
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
DeepOmicsAE: ייצוג מודולי איתות במחלת אלצהיימר עם ניתוח למידה עמוקה של פרוטאומיקה, מטבולומיקה ונתונים קליניים
In This Article
Summary
DeepOmicsAE היא זרימת עבודה המתמקדת ביישום שיטת למידה עמוקה (כלומר, מקודד אוטומטי) כדי להפחית את המימדיות של נתוני multi-omics, מתן בסיס למודלים חיזוי ומודולי איתות המייצגים שכבות מרובות של נתוני omics.
Abstract
מערכי נתונים גדולים של omics הופכים זמינים יותר ויותר למחקר על בריאות האדם. מאמר זה מציג את DeepOmicsAE, זרימת עבודה המותאמת לניתוח מערכי נתונים רב-אומיים, כולל פרוטאומיקה, מטבולומיקה ונתונים קליניים. זרימת עבודה זו משתמשת בסוג של רשת עצבית הנקראת autoencoder, כדי לחלץ קבוצה תמציתית של תכונות מנתוני הקלט הרב-מימדיים הגבוהים. יתר על כן, זרימת העבודה מספקת שיטה לייעול הפרמטרים העיקריים הדרושים ליישום המקודד האוטומטי. כדי להציג את זרימת העבודה הזו, נותחו נתונים קליניים מעוקבה של 142 אנשים שהיו בריאים או אובחנו עם מחלת אלצהיימר, יחד עם הפרוטאום והמטבוליזם של דגימות המוח שלהם לאחר המוות. התכונות המופקות מהשכבה הסמויה של המקודד האוטומטי שומרות על המידע הביולוגי המפריד בין חולים בריאים לחולים. בנוסף, התכונות הבודדות שחולצו מייצגות מודולי איתות מולקולרי נפרדים, שכל אחד מהם מקיים אינטראקציה ייחודית עם התכונות הקליניות של הפרטים, ומספק אמצעי לשילוב הפרוטאומיקה, המטבולומיקה והנתונים הקליניים.
Introduction
חלק הולך וגדל מהאוכלוסייה מזדקן והנטל של מחלות הקשורות לגיל, כגון ניוון עצבי, צפוי לעלות בחדות בעשורים הקרובים1. מחלת אלצהיימר היא הסוג הנפוץ ביותר של מחלות נוירודגנרטיביות2. ההתקדמות במציאת טיפול הייתה איטית בהתחשב בהבנה הלקויה שלנו של המנגנונים המולקולריים הבסיסיים המניעים את הופעת המחלה והתקדמותה. רוב המידע על מחלת האלצהיימר מתקבל לאחר המוות מבדיקת רקמת המוח, מה שהפך את ההבחנה בין סיבות ותוצאות למשימה קשה3. פרויקט מחקר המסדרים הדתיים / זיכרון והזדקנות (ROSMAP) הוא מאמץ שאפתני להשיג הבנה רחבה יותר של ניוון עצבי, הכולל מחקר של אלפי אנשים שהתחייבו לעבור בדיקות רפואיות ופסיכולוגיות מדי שנה ולתרום את מוחם למחקר לאחר מותם4. המחקר מתמקד במעבר מתפקוד תקין של המוח למחלת אלצהיימר2. במסגרת הפרויקט, דגימות מוח לאחר המוות נותחו עם שפע של גישות אומיקס, כולל גנומיקה, אפיגנומיקה, טרנסקריפטומיקס, פרוטאומיקה5 ומטבולומיקה.
טכנולוגיות Omics המציעות קריאות פונקציונליות של מצבים תאיים (כלומר, פרוטאומיקה ומטבולומיקה)6,7 הן המפתח לפענוח מחלה 8,9,10,11,12, בשל הקשר הישיר בין חלבון ושפע מטבוליטים ופעילויות תאיות. חלבונים הם המבצעים העיקריים של תהליכים תאיים, בעוד מטבוליטים הם המצע והתוצרים של תגובות ביוכימיות. ניתוח נתונים מולטי-אומיקה מציע את האפשרות להבין את היחסים המורכבים בין נתונים פרוטאומיים ומטאבולומיים במקום להעריך אותם בנפרד. מולטי-אומיקס היא דיסציפלינה החוקרת שכבות מרובות של נתונים ביולוגיים בממדים גבוהים, כולל נתונים מולקולריים (רצף גנום ומוטציות, תעתוק, פרוטום, מטבוליזם), נתוני הדמיה קלינית ותכונות קליניות. בפרט, ניתוח נתונים מולטי-אומיקס נועד לשלב שכבות כאלה של נתונים ביולוגיים, להבין את הוויסות ההדדי שלהם ואת דינמיקת האינטראקציה, ולספק הבנה הוליסטית של התפרצות המחלה והתקדמותה. עם זאת, שיטות לשילוב נתונים מולטי-אומיים נותרו בשלבים המוקדמים של הפיתוח13.
Autoencoders, סוג של רשת עצבית14 ללא פיקוח, הם כלי רב עוצמה לשילוב נתונים multi-omics. שלא כמו רשתות עצביות מפוקחות, מקודדים אוטומטיים אינם ממפים דגימות לערכי מטרה ספציפיים (כגון בריאים או חולים), והם אינם משמשים לחיזוי תוצאות. אחד היישומים העיקריים שלהם טמון בהפחתת ממדיות. עם זאת, מקודדים אוטומטיים מציעים מספר יתרונות על פני שיטות פשוטות יותר להפחתת ממדיות, כגון ניתוח רכיבים עיקריים (PCA), הטבעה של שכן סטוכסטי מבוזר t (tSNE), או קירוב סעפת אחידה והקרנה (UMAP). שלא כמו PCA, מקודדים אוטומטיים יכולים ללכוד קשרים לא ליניאריים בתוך הנתונים. שלא כמו tSNE ו- UMAP, הם יכולים לזהות יחסים היררכיים ורב-מודאליים בתוך הנתונים מכיוון שהם מסתמכים על שכבות מרובות של יחידות חישוביות שכל אחת מהן מכילה פונקציות הפעלה לא ליניאריות. לכן, הם מייצגים מודלים אטרקטיביים כדי ללכוד את המורכבות של נתונים multi-omics. לבסוף, בעוד היישום העיקרי של PCA, tSNE ו- UMAP הוא זה של קיבוץ הנתונים, מקודדים אוטומטיים דוחסים את נתוני הקלט לתכונות מחולצות המתאימות היטב למשימות חיזוי במורד הזרם15,16.
בקצרה, רשתות עצביות מורכבות מכמה שכבות, שכל אחת מהן מכילה יחידות חישוביות מרובות או "נוירונים". השכבה הראשונה והשכבה האחרונה נקראות שכבות הקלט והפלט, בהתאמה. מקודדים אוטומטיים הם רשתות עצביות בעלות מבנה שעון חול, המורכב משכבת קלט, ואחריה שכבה אחת עד שלוש שכבות נסתרות ושכבה "סמויה" קטנה המכילה בדרך כלל בין שניים לשישה נוירונים. חציו הראשון של מבנה זה מכונה מקודד והוא משולב עם מפענח המשקף את המקודד. המפענח מסתיים בשכבת פלט המכילה את אותו מספר תאי עצב כמו שכבת הקלט. מקודדים אוטומטיים לוקחים את הקלט דרך צוואר הבקבוק ומשחזרים אותו בשכבת הפלט, במטרה לייצר פלט המשקף את המידע המקורי קרוב ככל האפשר. זה מושג על ידי מזעור מתמטי של פרמטר המכונה "אובדן שחזור". הקלט מורכב מסט של תכונות, אשר ביישום המוצג כאן יהיו שפע חלבונים ומטבוליטים, ומאפיינים קליניים (כלומר, מין, השכלה וגיל בעת המוות). השכבה הסמויה מכילה ייצוג דחוס ועשיר במידע של הקלט, שניתן להשתמש בו ליישומים הבאים כגון מודלים לחיזוי17,18.
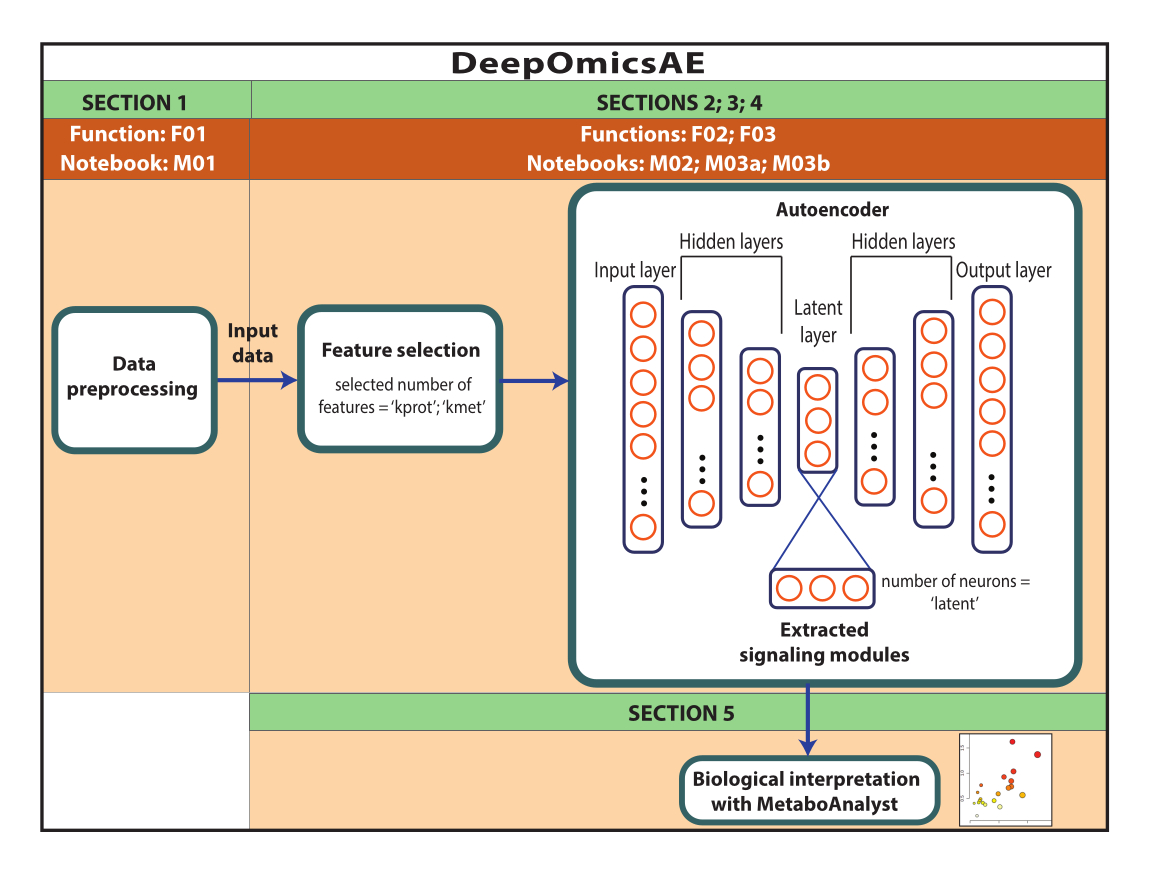
פרוטוקול זה מציג זרימת עבודה, DeepOmicsAE, הכוללת: 1) עיבוד מקדים של פרוטאומיקה, מטבולומיקה ונתונים קליניים (כלומר, נורמליזציה, קנה מידה, הסרה חריגה) כדי להשיג נתונים בקנה מידה עקבי לניתוח למידת מכונה; 2) בחירת תכונות קלט מקודד אוטומטי מתאימות, שכן עומס יתר על תכונות עלול לטשטש דפוסי מחלה רלוונטיים; 3) אופטימיזציה ואימון של המקודד האוטומטי, כולל קביעת המספר האופטימלי של חלבונים ומטבוליטים לבחירה, ושל נוירונים לשכבה הסמויה; 4) חילוץ תכונות מהשכבה הסמויה; ו-5) ניצול התכונות המחולצות לפרשנות ביולוגית על ידי זיהוי מודולי איתות מולקולרי והקשר שלהם עם תכונות קליניות.
פרוטוקול זה נועד להיות פשוט וישים על ידי ביולוגים עם ניסיון חישובי מוגבל שיש להם הבנה בסיסית של תכנות עם Python. הפרוטוקול מתמקד בניתוח נתונים מולטי-אומיקס, כולל פרוטאומיקה, מטבולומיקה ותכונות קליניות, אך ניתן להרחיב את השימוש בו לסוגים אחרים של נתוני ביטוי מולקולרי, כולל תעתוק. יישום חדשני חשוב אחד שהוצג על ידי פרוטוקול זה הוא מיפוי ציוני החשיבות של תכונות מקוריות על נוירונים בודדים בשכבה הסמויה. כתוצאה מכך, כל נוירון בשכבה הסמויה מייצג מודול איתות, המפרט את האינטראקציות בין שינויים מולקולריים ספציפיים לבין המאפיינים הקליניים של החולים. הפרשנות הביולוגית של מודולי האיתות המולקולרי מתקבלת באמצעות MetaboAnalyst, כלי זמין לציבור המשלב נתוני גנים/חלבונים ומטבוליטים כדי להפיק מסלולי איתות מטבוליים ותאים מועשרים17.
Protocol
הערה: הנתונים ששימשו כאן היו נתוני ROSMAP שהורדו מפורטל הידע של AD. אין צורך בהסכמה מדעת כדי להוריד את הנתונים ולעשות בהם שימוש חוזר. הפרוטוקול המוצג כאן משתמש בלמידה עמוקה כדי לנתח נתונים מולטי-אומיים ולזהות מודולי איתות המבחינים בין מטופלים ספציפיים או קבוצות מדגם המבוססות, למשל, על האבחנה שלהם. הפרוטוקול גם מספק קבוצה קטנה של תכונות שחולצו שמסכמות את הנתונים המקוריים בקנה מידה גדול, וניתן להשתמש בהן לניתוח נוסף, כגון אימון מודל חיזוי באמצעות אלגוריתמים של למידת מכונה (איור 1). עיין בקובץ משלים 1 ובטבלת החומרים לקבלת מידע אודות גישה לקוד והגדרת הסביבה החישובית לפני ביצוע הפרוטוקול. יש לבצע את השיטות לפי הסדר המפורט להלן.

איור 1: סכמה של זרימת העבודה של DeepOmicsAE. ייצוג סכמטי של זרימת העבודה לניתוח נתוני multi-omics באמצעות זרימת העבודה. בתיאור המקודד האוטומטי, מלבנים מייצגים שכבות של הרשת העצבית ומעגלים מייצגים נוירונים בתוך שכבות. אנא לחץ כאן כדי להציג גרסה גדולה יותר של איור זה.
{kind=link}
1. עיבוד מקדים של נתונים
הערה: מטרת סעיף זה היא לעבד מראש את הנתונים, כולל טיפול בנתונים חסרים; נרמול וקנה מידה של נתונים פרוטאומיים, מטבוליים וקליניים; והסרת חריגים. הפרוטוקול מיועד למערך נתונים הכולל נתוני פרוטאומיקה המבוטאים כיומן2(יחס); נתוני מטבולומיקה המתבטאים בשינוי קיפול; ומאפיינים קליניים הכוללים מאפיינים רציפים וקטגוריים. יש לקבץ את החולים או הדגימות על סמך אבחנה או פרמטרים דומים אחרים. דגימות או מטופלים צריכים להיות לאורך השורות והתכונות לאורך העמודות.
- כדי להתחיל מופע חדש של Jupyter Notebook בדפדפן, פתח חלון מסוף חדש, הקלד את הטקסט הבא והקש Enter.
מחברת יופיטר - בדף הבית של Jupyter בדפדפן, לחץ על המחברת M01 - expression data pre-processing.ipynb כדי לפתוח אותה בכרטיסייה חדשה (קובץ משלים 2, שלב 1.1).
- בתא השני של המחברת, הקלד את שם קובץ ערכת הנתונים במקום your_dataset_name.csv.
- בתא האחרון של המחברת, הקלד את השם הרצוי של קובץ נתוני הפלט במקום M01_output_data.csv.
- בתא החמישי של המחברת, ציין את מיקום העמודות עבור כל סוג נתונים באופן הבא: נתוני פרוטאומיקה (cols_prot), נתוני מטבולומיקה (cols_met), נתונים קליניים רציפים (לדוגמה, גיל) (cols_clin_con), נתונים קליניים בינאריים (לדוגמה, מין) (cols_clin_bin). הזן את אינדקס העמודות הראשון עבור כל סוג נתונים במקום col_start ואת אינדקס העמודות האחרונות במקום col_end; לדוגמה: cols_prot = פרוסה(0, 8817). ודאו שהערכים שצוינו בעצמי הפרוסה תואמים לאינדקסים של העמודה הראשונה והאחרונה המתאימים לכל סוג נתונים. השתמש בפקודה בתא הרביעי של אותה מחברת (df.iloc[:, :]) כדי לקבוע את מיקום ההתחלה והסיום עבור כל סוג נתונים (קובץ משלים 2, שלב 1.2).
- בחר תא | הפעל הכל משורת התפריטים ב- Jupyter כדי ליצור את קובץ נתוני הפלט בתיקייה שצוינה (קובץ משלים 2, שלב 1.3).
הערה: נתונים אלה ישמשו כקלט עבור הפרוטוקולים המתוארים בסעיפים 2, 3 או 4.
2. אופטימיזציה מותאמת אישית של זרימת העבודה (אופציונלי)
הערה: סעיף 2 הוא אופציונלי מכיוון שהוא דורש צורך רב במחשב. משתמשים צריכים לדלג ישירות לסעיף 4 אם הם מחליטים לא לבצע את סעיף 2. פרוטוקול זה ינחה את המשתמש דרך אופטימיזציה של זרימת העבודה באופן אוטומטי. באופן ספציפי, השיטה מזהה את הפרמטרים המספקים את הביצועים הטובים ביותר של המקודד האוטומטי במונחים של יצירת תכונות שחולצו שמפרידות היטב בין קבוצות המדגם. הפרמטרים הממוטבים הנוצרים כפלט כוללים את מספר התכונות לשימוש לבחירת תכונות (k_prot ו- k_met) ואת מספר תאי העצב בשכבה הסמויה של המקודד האוטומטי (סמוי). לאחר מכן ניתן להשתמש בפרמטרים אלה בפרוטוקול המתואר בסעיף 3 כדי ליצור את המודל.
- בדף הבית של Jupyter בדפדפן, לחץ על המחברת M02 - DeepOmicsAE model optimization.ipynb כדי לפתוח אותה בכרטיסייה חדשה (קובץ משלים 2, שלב 2.1).
- בתא השני של המחברת, הקלד את שם קובץ הקלט במקום M01_output_data.csv. הקלט לפונקציה זו הוא נתוני הפלט מסעיף 1.
- בתא החמישי של המחברת, ציין את מיקום העמודות עבור כל סוג נתונים באופן הבא: נתוני פרוטאומיקה (cols_X_prot), נתוני מטבולומיקה (cols_X_met), נתונים קליניים (cols_clin; כולל כל הנתונים הקליניים), כל נתוני הביטוי המולקולרי, כולל נתוני פרוטאומיקה ומטבולומיקה (cols_X_expr). הזן את אינדקס העמודות הראשון עבור כל סוג נתונים במקום col_start ואת אינדקס העמודות האחרונות במקום col_end; לדוגמה, cols_prot = פרוסה(0, 8817). ודא שהערכים שצוינו באובייקטי הפרוסה תואמים לאינדקס העמודות הראשונה והאחרונה המתאים לכל סוג נתונים, והשתמש בפקודות בתא השלישי והרביעי של המחברת כדי לחקור את הנתונים ולקבוע את מיקום ההתחלה והסיום של כל סוג נתונים. ציין את שם העמודה הכוללת את משתנה היעד במקום y_column_name כ- y_label (קובץ משלים 2, שלב 2.2).
הערה: הערכים של האינדקסים שצוינו ב- cols_X_prot, cols_X_met, cols_clin ו- cols_X_expr יהיו שונים מאלה המשמשים בסעיף 1 עקב העיצוב מחדש של מסגרת הנתונים המתרחש במהלך עיבוד מקדים של נתונים. - בתא השישי של המחברת, ציין כמה סבבי מיטוב יש לבצע על-ידי הקצאת ערך ל - n_comb. זמני העיבוד הם כ 4-5 דקות במשך 10 סיבובים; 20 דקות ל-50 סיבובים, ו-40 דקות ל-100 סיבובים (קובץ משלים 2, שלב 2.3).
- בחר תא | הפעל הכל משורת התפריטים ביופיטר.
הערה: משתני הפלט kprot, kmet ו - latent יאוחסנו וניתן יהיה לגשת אליהם מהמחברות האחרות, אשר ישמשו להמשך זרימת העבודה האנליטית. AE_optimization_plot.pdf העלילה ייווצר ויישמר בתיקייה המקומית (איור 2).
3. יישום זרימת עבודה עם פרמטרים מותאמים אישית
הערה: בצע פרוטוקול זה רק לאחר מיטוב השיטה (סעיף 2). אם משתמשים בוחרים שלא לבצע מיטוב שיטה, דלג ישירות לסעיף 4. פרוטוקול זה ינחה את המשתמש ביצירת מודל באמצעות הפרמטרים המותאמים אישית הנגזרים מסעיף 2. המקודד האוטומטי 1) ייצור קבוצה של תכונות שחולצו המשחזרות את הנתונים המקוריים ו-2) יזהה את התכונות החשובות המניעות כל נוירון בשכבה הסמויה, תוך ייצוג יעיל של מודולי איתות ייחודיים. מודולי האיתות יפורשו באמצעות הפרוטוקול המפורט בסעיף 5.
- בדף הבית של Jupyter בדפדפן, לחץ על המחברת M03a - יישום DeepOmicsAE עם פרמטרים מותאמים אישית.ipynb כדי לפתוח אותו בכרטיסייה חדשה (קובץ משלים 2, שלב 3.1).
- בתא השני של המחברת, הקלד את שם קובץ הקלט במקום M01_output_data.csv. הקלט לפונקציה זו הוא נתוני הפלט מסעיף 1.
- בתא החמישי של המחברת, ציין את מיקום העמודות עבור כל סוג נתונים באופן הבא: נתוני פרוטאומיקה (cols_prot), נתוני מטבולומיקה (cols_met), נתונים קליניים (cols_clin; כולל את כל הנתונים הקליניים). הזן את אינדקס העמודות הראשון עבור כל סוג נתונים במקום col_start ואת אינדקס העמודות האחרונות במקום col_end; לדוגמה: cols_prot = פרוסה(0, 8817). ודא שהערכים שצוינו באובייקטי הפרוסה תואמים לאינדקסים של העמודה הראשונה והאחרונה המתאימים לכל סוג נתונים, והשתמש בפקודות בתא השלישי והרביעי של המחברת כדי לחקור את הנתונים ולקבוע את מיקום ההתחלה והסיום של כל סוג נתונים. ציין את שם העמודה המכילה את משתנה היעד (לדוגמה, 0 או 1, המתאים לבריא או חולה) במקום y_column_name כ- y_label.
הערה: ערך האינדקסים המצוין ב- cols_X_prot, cols_X_met, cols_clin ו- cols_X_expr יהיה שונה מאלה המשמשים בסעיף 1 עקב העיצוב מחדש של מסגרת הנתונים המתרחש במהלך עיבוד מקדים של נתונים. - בחר תא | הריצו הכל משורת התפריטים בצדק כדי ליצור ולשמור את החלקות PCA_initial_data.pdf, PCA_extracted_features.pdf ו-distribution_important_feature_scores.pdf בתיקייה המקומית (איור 3 ואיור משלים S1). בנוסף, רשימות של תכונות חשובות עבור כל מודול איתות מזוהה יאוחסנו בקבצי טקסט בתיקייה המקומית, בשם module_n.txt, כאשר n יוחלף במספר המודול.
4. יישום זרימת עבודה עם פרמטרים מוגדרים מראש
- עיין בסעיף 3 לקבלת הוראות מפורטות כיצד להפעיל שיטה זו (קובץ משלים 2, שלב 4.1). ההבדל היחיד בין שני הפרוטוקולים האלה הוא שהפרמטרים kprot, kmet ו-latent (בתא השביעי של המחברת) נגזרים מתמטית בהתבסס על תוצאות האופטימיזציה שבוצעה כפי שמוצג באיור 2.
הערה: אם סעיף 4 מספק הפרדה גרועה בין קבוצות המדגם, דבר המצביע על ביצועי מודל לא אופטימליים, מומלץ לבצע אופטימיזציה של המודל (סעיף 2) באמצעות לפחות 15 חזרות, ואם אפשר, עד 50.
5. פרשנות ביולוגית באמצעות MetaboAnalyst
- פתח את הדפדפן ונווט לקישור למטה כדי לגשת לפונקציונליות ניתוח המסלול המשותף באתר MetaboAnalyst : https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml.
- גש לתיקייה שבה נשמרו קבצי הפלט משיטה 3 או שיטה 4 ופתח את קבצי הטקסט module_n.txt עבור כל מודול איתות n שנוצר על-ידי שיטה 3 או על-ידי שיטה 4.
- אתר את החלבונים בקובצי הטקסט והעתק אותם.
- הדבק את רשימת החלבונים בחלון גנים/חלבונים עם שינויי קיפול אופציונליים בדף האינטרנט של MetaboAnalyst.
- חזור על השלב לעיל עבור מטבוליטים והדבק אותם ברשימת התרכובות בחלון עם שינויי קיפול אופציונליים באותו דף אינטרנט.
- בחר את האורגניזם המתאים ואת סוג המזהה ולאחר מכן לחץ על שלח בתחתית הדף (קובץ משלים 2, שלב 5.1).
הערה: ודא שהמזהים מזוהים על-ידי MetaboAnalyst. מזהים מוכרים כוללים מזהה Entrez, סמלי גנים רשמיים ומזהה Uniprot לחלבונים; שם תרכובת, מזהה HMDB ומזהה KEGG עבור מטבוליטים. אם המזהים אינם מסוגים אלה, יש צורך בהמרה מתאימה לפני הניתוח. - בדף הבא, בדוק את מיפוי המזהים לפני שתלחץ על המשך כדי לוודא שהמזהים מזוהים.
- בדף הגדרת פרמטרים , בחר מסלולים מטבוליים (משולבים) או כל המסלולים (משולבים) כדי להציג בהתאמה את תרומת הקלט למסלולים מטבוליים בלבד או לכל מסלולי האיתות (קובץ משלים 2, שלב 5.2). בחלונית בחירת אלגוריתם , בחר Enrichment analysis: Hypergeometric test, Topology measure: degree centrality, and integration method: Combine p values (pathway-level). לחץ על שלח בתחתית הדף.
- העמוד האחרון הוא תצוגת התוצאות, המציגה את תוצאות ניתוח ההעשרה. מסלולים מועשרים מתווטים על פי השפעתם ומשמעותם, ורשימת המסלולים מסופקת גם בפורמט טבלאי.
תוצאות
כדי להציג את הפרוטוקול, ניתחנו מערך נתונים הכולל פרוטום, מטבוליזם ומידע קליני שמקורו במוחות לאחר המוות של 142 אנשים שהיו בריאים או אובחנו עם מחלת אלצהיימר.
לאחר ביצוע פרוטוקול סעיף 1 כדי לעבד מראש את הנתונים, מערך הנתונים כלל 6,497 חלבונים, 443 מטבוליטים, ושלושה מאפיינים קליניים (?...
Discussion
מבנה מערך הנתונים הוא קריטי להצלחת הפרוטוקול ויש לבדוק אותו בקפידה. יש לעצב את הנתונים כפי שמצוין בסעיף 1 של הפרוטוקול. הקצאה נכונה של מיקומי טורים היא גם קריטית להצלחת השיטה. נתוני פרוטאומיקה ומטבולומיקה מעובדים מראש באופן שונה ובחירת התכונות מתבצעת בנפרד בשל אופיים השונה של הנתונים. לכן...
Disclosures
המחבר מצהיר כי אין להם ניגודי עניינים.
Acknowledgements
עבודה זו נתמכה על ידי CA201402 המענקים של NIH ופרס Cornell Center for Vertebrate Genomics (CVG) Distinguished Scholar Award. התוצאות המתפרסמות כאן מבוססות במלואן או בחלקן על נתונים שהתקבלו מפורטל הידע של AD (https://adknowledgeportal.org). נתוני המחקר סופקו באמצעות השותפות להאצת הרפואה עבור אלצהיימר (U01AG046161 ו-U01AG061357) בהתבסס על דגימות שסופקו על ידי המרכז למחלות אלצהיימר ראש, המרכז הרפואי האוניברסיטאי ראש, שיקגו. איסוף הנתונים נתמך באמצעות מימון על ידי מענקי NIA P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, מחלקת בריאות הציבור של אילינוי ומכון המחקר לגנומיקה תרגומית. מערך הנתונים המטאבולומי נוצר במטאבולון ועובד מראש על ידי ADMC.
Materials
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
References
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved