
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
DeepOmicsAE:通过蛋白质组学、代谢组学和临床数据的深度学习分析来表示阿尔茨海默病中的信号转导模块
摘要
DeepOmicsAE是一个以应用深度学习方法(即自动编码器)为中心的工作流程,以降低多组学数据的维度,为代表多层组学数据的预测模型和信号转导模块提供基础。
摘要
大型组学数据集正越来越多地用于人类健康研究。本文介绍了 DeepOmicsAE,这是一种针对多组学数据集分析(包括蛋白质组学、代谢组学和临床数据)进行优化的工作流程。该工作流程采用一种称为自动编码器的神经网络,从高维多组学输入数据中提取一组简洁的特征。此外,该工作流还提供了一种优化实现自动编码器所需的关键参数的方法。为了展示这一工作流程,分析了142名健康或被诊断患有阿尔茨海默病的个体的临床数据,以及他们死后大脑样本的蛋白质组和代谢组。从自动编码器的潜在层提取的特征保留了区分健康和患病患者的生物信息。此外,单个提取的特征代表了不同的分子信号转导模块,每个模块都与个体的临床特征相互作用,为整合蛋白质组学、代谢组学和临床数据提供了一种手段。
引言
老龄化人口的比例越来越大,预计未来几十年神经退行性疾病等与年龄相关的疾病的负担将急剧增加1。阿尔茨海默病是最常见的神经退行性疾病类型 2.鉴于我们对驱动疾病发作和进展的基本分子机制知之甚少,寻找治疗方法的进展缓慢。关于阿尔茨海默病的大部分信息都是在死后通过脑组织检查获得的,这使得区分病因和后果成为一项艰巨的任务3.宗教秩序研究/记忆和衰老项目 (ROSMAP) 是一项雄心勃勃的努力,旨在更广泛地了解神经退行性疾病,其中包括对数千名致力于每年接受医学和心理检查并在他们去世后为研究贡献大脑的人的研究 4.该研究的重点是从大脑正常功能到阿尔茨海默病的过渡 2.在该项目中,使用多种组学方法分析了死后脑样本,包括基因组学、表观基因组学、转录组学、蛋白质组学5 和代谢组学。
由于蛋白质和代谢物丰度与细胞活性之间的直接关系,提供细胞状态功能读数的组学技术(即蛋白质组学和代谢组学)6,7是解释疾病8,9,10,11,12的关键。蛋白质是细胞过程的主要执行者,而代谢物是生化反应的底物和产物。多组学数据分析提供了理解蛋白质组学和代谢组学数据之间复杂关系的可能性,而不是孤立地理解它们。多组学是一门研究多层高维生物数据的学科,包括分子数据(基因组序列和突变、转录组、蛋白质组、代谢组)、临床影像数据和临床特征。特别是,多组学数据分析旨在整合这些生物数据层,了解它们的相互调控和相互作用动态,并提供对疾病发生和进展的整体理解。然而,整合多组学数据的方法仍处于开发的早期阶段13。
自动编码器是一种无监督神经网络14,是多组学数据集成的强大工具。与监督神经网络不同,自动编码器不会将样本映射到特定的目标值(例如健康或患病),也不会用于预测结果。它们的主要应用之一在于降维。然而,与主成分分析 (PCA)、t 分布随机邻域嵌入 (tSNE) 或均匀流形近似和投影 (UMAP) 等更简单的降维方法相比,自动编码器具有多项优势。与 PCA 不同,自动编码器可以捕获数据中的非线性关系。与 tSNE 和 UMAP 不同,它们可以检测数据中的分层和多模态关系,因为它们依赖于多层计算单元,每个单元都包含非线性激活函数。因此,它们代表了捕捉多组学数据复杂性的有吸引力的模型。最后,虽然 PCA、tSNE 和 UMAP 的主要应用是对数据进行聚类,但自动编码器将输入数据压缩为非常适合下游预测任务的提取特征15,16。
简而言之,神经网络由几层组成,每层包含多个计算单元或“神经元”。第一层和最后一层分别称为输入层和输出层。自动编码器是具有沙漏结构的神经网络,由一个输入层组成,然后是一到三个隐藏层和一个通常包含两到六个神经元的小“潜在”层。这种结构的前半部分称为编码器,并与镜像编码器的解码器结合使用。解码器以输出层结束,输出层包含与输入层相同数量的神经元。自动编码器将输入通过瓶颈,并在输出层中重建它,目的是生成尽可能接近原始信息的输出。这是通过数学上最小化称为“重建损失”的参数来实现的。输入由一组特征组成,在此展示的应用程序中,这些特征将是蛋白质和代谢物丰度,以及临床特征(即性别、教育和死亡年龄)。潜在层包含输入的压缩和信息丰富的表示,可用于后续应用,例如预测模型17,18。
该协议提出了一个工作流程 DeepOmicsAE,它涉及:1) 蛋白质组学、代谢组学和临床数据(即归一化、缩放、异常值去除)的预处理,以获得具有一致规模的数据,用于机器学习分析;2)选择适当的自动编码器输入特征,因为特征过载可能会掩盖相关的疾病模式;3)优化和训练自动编码器,包括确定要选择的蛋白质和代谢物的最佳数量,以及潜伏层的神经元数量;4)从潜层中提取特征;5)通过识别分子信号转导模块及其与临床特征的关系,利用提取的特征进行生物学解释。
该协议旨在简单易行,适用于计算经验有限且对 Python 编程有基本了解的生物学家。该协议侧重于分析多组学数据,包括蛋白质组学、代谢组学和临床特征,但其用途可以扩展到其他类型的分子表达数据,包括转录组学。该协议引入的一个重要新应用是将原始特征的重要性分数映射到潜伏层中的单个神经元上。因此,潜伏层中的每个神经元都代表一个信号模块,详细说明了特定分子改变与患者临床特征之间的相互作用。分子信号转导模块的生物学解释是通过使用 MetaboAnalyst 获得的,MetaboAnalyst 是一种公开可用的工具,它集成了基因/蛋白质和代谢物数据以推导富集的代谢和细胞信号通路17。
研究方案
注意:此处使用的数据是从 AD 知识门户下载的 ROSMAP 数据。下载和重复使用数据不需要知情同意。本文介绍的方案利用深度学习来分析多组学数据并识别信号转导模块,这些模块根据例如他们的诊断来区分特定的患者或样本组。该协议还提供了一小组提取的特征,这些特征汇总了原始的大规模数据,可用于进一步分析,例如使用机器学习算法训练预测模型(图 1)。有关在执行协议之前访问代码和设置计算环境的信息,请参阅 补充文件 1 和 材料表 。这些方法应按照下面指定的顺序执行。

图 1:DeepOmicsAE 工作流程示意图。 使用工作流程分析多组学数据的工作流程示意图。在自动编码器描述中,矩形表示神经网络的层,圆圈表示层内的神经元。 请点击这里查看此图的较大版本.
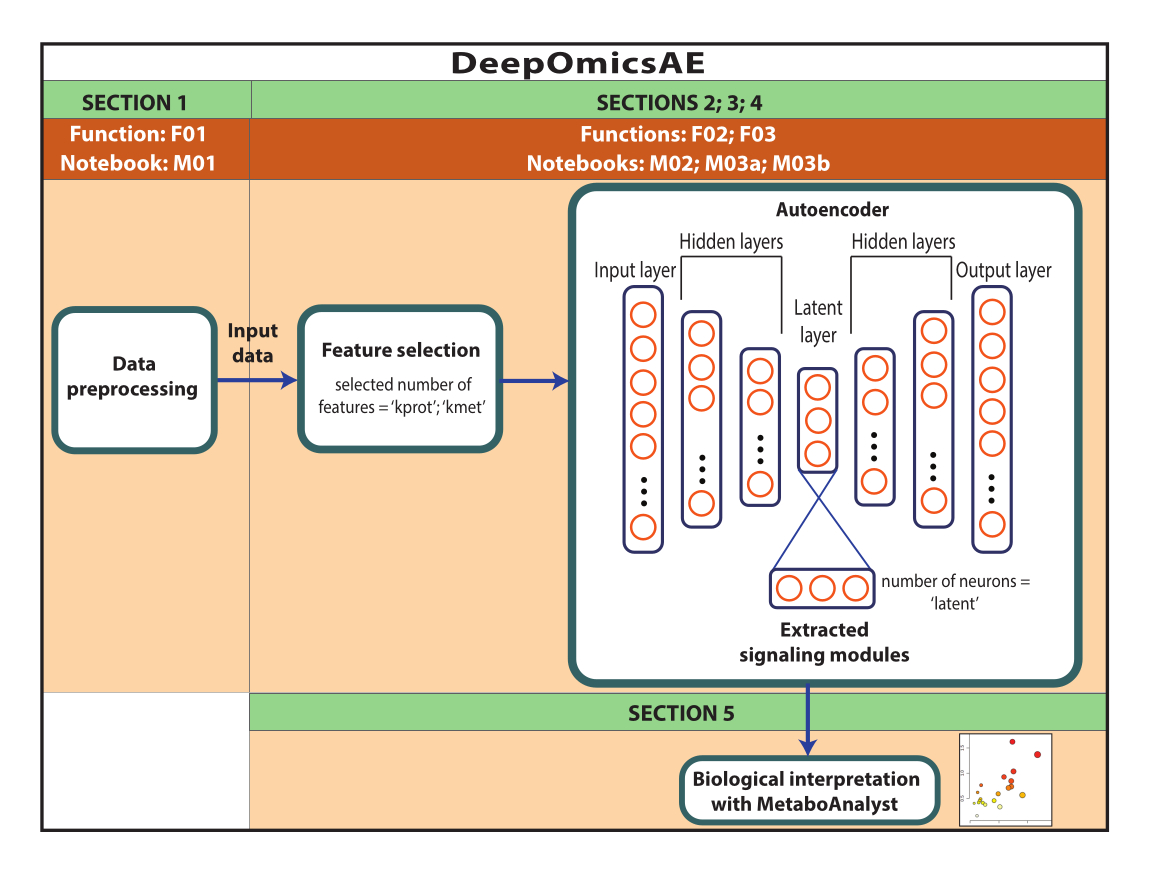{kind=link}
1. 数据预处理
注意:本节的目标是对数据进行预处理,包括处理缺失的数据;蛋白质组学、代谢组学表达和临床数据标准化和缩放;并删除异常值。该协议专为包含蛋白质组学数据的数据集而设计,该数据集表示为log2(ratio);代谢组学数据表示为倍数变化;以及临床特征,包括连续性和分类性特征。应根据诊断或其他类似参数对患者或样本进行分组。样本或患者应跨行,特征应跨列。
- 若要在浏览器中启动 Jupyter Notebook 的新实例,请打开新的终端窗口,键入以下内容并按 Enter。
Jupyter 笔记本 - 在浏览器的 Jupyter 主页中,单击笔记本 M01 - 表达式数据 pre-processing.ipynb 以在新选项卡中打开它(补充文件 2,步骤 1.1)。
- 在笔记本 的第二个单元格 中,键入数据集文件的名称来代替 your_dataset_name.csv。
- 在笔记本 的最后一个单元格 中,键入输出数据文件的所需名称来代替 M01_output_data.csv。
- 在笔记本 的第五个单元格 中,指定每种数据类型的列的位置,如下所示:蛋白质组学数据 (cols_prot)、代谢组学数据 (cols_met)、连续临床数据(例如年龄)(cols_clin_con)、二进制临床数据(例如性别)(cols_clin_bin)。输入每种数据类型的第一列索引来代替 col_start ,最后几列索引代替 col_end;例如: cols_prot = slice(0, 8817)。确保切片对象中指定的值对应于与每种数据类型对应的第一列和最后一列索引。使用同一笔记本的第四个单元格中的命令 (df.iloc[:, :]) 确定每种数据类型的开始和结束位置(补充文件 2,步骤 1.2)。
- 选择 单元格 | 从 Jupyter 中的菜单栏运行所有内容,以在指定文件夹中创建输出数据文件(补充文件 2,步骤 1.3)。
注意:这些数据将用作第 2、3 或 4 节中描述的协议的输入。
2. 自定义优化工作流程(可选)
注意:第 2 部分是可选的,因为它是计算机密集型的。如果用户决定不执行第 2 节,则应直接跳到第 4 节。该协议将指导用户以自动化方式优化工作流程。具体而言,该方法可识别在生成提取特征以很好地分离样品组方面提供自动编码器最佳性能的参数。作为输出生成的优化参数包括用于特征选择的特征数量(k_prot 和 k_met)以及自编码器潜伏层中的神经元数量(潜伏)。然后,可以在第 3 节中描述的协议中使用这些参数来生成模型。
- 在浏览器的 Jupyter 主页上,单击笔记本 M02 - DeepOmicsAE model optimization.ipynb 以在新选项卡中打开它(补充文件 2,步骤 2.1)。
- 在笔记本 的第二个 单元格中,键入输入文件 的名称 来代替 M01_output_data.csv。此函数的输入是第 1 节的输出数据。
- 在笔记本 的第五个 单元格中,指定每种数据类型的列的位置,如下所示:蛋白质组学数据 (cols_X_prot)、代谢组学数据 (cols_X_met)、临床数据 (cols_clin;包括所有临床数据)、所有分子表达数据,包括蛋白质组学和代谢组学数据 (cols_X_expr)。输入每种数据类型的第一列索引来代替 col_start ,最后几列索引代替 col_end;例如, cols_prot = slice(0, 8817)。确保切片对象中指定的值对应于与每种数据类型对应的第一列和最后一列索引,并使用笔记本的第三和第四单元格中的命令浏览数据并确定每种数据类型的开始和结束位置。指定包含目标变量的列的名称,以代替 y_column_name 作为 y_label (补充文件 2,步骤 2.2)。
注意:由于在数据预处理期间对数据帧进行重塑, 因此 cols_X_prot、 cols_X_met、 cols_clin 和 cols_X_expr 中指定的索引值将与第 1 节中使用的值不同。 - 在笔记本的第 六个 单元格中,通过为 n_comb 分配值来指定要执行的优化轮数。处理时间约为4-5分钟,共10轮;20 分钟 50 发,40 分钟 100 发(补充文件 2,步骤 2.3)。
- 选择 单元格 | 从 Jupyter 中的菜单栏运行所有内容。
注意:输出变量 kprot、 kmet 和 latent 将被存储,并可从其他笔记本访问,这些笔记本将用于继续分析工作流程。绘图 AE_optimization_plot.pdf 将生成并保存在本地文件夹中(图 2)。
3. 使用自定义优化参数实现工作流
注意:仅在方法优化(第 2 节)后执行此协议。如果用户选择不执行方法优化,请直接跳到第 4 节。该协议将指导用户使用从第 2 节派生的自定义优化参数生成模型。自动编码器将 1) 生成一组提取的特征,以概括原始数据,以及 2) 识别驱动潜在层中每个神经元的重要特征,有效地表示独特的信号模块。信令模块将使用第 5 节中提供的协议进行解释。
- 在浏览器的 Jupyter 主页上,单击 带有自定义优化参数的笔记本 M03a - DeepOmicsAE 实现.ipynb ,在新选项卡中打开它(补充文件 2,步骤 3.1)。
- 在笔记本 的第二个 单元格中,键入输入文件的名称来代替 M01_output_data.csv。此函数的输入是第 1 节的输出数据。
- 在笔记本 的第五个 单元格中,指定每种数据类型的列的位置,如下所示:蛋白质组学数据 (cols_prot)、代谢组学数据 (cols_met)、临床数据 (cols_clin;包括所有临床数据)。输入每种数据类型的第一列索引来代替 col_start ,最后几列索引代替 col_end;例如: cols_prot = slice(0, 8817)。确保切片对象中指定的值对应于对应于每种数据类型的第一列和最后一列索引,并使用笔记本的第三和第四单元格中的命令浏览数据并确定每种数据类型的开始和结束位置。指定包含目标变量的列的名称(例如, 0 或 1,对应 于健康 或 患病),而不是 y_column_name 作为 y_label。
注意:由于在数据预处理期间发生了数据帧的重塑, 因此 cols_X_prot、 cols_X_met、 cols_clin 和 cols_X_expr 中指定的索引值将与第 1 节中使用的索引值不同。 - 选择 单元格 | 从 Jupyter 中的菜单栏运行所有内容,以生成绘图 PCA_initial_data.pdf、PCA_extracted_features.pdf 和 distribution_important_feature_scores.pdf 并将其保存在本地文件夹中(图 3 和 补充图 S1)。此外,每个已识别信令模块的重要功能列表将存储在名为 module_n.txt 的本地文件夹的文本文件中,其中 n 将被 模块编号替换。
4. 使用预设参数实现工作流程
- 有关如何运行此方法的详细说明,请参阅第 3 节(补充文件 2,步骤 4.1)。这两个协议之间的唯一区别是参数 kprot、 kmet 和 latent (在笔记本 的第七个 单元格中)是根据执行的优化结果在数学上推导出来的,如 图 2 所示。
注意:如果第 4 节提供的样本组分离不佳,表明模型性能欠佳,则建议使用 至少 15 次迭代来执行模型优化(第 2 节),如果可能, 最多 50 次迭代。
5. 使用 MetaboAnalyst 进行生物学解释
- 打开浏览器并导航到下面的链接,以访问MetaboAnalyst网站上的联合通路分析功能:https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml。
- 访问保存方法 3 或方法 4 的输出文件的文件夹,并打开方法 3 或方法 4 生成的每个信令模块 n module_n.txt 的文本文件。
- 在文本文件中找到蛋白质并复制它们。
- 将蛋白质列表粘贴到 MetaboAnalyst 网页中 具有可选倍数变化的基因/蛋白质 窗口中。
- 对代谢物重复上述步骤,并将它们粘贴到同一网页上 具有可选折叠更改的窗口化合物列表中 。
- 选择适当的 生物体 和 ID 类型,然后单击页面底部 的提交 (补充文件 2,步骤 5.1)。
注意:确保 MetaboAnalyst 识别标识符。公认的标识符包括 Entrez ID、官方基因符号和蛋白质的 Uniprot ID;代谢物的化合物名称、HMDB ID 和 KEGG ID。如果标识符不是这些类型,则在分析之前需要进行适当的转换。 - 在下一页上,检查 ID 映射,然后单击 “继续 ”以验证标识符是否被识别。
- 在 “参数设置 ”页面中,选择“ 代谢通路(集成) ”或 “所有通路(集成) ”,以分别可视化输入对仅代谢通路或所有信号通路的贡献(补充文件 2,步骤 5.2)。在 “算法选择 ”面板中,选择“ 扩集分析:超几何检验”、“拓扑测量:度中心性”和 “积分方法:合并 p 值(路径级别)”。点击页面底部 的提交 。
- 最后一页是 “结果视图”,其中显示了富集分析的结果。富集路径根据其影响和重要性绘制,路径列表也以表格格式提供。
结果
为了展示该协议,我们分析了一个数据集,该数据集包括蛋白质组、代谢组和来自 142 名健康或被诊断患有阿尔茨海默病的个体的死后大脑的临床信息。
在执行协议第 1 部分对数据进行预处理后,数据集包括 6,497 种蛋白质、443 种代谢物和三个临床特征(性别、死亡年龄和教育程度)。目标特征是死亡时认知状态的临床共识诊断,编码为 cogdx,?...
讨论
数据集的结构对于协议的成功至关重要,应仔细检查。数据的格式应如协议第 1 节所示。正确分配列位置对于该方法的成功也至关重要。蛋白质组学和代谢组学数据的预处理方式不同,由于数据性质不同,特征选择是分开进行的。因此,在协议步骤 1.5、2.3 和 3.3 中正确分配色谱柱位置至关重要。
如果临床数据包含非数值(连续值或二进制值)的数据类型,则用户在运行协议?...
披露声明
提交人声明他们没有利益冲突。
致谢
这项工作得到了美国国立卫生研究院CA201402资助和康奈尔大学脊椎动物基因组学中心(CVG)杰出学者奖的支持。此处发布的结果全部或部分基于从 AD 知识门户 (https://adknowledgeportal.org) 获得的数据。研究数据是通过AD加速医学伙伴关系(U01AG046161和U01AG061357)提供的,基于芝加哥拉什大学医学中心拉什阿尔茨海默病中心提供的样本。数据收集得到了NIA赠款的支持,P30AG10161、R01AG15819、R01AG17917、R01AG30146、R01AG36836、U01AG32984、U01AG46152、伊利诺伊州公共卫生部和转化基因组学研究所。代谢组学数据集在 Metabolon 生成,并由 ADMC 进行预处理。
材料
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
参考文献
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。