
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
DeepOmicsAE: Representando Módulos de Sinalização na Doença de Alzheimer com Análise de Aprendizagem Profunda de Proteômica, Metabolômica e Dados Clínicos
Neste Artigo
Resumo
DeepOmicsAE é um fluxo de trabalho centrado na aplicação de um método de aprendizagem profunda (ou seja, um autoencoder) para reduzir a dimensionalidade de dados multi-ômicos, fornecendo uma base para modelos preditivos e módulos de sinalização que representam várias camadas de dados ômicos.
Resumo
Grandes conjuntos de dados ômicos estão se tornando cada vez mais disponíveis para pesquisas em saúde humana. Este artigo apresenta o DeepOmicsAE, um fluxo de trabalho otimizado para a análise de conjuntos de dados multi-ômicos, incluindo proteômica, metabolômica e dados clínicos. Esse fluxo de trabalho emprega um tipo de rede neural chamada autoencoder, para extrair um conjunto conciso de recursos dos dados de entrada multi-ômicos de alta dimensão. Além disso, o fluxo de trabalho fornece um método para otimizar os principais parâmetros necessários para implementar o autoencoder. Para mostrar esse fluxo de trabalho, dados clínicos foram analisados de uma coorte de 142 indivíduos saudáveis ou diagnosticados com doença de Alzheimer, juntamente com o proteoma e o metaboloma de suas amostras cerebrais post-mortem. As características extraídas da camada latente do autoencoder retêm a informação biológica que separa pacientes saudáveis e doentes. Além disso, as características individuais extraídas representam módulos de sinalização molecular distintos, cada um dos quais interage de forma única com as características clínicas dos indivíduos, fornecendo um meio para integrar os dados proteômicos, metabolômicos e clínicos.
Introdução
Uma proporção cada vez maior da população está envelhecendo e espera-se que a carga de doenças relacionadas à idade, como a neurodegeneração, aumente acentuadamente nas próximas décadas1. A doença de Alzheimer é o tipo mais comum de doença neurodegenerativa2. O progresso na busca de um tratamento tem sido lento, dada a nossa fraca compreensão dos mecanismos moleculares fundamentais que impulsionam o início e o progresso da doença. A maioria das informações sobre a doença de Alzheimer é obtida post-mortem a partir do exame do tecido cerebral, o que tornou a distinção entre causas e consequências uma tarefa difícil3. O Religious Orders Study/Memory and Aging Project (ROSMAP) é um esforço ambicioso para obter uma compreensão mais ampla da neurodegeneração, que envolve o estudo de milhares de indivíduos que se comprometeram a submeter-se a exames médicos e psicológicos anualmente e a contribuir com seus cérebros para pesquisas após sua morte4. O estudo se concentra na transição do funcionamento normal do cérebro para a doença de Alzheimer2. Dentro do projeto, amostras cerebrais post-mortem foram analisadas com uma infinidade de abordagens ômicas, incluindo genômica, epigenômica, transcriptômica, proteômica5 e metabolômica.
Tecnologias ômicas que oferecem leituras funcionais de estados celulares (i.e., proteômica e metabolômica)6,7 são fundamentais para a interpretação da doença8,9,10,11,12, devido à relação direta entre a abundância de proteínas e metabólitos e as atividades celulares. As proteínas são os principais executores dos processos celulares, enquanto os metabólitos são os substratos e produtos para as reações bioquímicas. A análise de dados multi-ômicos oferece a possibilidade de compreender as complexas relações entre dados proteômicos e metabolômicos em vez de apreciá-los isoladamente. Multi-omics é uma disciplina que estuda múltiplas camadas de dados biológicos de alta dimensão, incluindo dados moleculares (sequência e mutações do genoma, transcriptoma, proteoma, metaboloma), dados de imagem clínica e características clínicas. Particularmente, a análise de dados multi-ômicos visa integrar essas camadas de dados biológicos, entender sua regulação recíproca e dinâmica de interação e fornecer uma compreensão holística do início e progressão da doença. No entanto, os métodos de integração de dados multi-ômicos permanecem nos estágios iniciais de desenvolvimento13.
Os autoencoders, um tipo de rede neural não supervisionada14, são uma ferramenta poderosa para a integração de dados multi-ômicos. Ao contrário das redes neurais supervisionadas, os autoencoders não mapeiam amostras para valores-alvo específicos (como saudáveis ou doentes), nem são usados para prever resultados. Uma de suas principais aplicações está na redução da dimensionalidade. No entanto, os autoencoders oferecem várias vantagens em relação a métodos mais simples de redução de dimensionalidade, como análise de componentes principais (PCA), incorporação de vizinhos estocásticos distribuídos em t (tSNE) ou aproximação e projeção de variedade uniforme (UMAP). Ao contrário do PCA, os codificadores automáticos podem capturar relações não lineares dentro dos dados. Ao contrário de tSNE e UMAP, eles podem detectar relações hierárquicas e multimodais dentro dos dados, uma vez que dependem de várias camadas de unidades computacionais, cada uma contendo funções de ativação não lineares. Portanto, representam modelos atrativos para capturar a complexidade de dados multi-ômicos. Finalmente, enquanto a principal aplicação do PCA, tSNE e UMAP é a de agrupar os dados, os codificadores automáticos compactam os dados de entrada em recursos extraídos que são adequados para tarefas preditivas a jusante15,16.
Resumidamente, as redes neurais compreendem várias camadas, cada uma contendo múltiplas unidades computacionais ou "neurônios". A primeira e a última camadas são chamadas de camadas de entrada e saída, respectivamente. Autoencoders são redes neurais com uma estrutura ampulheta, consistindo de uma a três camadas de entrada, seguidas por uma a três camadas ocultas e uma pequena camada "latente" tipicamente contendo entre dois e seis neurônios. A primeira metade dessa estrutura é conhecida como codificador e é combinada com um decodificador espelhando o codificador. O decodificador termina com uma camada de saída contendo o mesmo número de neurônios que a camada de entrada. Os codificadores automáticos levam a entrada através do gargalo e a reconstroem na camada de saída, com o objetivo de gerar uma saída que espelhe as informações originais o mais próximo possível. Isso é conseguido minimizando matematicamente um parâmetro denominado "perda de reconstrução". A entrada consiste em um conjunto de características, que no aplicativo apresentado aqui serão abundâncias de proteínas e metabólitos e características clínicas (ou seja, sexo, educação e idade ao morrer). A camada latente contém uma representação compactada e rica em informações da entrada, que pode ser usada para aplicações subsequentes, como modelos preditivos17,18.
Este protocolo apresenta um fluxo de trabalho, DeepOmicsAE, que envolve: 1) pré-processamento de dados proteômicos, metabolômicos e clínicos (ou seja, normalização, escala, remoção de outliers) para obter dados com uma escala consistente para análise de aprendizado de máquina; 2) selecionar recursos de entrada apropriados do autoencoder, uma vez que a sobrecarga de recursos pode obscurecer padrões de doenças relevantes; 3) otimizar e treinar o autoencoder, incluindo a determinação do número ótimo de proteínas e metabólitos a serem selecionados, e de neurônios para a camada latente; 4) extrair feições da camada latente; e 5) utilizar as características extraídas para interpretação biológica, identificando módulos de sinalização molecular e sua relação com as características clínicas.
Este protocolo pretende ser simples e aplicável por biólogos com experiência computacional limitada que tenham um conhecimento básico de programação com Python. O protocolo se concentra na análise de dados multi-ômicos, incluindo proteômica, metabolômica e características clínicas, mas seu uso pode ser estendido a outros tipos de dados de expressão molecular, incluindo transcriptômica. Uma importante aplicação nova introduzida por este protocolo é o mapeamento dos escores de importância das características originais em neurônios individuais na camada latente. Como resultado, cada neurônio da camada latente representa um módulo de sinalização, detalhando as interações entre alterações moleculares específicas e as características clínicas dos pacientes. A interpretação biológica dos módulos de sinalização molecular é obtida usando o MetaboAnalyst, uma ferramenta publicamente disponível que integra dados de genes/proteínas e metabólitos para derivar vias metabólicas e de sinalização celular enriquecidas17.
Protocolo
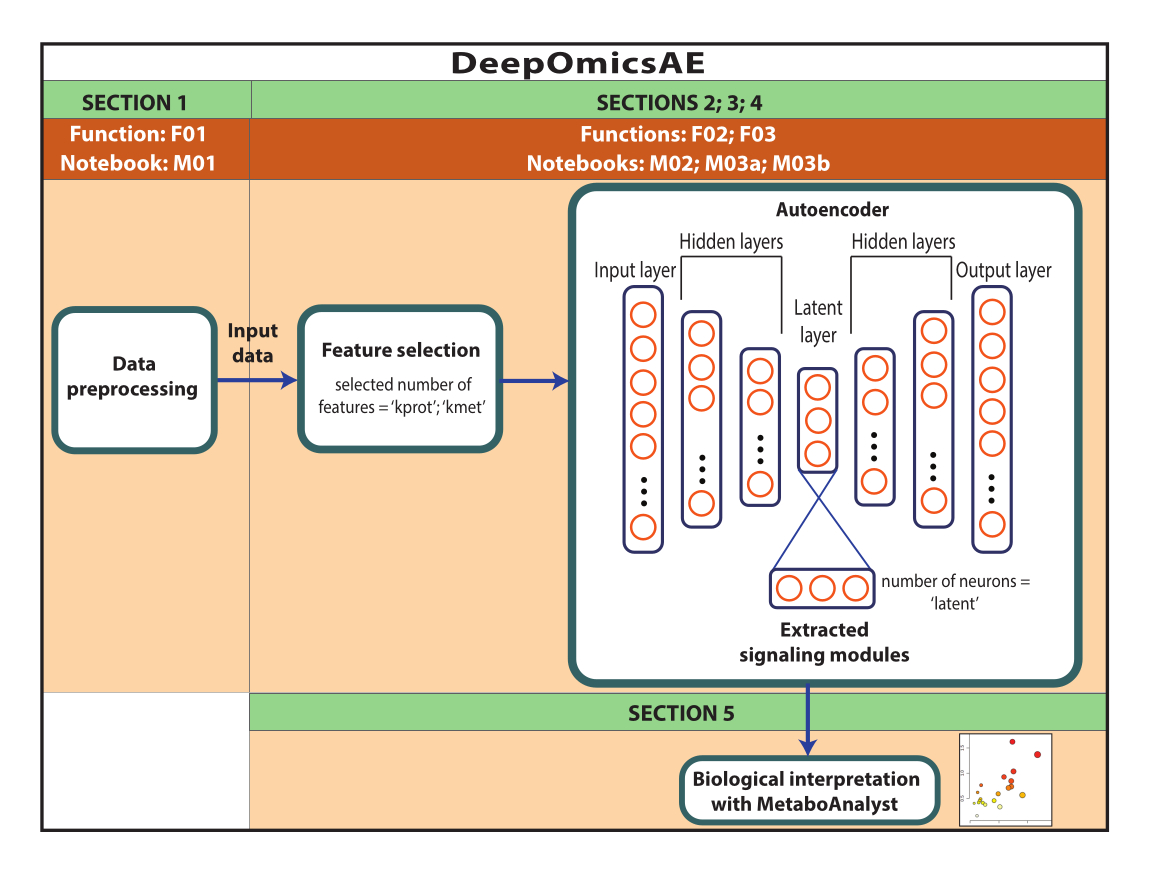
Observação : os dados usados aqui foram dados ROSMAP baixados do portal de conhecimento do AD. Não é necessário consentimento informado para baixar e reutilizar os dados. O protocolo aqui apresentado utiliza deep learning para analisar dados multi-ômicos e identificar módulos de sinalização que distinguem pacientes específicos ou grupos amostrais baseados, por exemplo, em seu diagnóstico. O protocolo também fornece um pequeno conjunto de recursos extraídos que resumem os dados originais em grande escala e podem ser usados para análises adicionais, como o treinamento de um modelo preditivo usando algoritmos de aprendizado de máquina (Figura 1). Consulte o Arquivo Suplementar 1 e a Tabela de Materiais para obter informações sobre como acessar o código e configurar o ambiente computacional antes de executar o protocolo. Os métodos devem ser executados seguindo a ordem especificada abaixo.

Figura 1: Esquema do fluxo de trabalho do DeepOmicsAE. Representação esquemática do fluxo de trabalho para analisar dados multi-ômicos usando o fluxo de trabalho. Na representação do autoencoder, retângulos representam camadas da rede neural e círculos representam neurônios dentro de camadas. Clique aqui para ver uma versão maior desta figura.
{kind=link}
1. Pré-processamento de dados
NOTA: O objetivo desta seção é pré-processar os dados, incluindo o tratamento de dados ausentes; normalização e escalonamento de dados proteômicos, metabolômicos e clínicos; e remoção de outliers. O protocolo é projetado para um conjunto de dados que inclui dados proteômicos expressos como log2 (razão); dados metabolômicos expressos como mudança de dobra; e características clínicas, incluindo características contínuas e categóricas. Os pacientes ou amostras devem ser agrupados com base no diagnóstico ou em outros parâmetros semelhantes. As amostras ou os pacientes devem estar nas linhas e nos recursos nas colunas.
- Para iniciar uma nova instância do Jupyter Notebook no navegador, abra uma nova janela do terminal, digite o seguinte e pressione Enter.
Caderno Jupyter - Na página inicial do Jupyter no navegador, clique no notebook M01 - expression data pre-processing.ipynb para abri-lo em uma nova aba (Arquivo Suplementar 2, Passo 1.1).
- Na segunda célula do bloco de anotações, digite o nome do arquivo de conjunto de dados no lugar de your_dataset_name.csv.
- Na última célula do bloco de anotações, digite o nome desejado do arquivo de dados de saída no lugar de M01_output_data.csv.
- Na quinta célula do caderno, especifique a posição das colunas para cada tipo de dados da seguinte forma: dados proteômicos (cols_prot), dados metabolômicos (cols_met), dados clínicos contínuos (por exemplo, idade) (cols_clin_con), dados clínicos binários (por exemplo, sexo) (cols_clin_bin). Insira o índice da primeira coluna para cada tipo de dados no lugar de col_start e o índice das últimas colunas no lugar de col_end; por exemplo: cols_prot = slice(0, 8817). Verifique se os valores especificados nos objetos de fatia correspondem aos índices da primeira e da última colunas correspondentes a cada tipo de dados. Use o comando na quarta célula do mesmo bloco de anotações (df.iloc[:, :]) para determinar a posição inicial e final para cada tipo de dados (Arquivo Suplementar 2, Etapa 1.2).
- Selecionar célula | Execute tudo a partir da barra de menus no Jupyter para criar o arquivo de dados de saída na pasta especificada (Arquivo Suplementar 2, Etapa 1.3).
NOTA: Esses dados serão usados como entrada para os protocolos descritos nas seções 2, 3 ou 4.
2. Otimização personalizada do fluxo de trabalho (opcional)
Observação : seção 2 é opcional porque ele é computador intensivo. Os usuários devem pular diretamente para a seção 4 se decidirem não executar a seção 2. Esse protocolo guiará o usuário pela otimização do fluxo de trabalho de forma automatizada. Especificamente, o método identifica os parâmetros que oferecem o melhor desempenho do autoencoder em termos de geração de recursos extraídos que separam bem os grupos de amostra. Os parâmetros otimizados gerados como saída incluem o número de recursos a serem usados para seleção de feições (k_prot e k_met) e o número de neurônios na camada latente do autoencoder (latente). Esses parâmetros podem então ser usados no protocolo descrito na seção 3 para gerar o modelo.
- Na página inicial do Jupyter no navegador, clique no notebook M02 - DeepOmicsAE model optimization.ipynb para abri-lo em uma nova aba (Arquivo Suplementar 2, Passo 2.1).
- Na segunda célula do bloco de anotações, digite o nome do arquivo de entrada no lugar de M01_output_data.csv. A entrada para esta função são os dados de saída da seção 1.
- Na quinta célula do caderno, especifique a posição das colunas para cada tipo de dados da seguinte forma: dados proteômicos (cols_X_prot), dados metabolômicos (cols_X_met), dados clínicos (cols_clin; inclui todos os dados clínicos), todos os dados de expressão molecular, incluindo dados proteômicos e metabolômicos (cols_X_expr). Insira o índice da primeira coluna para cada tipo de dados no lugar de col_start e o índice das últimas colunas no lugar de col_end; por exemplo, cols_prot = fatia(0, 8817). Certifique-se de que os valores especificados nos objetos de fatia correspondam ao índice da primeira e da última colunas correspondentes a cada tipo de dados e use os comandos na terceira e quarta células do bloco de anotações para explorar os dados e determinar as posições inicial e final para cada tipo de dados. Especifique o nome da coluna que contém a variável de destino no lugar de y_column_name como y_label (Arquivo Suplementar 2, Etapa 2.2).
Observação : os valores dos índices especificados em cols_X_prot, cols_X_met, cols_clin e cols_X_expr serão diferentes daqueles usados na seção 1 devido à remodelagem do dataframe que ocorre durante o pré-processamento de dados. - Na sexta célula do bloco de anotações, especifique quantas rodadas de otimização devem ser executadas atribuindo um valor a n_comb. Os tempos de processamento são de aproximadamente 4-5 min para 10 rodadas; 20 min para 50 rodadas e 40 min para 100 rodadas (Arquivo Suplementar 2, Passo 2.3).
- Selecionar célula | Execute tudo a partir da barra de menus no Jupyter.
NOTA: As variáveis de saída kprot, kmet e latent serão armazenadas e podem ser acessadas a partir dos outros blocos de anotações, que serão usados para continuar o fluxo de trabalho analítico. O AE_optimization_plot.pdf do gráfico será gerado e salvo na pasta local (Figura 2).
3. Implementação do fluxo de trabalho com parâmetros otimizados sob medida
Observação : execute esse protocolo somente após a otimização do método (seção 2). Se os usuários optarem por não executar a otimização do método, pule diretamente para a seção 4. Este protocolo guiará o usuário através da geração de um modelo usando os parâmetros otimizados personalizados derivados da seção 2. O autoencoder irá 1) gerar um conjunto de características extraídas que recapitulam os dados originais e 2) identificar as características importantes que conduzem cada neurônio na camada latente, representando efetivamente módulos de sinalização únicos. Os módulos de sinalização serão interpretados utilizando o protocolo previsto na seção 5.
- Na página inicial do Jupyter no navegador, clique no notebook M03a - implementação DeepOmicsAE com parâmetros otimizados personalizados.ipynb para abri-lo em uma nova aba (Arquivo Suplementar 2, Passo 3.1).
- Na segunda célula do bloco de anotações, digite o nome do arquivo de entrada no lugar de M01_output_data.csv. A entrada para esta função são os dados de saída da seção 1.
- Na quinta célula do caderno, especifique a posição das colunas para cada tipo de dados da seguinte forma: dados proteômicos (cols_prot), dados metabolômicos (cols_met), dados clínicos (cols_clin; inclui todos os dados clínicos). Insira o índice da primeira coluna para cada tipo de dados no lugar de col_start e o índice das últimas colunas no lugar de col_end; por exemplo: cols_prot = fatia(0, 8817). Certifique-se de que os valores especificados nos objetos de fatia correspondam aos índices da primeira e da última colunas correspondentes a cada tipo de dados e use os comandos na terceira e quarta células do bloco de anotações para explorar os dados e determinar as posições inicial e final para cada tipo de dados. Especifique o nome da coluna que contém a variável de destino (por exemplo, 0 ou 1, correspondente a saudável ou doente) no lugar de y_column_name como y_label.
Observação : o valor dos índices especificados em cols_X_prot, cols_X_met, cols_clin e cols_X_expr será diferente daqueles usados na seção 1 devido à remodelagem do dataframe que ocorre durante o pré-processamento de dados. - Selecionar célula | Execute tudo a partir da barra de menus no Jupyter para gerar e salvar os gráficos PCA_initial_data.pdf, PCA_extracted_features.pdf e distribution_important_feature_scores.pdf na pasta local (Figura 3 e Figura Suplementar S1). Adicionalmente, listas de características importantes para cada módulo de sinalização identificado serão armazenadas em arquivos de texto na pasta local, denominada module_n.txt, onde n será substituído pelo número do módulo.
4. Implementação do fluxo de trabalho com parâmetros predefinidos
- Consulte a seção 3 para obter instruções detalhadas sobre como executar esse método (Arquivo Suplementar 2, Etapa 4.1). A única diferença entre esses dois protocolos é que os parâmetros kprot, kmet e latente (na sétima célula do notebook) são derivados matematicamente com base nos resultados da otimização realizada como mostra a Figura 2.
NOTA: Se a seção 4 fornecer uma separação ruim dos grupos de amostra, indicando desempenho de modelo abaixo do ideal, recomenda-se executar a otimização do modelo (seção 2) usando pelo menos 15 iterações e, se possível, até 50.
5. Interpretação biológica usando MetaboAnalyst
- Abra o navegador e navegue até o link abaixo para acessar a funcionalidade Joint Pathway Analysis no site do MetaboAnalyst : https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml.
- Acesse a pasta onde os arquivos de saída do Método 3 ou Método 4 foram salvos e abra os arquivos de texto module_n.txt para cada módulo de sinalização n gerado pelo Método 3 ou pelo Método 4.
- Localize as proteínas nos arquivos de texto e copie-as.
- Cole a lista de proteínas na janela Genes/proteínas com alterações de dobra opcionais na página da Web MetaboAnalyst.
- Repita a etapa acima para metabólitos e cole-os na janela Lista de compostos com alterações de dobra opcionais na mesma página da Web.
- Selecione o organismo apropriado e o tipo de ID e clique em Enviar na parte inferior da página (Arquivo Suplementar 2, Etapa 5.1).
NOTA: Certifique-se de que os identificadores são reconhecidos pelo MetaboAnalyst. Os identificadores reconhecidos incluem Entrez ID, símbolos oficiais de genes e Uniprot ID para proteínas; nome do composto, ID HMDB e ID KEGG para metabólitos. Se os identificadores forem diferentes desses tipos, a conversão apropriada será necessária antes da análise. - Na página seguinte, verifique o mapeamento de ID antes de clicar em Continuar para verificar se os identificadores estão sendo reconhecidos.
- Na página Configuração de parâmetros , selecione Vias metabólicas (integradas) ou Todas as vias (integradas) para visualizar, respectivamente, a contribuição da entrada apenas para as vias metabólicas ou para todas as vias de sinalização (Arquivo Suplementar 2, Etapa 5.2). No painel Seleção de algoritmo , escolha Análise de enriquecimento: Teste hipergeométrico, Medida de topologia: Centralidade de grau e Método de integração: Combinar valores de p (nível de caminho). Clique em Enviar na parte inferior da página.
- A última página é a Visualização de Resultados, que apresenta os resultados da análise de enriquecimento. Os caminhos enriquecidos são plotados com base em seu impacto e significado, e a lista de caminhos também é fornecida em formato tabular.
Resultados
Para mostrar o protocolo, analisamos um conjunto de dados que compreende o proteoma, metaboloma e informações clínicas derivadas de cérebros post-mortem de 142 indivíduos saudáveis ou diagnosticados com doença de Alzheimer.
Após a realização da seção 1 do protocolo para pré-processar os dados, o conjunto de dados incluiu 6.497 proteínas, 443 metabólitos e três características clínicas (sexo, idade ao morrer e educação). A característica alvo é o diagnóstico de co...
Discussão
A estrutura do conjunto de dados é crítica para o sucesso do protocolo e deve ser cuidadosamente verificada. Os dados devem ser formatados conforme indicado na seção 1 do protocolo. A atribuição correta de posições de coluna também é fundamental para o sucesso do método. Os dados proteômicos e metabolômicos são pré-processados de forma diferente e a seleção de características é conduzida separadamente devido à natureza diferente dos dados. Portanto, é fundamental atribuir posições de coluna correta...
Divulgações
O autor declara não haver conflitos de interesse.
Agradecimentos
Este trabalho foi apoiado pelo NIH grant CA201402 e pelo Cornell Center for Vertebrate Genomics (CVG) Distinguished Scholar Award. Os resultados aqui publicados são, no todo ou em parte, baseados em dados obtidos do Portal de Conhecimento do AD (https://adknowledgeportal.org). Os dados do estudo foram fornecidos através da Accelerating Medicine Partnership for AD (U01AG046161 e U01AG061357) com base em amostras fornecidas pelo Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago. A coleta de dados foi apoiada por meio de financiamento de subsídios do NIA P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, do Departamento de Saúde Pública de Illinois e do Instituto de Pesquisa Genômica Translacional. O conjunto de dados metabolômicos foi gerado no Metabolon e pré-processado pelo ADMC.
Materiais
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
Referências
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados