
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
DeepOmicsAE: Darstellung von Signalmodulen bei der Alzheimer-Krankheit mit Deep-Learning-Analyse von Proteomik, Metabolomik und klinischen Daten
In diesem Artikel
Zusammenfassung
DeepOmicsAE ist ein Workflow, der sich auf die Anwendung einer Deep-Learning-Methode (d. h. eines Autoencoders) konzentriert, um die Dimensionalität von Multi-Omics-Daten zu reduzieren, und bietet eine Grundlage für Vorhersagemodelle und Signalisierungsmodule, die mehrere Schichten von Omics-Daten darstellen.
Zusammenfassung
Große Omics-Datensätze werden zunehmend für die Erforschung der menschlichen Gesundheit verfügbar. In diesem Artikel wird DeepOmicsAE vorgestellt, ein Workflow, der für die Analyse von Multi-Omics-Datensätzen, einschließlich Proteomik, Metabolomik und klinischer Daten, optimiert ist. Dieser Workflow verwendet eine Art neuronales Netzwerk namens Autoencoder, um einen präzisen Satz von Features aus den hochdimensionalen Multi-Omics-Eingabedaten zu extrahieren. Darüber hinaus bietet der Workflow eine Methode zur Optimierung der Schlüsselparameter, die für die Implementierung des Autoencoders erforderlich sind. Um diesen Arbeitsablauf zu veranschaulichen, wurden klinische Daten aus einer Kohorte von 142 Personen analysiert, die entweder gesund waren oder bei denen Alzheimer diagnostiziert wurde, zusammen mit dem Proteom und Metabolom ihrer postmortalen Gehirnproben. Die aus der latenten Schicht des Autoencoders extrahierten Merkmale behalten die biologischen Informationen bei, die gesunde und kranke Patienten trennen. Darüber hinaus stellen die einzelnen extrahierten Merkmale unterschiedliche molekulare Signalmodule dar, von denen jedes einzigartig mit den klinischen Merkmalen der Individuen interagiert und ein Mittel zur Integration der Proteomik, Metabolomik und klinischen Daten bietet.
Einleitung
Ein immer größerer Teil der Bevölkerung altert und die Belastung durch altersbedingte Krankheiten wie Neurodegeneration wird in den kommenden Jahrzehnten voraussichtlich stark zunehmen1. Die Alzheimer-Krankheit ist die häufigste Form der neurodegenerativen Erkrankung2. Der Fortschritt bei der Suche nach einer Behandlung war langsam, da wir die grundlegenden molekularen Mechanismen, die den Ausbruch und das Fortschreiten der Krankheit bestimmen, nur unzureichend verstehen. Der Großteil der Informationen über die Alzheimer-Krankheit wird post mortem aus der Untersuchung von Hirngewebe gewonnen, was die Unterscheidung von Ursachen und Folgen zu einer schwierigen Aufgabe gemacht hat3. Das Religious Orders Study/Memory and Aging Project (ROSMAP) ist ein ehrgeiziges Vorhaben, ein breiteres Verständnis der Neurodegeneration zu erlangen, das die Untersuchung von Tausenden von Personen beinhaltet, die sich verpflichtet haben, sich jährlich medizinischen und psychologischen Untersuchungen zu unterziehen und ihr Gehirn nach ihrem Tod für die Forschung zur Verfügung zu stellen4. Die Studie konzentriert sich auf den Übergang von der normalen Funktion des Gehirns zur Alzheimer-Krankheit2. Im Rahmen des Projekts wurden postmortale Gehirnproben mit einer Vielzahl von Omics-Ansätzen analysiert, darunter Genomik, Epigenomik, Transkriptomik, Proteomik5 und Metabolomik.
Omics-Technologien, die funktionelle Auslesungen zellulärer Zustände ermöglichen (d. h. Proteomik und Metabolomik)6,7 sind aufgrund des direkten Zusammenhangs zwischen Protein- und Metabolitenhäufigkeit und zellulären Aktivitäten der Schlüssel zur Interpretation von Krankheiten 8,9,10,11,12. Proteine sind die primären Exekutoren zellulärer Prozesse, während Metaboliten die Substrate und Produkte für biochemische Reaktionen sind. Die Multi-Omics-Datenanalyse bietet die Möglichkeit, die komplexen Zusammenhänge zwischen Proteomics- und Metabolomics-Daten zu verstehen, anstatt sie isoliert zu betrachten. Multi-Omics ist eine Disziplin, die mehrere Schichten hochdimensionaler biologischer Daten untersucht, einschließlich molekularer Daten (Genomsequenz und Mutationen, Transkriptom, Proteom, Metabolom), klinischer Bildgebungsdaten und klinischer Merkmale. Insbesondere zielt die Multi-Omics-Datenanalyse darauf ab, solche Schichten biologischer Daten zu integrieren, ihre wechselseitige Regulation und Interaktionsdynamik zu verstehen und ein ganzheitliches Verständnis des Auftretens und Fortschreitens von Krankheiten zu liefern. Methoden zur Integration von Multi-Omics-Daten befinden sich jedoch noch in einem frühen Entwicklungsstadium13.
Autoencoder, eine Art unüberwachtes neuronales Netzwerk14, sind ein leistungsfähiges Werkzeug für die Multi-Omics-Datenintegration. Im Gegensatz zu überwachten neuronalen Netzen ordnen Autoencoder Proben weder bestimmten Zielwerten (z. B. gesund oder krank) zu, noch werden sie zur Vorhersage von Ergebnissen verwendet. Eine ihrer Hauptanwendungen liegt in der Dimensionalitätsreduktion. Autoencoder bieten jedoch mehrere Vorteile gegenüber einfacheren Methoden zur Dimensionalitätsreduktion wie Hauptkomponentenanalyse (PCA), t-verteilter stochastischer Nachbareinbettung (tSNE) oder gleichmäßiger Mannigfaltigkeitsapproximation und Projektion (UMAP). Im Gegensatz zu PCA können Autoencoder nichtlineare Beziehungen innerhalb der Daten erfassen. Im Gegensatz zu tSNE und UMAP können sie hierarchische und multimodale Beziehungen innerhalb der Daten erkennen, da sie auf mehreren Schichten von Recheneinheiten beruhen, die jeweils nichtlineare Aktivierungsfunktionen enthalten. Daher stellen sie attraktive Modelle dar, um die Komplexität von Multi-Omics-Daten zu erfassen. Während die primäre Anwendung von PCA, tSNE und UMAP das Clustering der Daten ist, komprimieren Autoencoder die Eingabedaten in extrahierte Merkmale, die sich gut für nachgelagerte Vorhersageaufgaben eignen15,16.
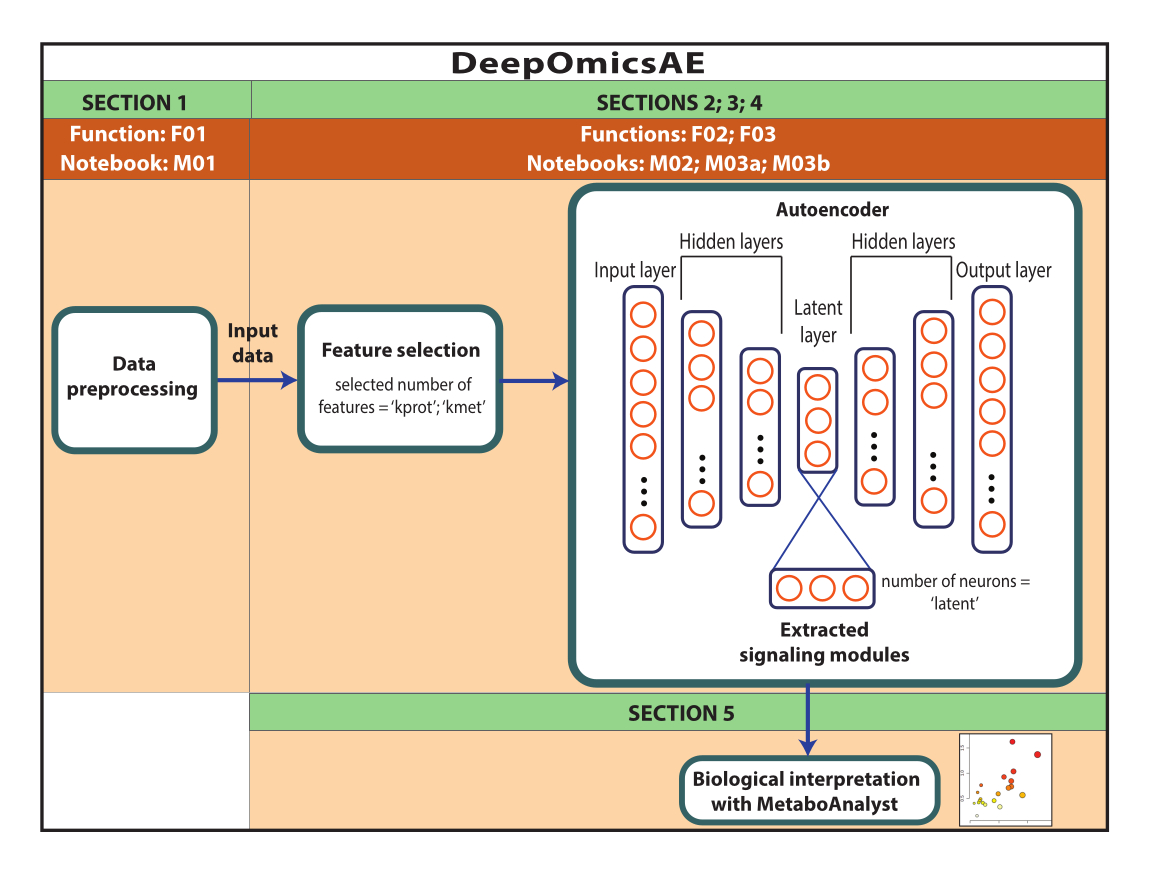
Kurz gesagt, neuronale Netze bestehen aus mehreren Schichten, die jeweils mehrere Recheneinheiten oder "Neuronen" enthalten. Die erste und letzte Schicht werden als Eingabe- bzw. Ausgabeschicht bezeichnet. Autoencoder sind neuronale Netze mit einer Sanduhrstruktur, bestehend aus einer Eingabeschicht, gefolgt von ein bis drei versteckten Schichten und einer kleinen "latenten" Schicht, die typischerweise zwischen zwei und sechs Neuronen enthält. Die erste Hälfte dieser Struktur wird als Encoder bezeichnet und ist mit einem Decoder kombiniert, der den Encoder spiegelt. Der Decoder endet mit einer Ausgabeschicht, die die gleiche Anzahl von Neuronen wie die Eingabeschicht enthält. Autoencoder nehmen die Eingabe durch den Flaschenhals und rekonstruieren sie in der Ausgabeschicht, mit dem Ziel, eine Ausgabe zu erzeugen, die die ursprünglichen Informationen so genau wie möglich widerspiegelt. Dies wird durch die mathematische Minimierung eines Parameters erreicht, der als "Rekonstruktionsverlust" bezeichnet wird. Die Eingabe besteht aus einer Reihe von Merkmalen, die in der hier gezeigten Anwendung Protein- und Metabolitenhäufigkeiten und klinische Merkmale (d. h. Geschlecht, Bildung und Alter zum Zeitpunkt des Todes) sein werden. Die latente Schicht enthält eine komprimierte und informationsreiche Darstellung der Eingabe, die für nachfolgende Anwendungen wie Vorhersagemodelle verwendet werden kann17,18.
Dieses Protokoll stellt einen Workflow, DeepOmicsAE, dar, der Folgendes umfasst: 1) Vorverarbeitung von Proteomik-, Metabolomik- und klinischen Daten (d. h. Normalisierung, Skalierung, Entfernung von Ausreißern), um Daten mit einer konsistenten Skala für die Analyse des maschinellen Lernens zu erhalten; 2) Auswahl geeigneter Autoencoder-Eingangsmerkmale, da eine Überlastung der Merkmale relevante Krankheitsmuster verschleiern kann; 3) Optimierung und Training des Autoencoders, einschließlich der Bestimmung der optimalen Anzahl von Proteinen und Metaboliten für die Selektion und von Neuronen für die latente Schicht; 4) Extrahieren von Merkmalen aus der latenten Schicht; und 5) Nutzung der extrahierten Merkmale für die biologische Interpretation durch Identifizierung molekularer Signalmodule und ihrer Beziehung zu klinischen Merkmalen.
Dieses Protokoll soll einfach und für Biologen mit begrenzter Computererfahrung anwendbar sein, die über ein grundlegendes Verständnis der Programmierung mit Python verfügen. Das Protokoll konzentriert sich auf die Analyse von Multi-Omics-Daten, einschließlich Proteomik, Metabolomik und klinischer Merkmale, aber seine Verwendung kann auf andere Arten von molekularen Expressionsdaten, einschließlich Transkriptomik, ausgeweitet werden. Eine wichtige neue Anwendung, die durch dieses Protokoll eingeführt wurde, ist die Zuordnung der Wichtigkeitswerte ursprünglicher Merkmale auf einzelne Neuronen in der latenten Schicht. Infolgedessen stellt jedes Neuron in der latenten Schicht ein Signalmodul dar, das die Wechselwirkungen zwischen spezifischen molekularen Veränderungen und den klinischen Merkmalen der Patienten detailliert beschreibt. Die biologische Interpretation der molekularen Signalmodule erfolgt mit MetaboAnalyst, einem öffentlich zugänglichen Werkzeug, das Gen-/Protein- und Metabolitendaten integriert, um angereicherte Stoffwechsel- und Zellsignalwege abzuleiten17.
Protokoll
HINWEIS: Bei den hier verwendeten Daten handelt es sich um ROSMAP-Daten, die vom AD Knowledge-Portal heruntergeladen wurden. Für das Herunterladen und Wiederverwenden der Daten ist keine Einwilligung nach Aufklärung erforderlich. Das hierin vorgestellte Protokoll verwendet Deep Learning, um Multi-Omics-Daten zu analysieren und Signalmodule zu identifizieren, die bestimmte Patienten- oder Stichprobengruppen beispielsweise basierend auf ihrer Diagnose unterscheiden. Das Protokoll liefert auch einen kleinen Satz extrahierter Merkmale, die die ursprünglichen großen Daten zusammenfassen und für weitere Analysen verwendet werden können, z. B. zum Trainieren eines Vorhersagemodells mit Algorithmen des maschinellen Lernens (Abbildung 1). In der Zusatzdatei 1 und in der Materialtabelle finden Sie Informationen zum Zugriff auf den Code und zum Einrichten der Rechenumgebung vor dem Ausführen des Protokolls. Die Methoden sollten in der unten angegebenen Reihenfolge ausgeführt werden.

Abbildung 1: Schematische Darstellung des DeepOmicsAE-Workflows. Schematische Darstellung des Workflows zur Analyse von Multi-Omics-Daten mithilfe des Workflows. In der Autoencoder-Darstellung stellen Rechtecke Schichten des neuronalen Netzwerks und Kreise Neuronen innerhalb von Schichten dar. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
1. Datenvorverarbeitung
HINWEIS: Das Ziel dieses Abschnitts ist die Vorverarbeitung der Daten, einschließlich der Behandlung fehlender Daten. Normalisierung und Skalierung von proteomischen, metabolomischen Expressions- und klinischen Daten; und das Entfernen von Ausreißern. Das Protokoll ist für einen Datensatz konzipiert, der Proteomikdaten enthält, die als log2(ratio) ausgedrückt werden; Metabolomik-Daten, ausgedrückt als Faltungsänderung; und klinische Merkmale, einschließlich kontinuierlicher und kategorischer Merkmale. Die Patienten oder Proben sollten auf der Grundlage der Diagnose oder anderer ähnlicher Parameter gruppiert werden. Proben oder Patienten sollten sich über die Zeilen und Features über die Spalten erstrecken.
- Um eine neue Instanz von Jupyter Notebook im Browser zu starten, öffnen Sie ein neues Terminalfenster, geben Sie Folgendes ein und drücken Sie die Eingabetaste.
Jupyter Notebook - Klicken Sie auf der Jupyter-Startseite im Browser auf das Notebook M01 - expression data pre-processing.ipynb , um es in einer neuen Registerkarte zu öffnen (Ergänzungsdatei 2, Schritt 1.1).
- Geben Sie in der zweiten Zelle des Notebooks den Namen der Datasetdatei anstelle von your_dataset_name.csv ein.
- Geben Sie in der letzten Zelle des Notebooks anstelle von M01_output_data.csv den gewünschten Namen der Ausgabedatendatei ein.
- Geben Sie in der fünften Zelle des Notebooks die Position der Spalten für jeden Datentyp wie folgt an: Proteomikdaten (cols_prot), Metabolomikdaten (cols_met), kontinuierliche klinische Daten (z. B. Alter) (cols_clin_con), binäre klinische Daten (z. B. Geschlecht) (cols_clin_bin). Geben Sie den Index der ersten Spalte für jeden Datentyp anstelle von col_start und den Index der letzten Spalten anstelle von col_end ein. Beispiel: cols_prot = slice(0, 8817). Stellen Sie sicher, dass die in den Segmentobjekten angegebenen Werte den Indizes der ersten und letzten Spalte entsprechen, die jedem Datentyp entsprechen. Verwenden Sie den Befehl in der vierten Zelle desselben Notebooks (df.iloc[:, :]), um die Start- und Endposition für jeden Datentyp zu bestimmen (Ergänzungsdatei 2, Schritt 1.2).
- Zelle auswählen | Führen Sie alle über die Menüleiste in Jupyter aus, um die Ausgabedatendatei im angegebenen Ordner zu erstellen (Ergänzende Datei 2, Schritt 1.3).
HINWEIS: Diese Daten werden als Eingabe für die in den Abschnitten 2, 3 oder 4 beschriebenen Protokolle verwendet.
2. Individuelle Optimierung des Workflows (optional)
HINWEIS: Abschnitt 2 ist optional, da er rechenintensiv ist. Benutzer sollten direkt zu Abschnitt 4 springen, wenn sie sich entscheiden, Abschnitt 2 nicht auszuführen. Dieses Protokoll führt den Benutzer automatisiert durch die Optimierung des Workflows. Insbesondere identifiziert die Methode die Parameter, die die beste Leistung des Autoencoders in Bezug auf die Generierung extrahierter Features liefern, die die Stichprobengruppen gut trennen. Zu den optimierten Parametern, die als Ausgabe generiert werden, gehören die Anzahl der Merkmale, die für die Merkmalsauswahl verwendet werden sollen (k_prot und k_met) und die Anzahl der Neuronen in der latenten Schicht des Autoencoders (latent). Diese Parameter können dann in dem in Abschnitt 3 beschriebenen Protokoll verwendet werden, um das Modell zu generieren.
- Klicken Sie auf der Jupyter-Startseite im Browser auf das Notebook M02 - DeepOmicsAE model optimization.ipynb , um es in einem neuen Tab zu öffnen (Supplemental File 2, Step 2.1).
- Geben Sie in der zweiten Zelle des Notebooks anstelle von M01_output_data.csv den Namen der Eingabedatei ein. Die Eingabe für diese Funktion sind die Ausgabedaten aus Abschnitt 1.
- Geben Sie in der fünften Zelle des Notebooks die Position der Spalten für jeden Datentyp wie folgt an: Proteomikdaten (cols_X_prot), Metabolomikdaten (cols_X_met), klinische Daten (cols_clin; enthält alle klinischen Daten), alle molekularen Expressionsdaten, einschließlich Proteomics- und Metabolomics-Daten (cols_X_expr). Geben Sie den ersten Spaltenindex für jeden Datentyp anstelle von col_start und den Index der letzten Spalten anstelle von col_end ein. Beispiel: cols_prot = slice(0, 8817). Stellen Sie sicher, dass die in den Segmentobjekten angegebenen Werte dem Index der ersten und letzten Spalte entsprechen, der jedem Datentyp entspricht, und verwenden Sie die Befehle in der dritten und vierten Zelle des Notebooks, um die Daten zu untersuchen und die Start- und Endpositionen für jeden Datentyp zu bestimmen. Geben Sie den Namen der Spalte, die die Zielvariable enthält, anstelle von y_column_name wie y_label an (Zusatzdatei 2, Schritt 2.2).
HINWEIS: Die Werte der in cols_X_prot, cols_X_met, cols_clin und cols_X_expr angegebenen Indizes unterscheiden sich von den in Abschnitt 1 verwendeten Werten, da der Datenrahmen während der Datenvorverarbeitung umgeformt wird. - Geben Sie in der sechsten Zelle des Notebooks an, wie viele Optimierungsrunden durchgeführt werden sollen, indem Sie n_comb einen Wert zuweisen. Die Bearbeitungszeiten betragen ca. 4-5 Minuten für 10 Runden; 20 Minuten für 50 Runden und 40 Minuten für 100 Runden (Ergänzungsdatei 2, Schritt 2.3).
- Zelle auswählen | Führen Sie alles über die Menüleiste in Jupyter aus.
HINWEIS: Die Ausgabevariablen kprot, kmet und latent werden gespeichert und können von den anderen Notebooks aus aufgerufen werden, die zum Fortsetzen des analytischen Workflows verwendet werden. Der Plot AE_optimization_plot.pdf wird generiert und im lokalen Ordner gespeichert (Abbildung 2).
3. Workflow-Implementierung mit individuell optimierten Parametern
HINWEIS: Führen Sie dieses Protokoll nur nach der Methodenoptimierung (Abschnitt 2) durch. Wenn Benutzer keine Methodenoptimierung durchführen möchten, fahren Sie direkt mit Abschnitt 4 fort. Dieses Protokoll führt den Benutzer durch die Generierung eines Modells mit den benutzerdefinierten optimierten Parametern, die aus Abschnitt 2 abgeleitet wurden. Der Autoencoder erzeugt 1) eine Reihe extrahierter Merkmale, die die Originaldaten rekapitulieren, und 2) identifiziert die wichtigen Merkmale, die jedes Neuron in der latenten Schicht antreiben und effektiv einzigartige Signalmodule darstellen. Die Signalisierungsmodule werden unter Verwendung des in Abschnitt 5 bereitgestellten Protokolls interpretiert.
- Klicken Sie auf der Jupyter-Startseite im Browser auf das Notebook M03a - DeepOmicsAE implementation with custom-optimized parameters.ipynb , um es in einem neuen Tab zu öffnen (Supplemental File 2, Step 3.1).
- Geben Sie in der zweiten Zelle des Notebooks anstelle von M01_output_data.csv den Namen der Eingabedatei ein. Die Eingabe für diese Funktion sind die Ausgabedaten aus Abschnitt 1.
- Geben Sie in der fünften Zelle des Notebooks die Position der Spalten für jeden Datentyp wie folgt an: Proteomikdaten (cols_prot), Metabolomikdaten (cols_met), klinische Daten (cols_clin; enthält alle klinischen Daten). Geben Sie den ersten Spaltenindex für jeden Datentyp anstelle von col_start und den Index der letzten Spalten anstelle von col_end ein. Beispiel: cols_prot = slice(0, 8817). Stellen Sie sicher, dass die in den Segmentobjekten angegebenen Werte den Indizes der ersten und letzten Spalte entsprechen, die jedem Datentyp entsprechen, und verwenden Sie die Befehle in der dritten und vierten Zelle des Notebooks, um die Daten zu untersuchen und die Start- und Endpositionen für jeden Datentyp zu bestimmen. Geben Sie den Namen der Spalte an, die die Zielvariable enthält (z. B. 0 oder 1, entsprechend gesund oder krank), anstelle von y_column_name wie y_label.
HINWEIS: Der Wert der in cols_X_prot, cols_X_met, cols_clin und cols_X_expr angegebenen Indizes unterscheidet sich von den in Abschnitt 1 verwendeten Indizes, da der Datenrahmen während der Datenvorverarbeitung umgeformt wird. - Zelle auswählen | Führen Sie alles über die Menüleiste in Jupyter aus, um die Diagramme PCA_initial_data.pdf, PCA_extracted_features.pdf und distribution_important_feature_scores.pdf im lokalen Ordner zu generieren und zu speichern (Abbildung 3 und ergänzende Abbildung S1). Zusätzlich werden Listen wichtiger Merkmale für jedes identifizierte Signalisierungsmodul in Textdateien im lokalen Ordner mit dem Namen module_n.txt gespeichert, wobei n durch die Modulnummer ersetzt wird.
4. Workflow-Implementierung mit voreingestellten Parametern
- In Abschnitt 3 finden Sie detaillierte Anweisungen zum Ausführen dieser Methode (Ergänzungsdatei 2, Schritt 4.1). Der einzige Unterschied zwischen diesen beiden Protokollen besteht darin, dass die Parameter kprot, kmet und latent (in der siebten Zelle des Notebooks) mathematisch basierend auf den Ergebnissen der wie in Abbildung 2 dargestellten Optimierung abgeleitet werden.
HINWEIS: Wenn Abschnitt 4 eine schlechte Trennung der Stichprobengruppen liefert, was auf eine suboptimale Modellleistung hinweist, wird empfohlen, die Modelloptimierung (Abschnitt 2) mit mindestens 15 Iterationen und wenn möglich bis zu 50 durchzuführen.
5. Biologische Interpretation mit MetaboAnalyst
- Öffnen Sie den Browser und navigieren Sie zum folgenden Link, um auf die Funktion Joint Pathway Analysis auf der MetaboAnalyst-Website zuzugreifen: https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml.
- Greifen Sie auf den Ordner zu, in dem die Ausgabedateien von Methode 3 oder Methode 4 gespeichert wurden, und öffnen Sie die Textdateien module_n.txt für jedes Signalisierungsmodul n, das von Methode 3 oder Methode 4 generiert wurde.
- Suchen Sie die Proteine in den Textdateien und kopieren Sie sie.
- Fügen Sie die Liste der Proteine in das Fenster Gene/Proteine mit optionalen Faltungsänderungen auf der MetaboAnalyst-Webseite ein.
- Wiederholen Sie den obigen Schritt für Metaboliten und fügen Sie sie in das Fenster Verbindungsliste mit optionalen Faltungsänderungen auf derselben Webseite ein.
- Wählen Sie den entsprechenden Organismus und ID-Typ aus und klicken Sie dann unten auf der Seite auf Senden (Zusatzdatei 2, Schritt 5.1).
HINWEIS: Stellen Sie sicher, dass die Bezeichner von MetaboAnalyst erkannt werden. Zu den anerkannten Identifikatoren gehören die Entrez-ID, offizielle Gensymbole und Uniprot-ID für Proteine; Verbindungsname, HMDB-ID und KEGG-ID für Metaboliten. Wenn es sich bei den Identifikatoren nicht um diese Typen handelt, ist vor der Analyse eine entsprechende Konvertierung erforderlich. - Überprüfen Sie auf der folgenden Seite die ID-Zuordnung, bevor Sie auf Fortfahren klicken, um zu überprüfen, ob die IDs erkannt werden.
- Wählen Sie auf der Seite Parametereinstellung die Option Stoffwechselwege (integriert) oder Alle Wege (integriert), um den Beitrag der Eingabe nur zu Stoffwechselwegen oder zu allen Signalwegen zu visualisieren (Zusatzdatei 2, Schritt 5.2). Wählen Sie im Auswahlbereich Algorithmus die Optionen Anreicherungsanalyse: Hypergeometrischer Test, Topologiemeasure: Gradzentralität und Integrationsmethode: Kombinieren von p-Werten (Pfadebene) aus. Klicken Sie unten auf der Seite auf Senden .
- Die letzte Seite ist die Ergebnisansicht, in der die Ergebnisse der Anreicherungsanalyse dargestellt werden. Angereicherte Pfade werden basierend auf ihrer Wirkung und Bedeutung dargestellt, und die Liste der Pfade wird auch in Tabellenform bereitgestellt.
Ergebnisse
Um das Protokoll zu präsentieren, analysierten wir einen Datensatz, der das Proteom, das Metabolom und klinische Informationen aus postmortalen Gehirnen von 142 Personen umfasste, die entweder gesund waren oder bei denen Alzheimer diagnostiziert wurde.
Nach Durchführung des Protokollabschnitts 1 zur Vorverarbeitung der Daten enthielt der Datensatz 6.497 Proteine, 443 Metaboliten und drei klinische Merkmale (Geschlecht, Sterbealter und Bildung). Das Zielmerkmal ist die klinische Konse...
Diskussion
Die Struktur des Datensatzes ist entscheidend für den Erfolg des Protokolls und sollte sorgfältig überprüft werden. Die Daten sollten wie in Protokollabschnitt 1 angegeben formatiert sein. Auch die korrekte Zuordnung von Spaltenpositionen ist entscheidend für den Erfolg der Methode. Proteomik- und Metabolomik-Daten werden unterschiedlich vorverarbeitet und die Merkmalsauswahl wird aufgrund der unterschiedlichen Art der Daten separat durchgeführt. Daher ist es wichtig, die Spaltenpositionen in den Protokollschritten...
Offenlegungen
Der Autor erklärt, dass er keine Interessenkonflikte hat.
Danksagungen
Diese Arbeit wurde durch NIH Grant CA201402 und den Cornell Center for Vertebrate Genomics (CVG) Distinguished Scholar Award unterstützt. Die hier veröffentlichten Ergebnisse basieren ganz oder teilweise auf Daten aus dem AD Knowledge Portal (https://adknowledgeportal.org). Die Studiendaten wurden von der Accelerating Medicine Partnership for AD (U01AG046161 und U01AG061357) auf der Grundlage von Proben bereitgestellt, die vom Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago, zur Verfügung gestellt wurden. Die Datenerhebung wurde durch NIA-Zuschüsse P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, das Illinois Department of Public Health und das Translational Genomics Research Institute unterstützt. Der Metabolomics-Datensatz wurde bei Metabolon generiert und vom ADMC vorverarbeitet.
Materialien
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
Referenzen
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten