Par exemple : cols_prot = tranche(0, 8817). Assurez-vous que les valeurs spécifiées dans les objets de tranche correspondent aux index de la première et de la dernière colonne correspondant à chaque type de données. Utilisez la commande dans la quatrième cellule du même bloc-notes (df.iloc[ :, :]) pour déterminer la position de début et de fin de chaque type de données (Fichier supplémentaire 2, étape 1.2).
- Sélectionner une cellule | Exécutez tout à partir de la barre de menus dans Jupyter pour créer le fichier de données de sortie dans le dossier spécifié (Fichier supplémentaire 2, étape 1.3).
REMARQUE : Ces données seront utilisées comme données d’entrée pour les protocoles décrits aux sections 2, 3 ou 4.
2. Optimisation personnalisée du flux de travail (facultatif)
REMARQUE : La section 2 est facultative car elle est gourmande en ressources informatiques. Les utilisateurs doivent passer directement à la section 4 s’ils décident de ne pas effectuer la section 2. Ce protocole guidera l’utilisateur dans l’optimisation du flux de travail de manière automatisée. Plus précisément, la méthode identifie les paramètres qui offrent les meilleures performances de l’auto-encodeur en termes de génération de caractéristiques extraites qui séparent bien les groupes d’échantillons. Les paramètres optimisés générés en sortie incluent le nombre de caractéristiques à utiliser pour la sélection des caractéristiques (k_prot et k_met) et le nombre de neurones dans la couche latente de l’auto-encodeur (latente). Ces paramètres peuvent ensuite être utilisés dans le protocole décrit à la section 3 pour générer le modèle.
- Sur la page d’accueil de Jupyter dans le navigateur, cliquez sur le notebook M02 - DeepOmicsAE model optimization.ipynb pour l’ouvrir dans un nouvel onglet (Fichier supplémentaire 2, étape 2.1).
- Dans la deuxième cellule du bloc-notes, tapez le nom du fichier d’entrée à la place de M01_output_data.csv. L’entrée de cette fonction est les données de sortie de la section 1.
- Dans la cinquième cellule du carnet, spécifiez la position des colonnes pour chaque type de données comme suit : données protéomiques (cols_X_prot), données métabolomiques (cols_X_met), données cliniques (cols_clin ; comprend toutes les données cliniques), toutes les données d’expression moléculaire, y compris les données protéomiques et métabolomiques (cols_X_expr). Entrez le premier index de colonne pour chaque type de données à la place de col_start et l’index des dernières colonnes à la place de col_end ; par exemple, cols_prot = slice(0, 8817). Assurez-vous que les valeurs spécifiées dans les objets de tranche correspondent à l’index de la première et de la dernière colonne correspondant à chaque type de données, et utilisez les commandes dans les troisième et quatrième cellules du bloc-notes pour explorer les données et déterminer les positions de début et de fin pour chaque type de données. Spécifiez le nom de la colonne contenant la variable cible à la place de y_column_name comme y_label (Fichier supplémentaire 2, étape 2.2).
REMARQUE : Les valeurs des index spécifiés dans cols_X_prot, cols_X_met, cols_clin et cols_X_expr seront différentes de celles utilisées dans la section 1 en raison du remodelage de la trame de données qui se produit pendant le prétraitement des données. - Dans la sixième cellule du bloc-notes, spécifiez le nombre de cycles d’optimisation à effectuer en attribuant une valeur à n_comb. Les temps de traitement sont d’environ 4 à 5 minutes pour 10 tours ; 20 min pour 50 rounds et 40 min pour 100 rounds (Fichier supplémentaire 2, étape 2.3).
- Sélectionner une cellule | Exécutez tout à partir de la barre de menus dans Jupyter.
REMARQUE : Les variables de sortie kprot, kmet et latent seront stockées et accessibles à partir des autres blocs-notes, qui seront utilisés pour poursuivre le flux de travail analytique. Le AE_optimization_plot.pdf de tracé sera généré et enregistré dans le dossier local (Figure 2).
3. Mise en œuvre du flux de travail avec des paramètres optimisés sur mesure
REMARQUE : N’effectuez ce protocole qu’après l’optimisation de la méthode (section 2). Si les utilisateurs choisissent de ne pas effectuer d’optimisation de méthode, passez directement à la section 4. Ce protocole guidera l’utilisateur dans la génération d’un modèle à l’aide des paramètres optimisés sur mesure dérivés de la section 2. L’auto-encodeur va 1) générer un ensemble de caractéristiques extraites qui récapitulent les données originales et 2) identifier les caractéristiques importantes pilotant chaque neurone de la couche latente, représentant efficacement des modules de signalisation uniques. Les modules de signalisation seront interprétés à l’aide du protocole fourni à la section 5.
- Sur la page d’accueil Jupyter du navigateur, cliquez sur le notebook M03a - Implémentation DeepOmicsAE avec des paramètres optimisés personnalisés.ipynb pour l’ouvrir dans un nouvel onglet (Fichier supplémentaire 2, étape 3.1).
- Dans la deuxième cellule du bloc-notes, tapez le nom du fichier d’entrée à la place de M01_output_data.csv. L’entrée de cette fonction est les données de sortie de la section 1.
- Dans la cinquième cellule du bloc-notes, spécifiez la position des colonnes pour chaque type de données comme suit : données protéomiques (cols_prot), données métabolomiques (cols_met), données cliniques (cols_clin ; comprend toutes les données cliniques). Entrez le premier index de colonne pour chaque type de données à la place de col_start et l’index des dernières colonnes à la place de col_end ; Par exemple : cols_prot = tranche(0, 8817). Assurez-vous que les valeurs spécifiées dans les objets de tranche correspondent aux index de la première et de la dernière colonne correspondant à chaque type de données, et utilisez les commandes des troisième et quatrième cellules du notebook pour explorer les données et déterminer les positions de début et de fin de chaque type de données. Spécifiez le nom de la colonne contenant la variable cible (par exemple, 0 ou 1, correspondant à sain ou malade) à la place de y_column_name comme y_label.
REMARQUE : La valeur des index spécifiés dans cols_X_prot, cols_X_met, cols_clin et cols_X_expr sera différente de celle utilisée dans la section 1 en raison du remodelage de la trame de données qui se produit pendant le prétraitement des données. - Sélectionner une cellule | Exécutez tout à partir de la barre de menus dans Jupyter pour générer et enregistrer les tracés PCA_initial_data.pdf, PCA_extracted_features.pdf et distribution_important_feature_scores.pdf dans le dossier local (Figure 3 et Figure S1 supplémentaire). De plus, les listes des caractéristiques importantes pour chaque module de signalisation identifié seront stockées dans des fichiers texte dans le dossier local, nommé module_n.txt, où n sera remplacé par le numéro du module.
4. Mise en œuvre du flux de travail avec des paramètres prédéfinis
- Reportez-vous à la section 3 pour obtenir des instructions détaillées sur la façon d’exécuter cette méthode (Fichier supplémentaire 2, étape 4.1). La seule différence entre ces deux protocoles est que les paramètres kprot, kmet et latent (dans la septième cellule du carnet) sont dérivés mathématiquement sur la base des résultats de l’optimisation effectuée comme le montre la figure 2.
REMARQUE : Si la section 4 fournit une mauvaise séparation des groupes d’échantillons, indiquant des performances de modèle sous-optimales, il est recommandé d’exécuter l’optimisation du modèle (section 2) en utilisant au moins 15 itérations, et si possible, jusqu’à 50.
5. Interprétation biologique à l’aide de MetaboAnalyst
- Ouvrez le navigateur et accédez au lien ci-dessous pour accéder à la fonctionnalité d’analyse des voies articulaires sur le site Web de MetaboAnalyst : https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml.
- Accédez au dossier dans lequel les fichiers de sortie de la méthode 3 ou de la méthode 4 ont été enregistrés et ouvrez les fichiers texte module_n.txt pour chaque module de signalisation n généré par la méthode 3 ou par la méthode 4.
- Localisez les protéines dans les fichiers texte et copiez-les.
- Collez la liste des protéines dans la fenêtre Gènes/protéines avec des changements de pliage facultatifs dans la page Web MetaboAnalyst.
- Répétez l’étape ci-dessus pour les métabolites et collez-les dans la liste des composés de la fenêtre avec des changements de pliage facultatifs sur la même page Web.
- Sélectionnez l’organisme et le type d’identification appropriés, puis cliquez sur Soumettre au bas de la page (Fichier supplémentaire 2, étape 5.1).
REMARQUE : Assurez-vous que les identificateurs sont reconnus par MetaboAnalyst. Les identifiants reconnus comprennent l’identifiant Entre, les symboles officiels des gènes et l’identifiant Uniprot pour les protéines ; nom du composé, identifiant HMDB et identifiant KEGG pour les métabolites. Si les identificateurs sont d’autres types que ces types, une conversion appropriée est nécessaire avant l’analyse. - Sur la page suivante, vérifiez le mappage d’ID avant de cliquer sur Continuer pour vérifier que les identificateurs sont reconnus.
- Dans la page Paramétrage , sélectionnez Voies métaboliques (intégrées) ou Toutes les voies (intégrées) pour visualiser respectivement la contribution de l’entrée aux voies métaboliques uniquement ou à toutes les voies de signalisation (Fichier supplémentaire 2, étape 5.2). Dans le panneau Sélection de l’algorithme , choisissez Analyse d’enrichissement : Test hypergéométrique, Mesure topologique : Centralité de degré et Méthode d’intégration : Combiner p valeurs (au niveau de la voie). Cliquez sur Soumettre en bas de la page.
- La dernière page est la vue des résultats, qui présente les résultats de l’analyse d’enrichissement. Les parcours enrichis sont tracés en fonction de leur impact et de leur importance, et la liste des parcours est également fournie sous forme de tableau.
Résultats
Pour présenter le protocole, nous avons analysé un ensemble de données comprenant le protéome, le métabolome et les informations cliniques dérivées des cerveaux post-mortem de 142 personnes en bonne santé ou diagnostiquées avec la maladie d’Alzheimer.
Après avoir effectué la section 1 du protocole pour prétraiter les données, l’ensemble de données comprenait 6 497 protéines, 443 métabolites et trois caractéristiques cliniques (sexe, âge au décès et éducation). La carac...
Discussion
La structure de l’ensemble de données est essentielle au succès du protocole et doit être soigneusement vérifiée. Les données doivent être formatées comme indiqué dans la section 1 du protocole. L’affectation correcte des positions des colonnes est également essentielle au succès de la méthode. Les données protéomiques et métabolomiques sont prétraitées différemment et la sélection des caractéristiques est effectuée séparément en raison de la nature différente des données. Par conséquent, il...
Déclarations de divulgation
L’auteur déclare qu’ils n’ont aucun conflit d’intérêts.
Remerciements
Ce travail a été soutenu par des subventions des NIH CA201402 et le prix Distinguished Scholar du Cornell Center for Vertebrate Genomics (CVG). Les résultats publiés ici sont en tout ou en partie basés sur les données obtenues à partir du portail de connaissances AD (https://adknowledgeportal.org). Les données de l’étude ont été fournies par l’intermédiaire de l’Accelerating Medicine Partnership for AD (U01AG046161 et U01AG061357) sur la base d’échantillons fournis par le Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. La collecte de données a été financée par des subventions NIA P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, le département de la santé publique de l’Illinois et l’Institut de recherche en génomique translationnelle. L’ensemble de données métabolomiques a été généré à Metabolon et prétraité par l’ADMC.
matériels
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
Références
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.