
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
DeepOmicsAE:プロテオミクス、メタボロミクス、臨床データの深層学習解析によるアルツハイマー病のシグナル伝達モジュールの表現
要約
DeepOmicsAEは、深層学習手法(オートエンコーダー)の適用を中心としたワークフローで、マルチオミクスデータの次元を縮小し、オミクスデータの多層化を表す予測モデルとシグナリングモジュールの基盤を提供します。
要約
大規模なオミクスデータセットは、人間の健康に関する研究にますます利用されるようになっています。本稿では、プロテオミクス、メタボロミクス、臨床データを含むマルチオミクスデータセットの解析に最適化されたワークフローであるDeepOmicsAEについて紹介します。このワークフローでは、オートエンコーダーと呼ばれるニューラルネットワークの一種を使用して、高次元マルチオミクス入力データから簡潔な特徴セットを抽出します。さらに、このワークフローは、オートエンコーダーの実装に必要な主要なパラメーターを最適化する方法を提供します。このワークフローを紹介するために、健康な人またはアルツハイマー病と診断された142人のコホートから、死後の脳サンプルのプロテオームとメタボロームとともに臨床データを分析しました。オートエンコーダーの潜在層から抽出された特徴は、健康な患者と病気の患者を区別する生物学的情報を保持します。さらに、抽出された個々の特徴は、それぞれが個人の臨床的特徴と一意に相互作用する異なる分子シグナル伝達モジュールを表し、プロテオミクス、メタボロミクス、および臨床データを統合する手段を提供します。
概要
人口の高齢化が進む割合はますます大きくなっており、神経変性などの加齢性疾患の負担は今後数十年で急激に増加すると予想されています1。アルツハイマー病は、神経変性疾患の最も一般的なタイプです2。病気の発症と進行を促進する基本的な分子メカニズムの理解が不十分なため、治療法の発見の進歩は遅々として進んでいません。アルツハイマー病に関する情報の大部分は、脳組織の検査から死後得られるため、原因と結果を区別することは困難な作業となっています3。Religious Orders Study/Memory and Aging Project(ROSMAP)は、神経変性をより広く理解するための野心的な取り組みであり、毎年医学的および心理学的検査を受け、死後の研究のために脳を提供することを約束した何千人もの個人の研究が含まれます4。この研究は、脳の正常な機能からアルツハイマー病への移行に焦点を当てています2。このプロジェクトでは、ゲノミクス、エピゲノミクス、トランスクリプトミクス、プロテオミクス5、メタボロミクスなど、多数のオミクスアプローチを用いて死後の脳サンプルを分析しました。
細胞の状態を機能的に読み取るオミクス技術(プロテオミクスやメタボロミクス)6,7は、タンパク質や代謝物の存在量と細胞活性との直接的な関係から、疾患を解釈する上で鍵となります8,9,10,11,12。タンパク質は細胞プロセスの主要な実行因子であり、代謝産物は生化学反応の基質および生成物です。マルチオミクスデータ解析は、プロテオミクスとメタボロミクスのデータとを単独で評価するのではなく、それらの間の複雑な関係を理解する可能性を提供します。マルチオミクスは、分子データ(ゲノム配列と変異、トランスクリプトーム、プロテオーム、メタボローム)、臨床画像データ、臨床的特徴など、高次元の生物学的データを多層的に研究する分野です。特に、マルチオミクスデータ解析は、このような生体データの層を統合し、それらの相互制御と相互作用のダイナミクスを理解し、疾患の発症と進行の全体像を理解することを目的としています。しかし、マルチオミクスデータを統合する方法は、まだ開発の初期段階にとどまっています13。
教師なしニューラルネットワーク14の一種であるオートエンコーダは、マルチオミクスデータ統合のための強力なツールである。教師ありニューラルネットワークとは異なり、オートエンコーダーはサンプルを特定のターゲット値(健康や病気の値など)にマッピングしたり、結果の予測に使用したりしません。その主な用途の1つは、次元削減にあります。ただし、自己符号化器には、主成分分析 (PCA)、t 分布確率的近傍埋め込み (tSNE)、一様多様体近似および射影 (UMAP) などの単純な次元削減法に比べていくつかの利点があります。PCA とは異なり、自己符号化器はデータ内の非線形関係をキャプチャできます。tSNEやUMAPとは異なり、それぞれが非線形活性化関数を含む計算ユニットの複数のレイヤーに依存しているため、データ内の階層的およびマルチモーダルな関係を検出できます。したがって、マルチオミクスデータの複雑さを捉えるための魅力的なモデルとなります。最後に、PCA、tSNE、およびUMAPの主な用途はデータのクラスタリングですが、オートエンコーダーは入力データを圧縮して、下流の予測タスクに適した抽出された特徴量にします15,16。
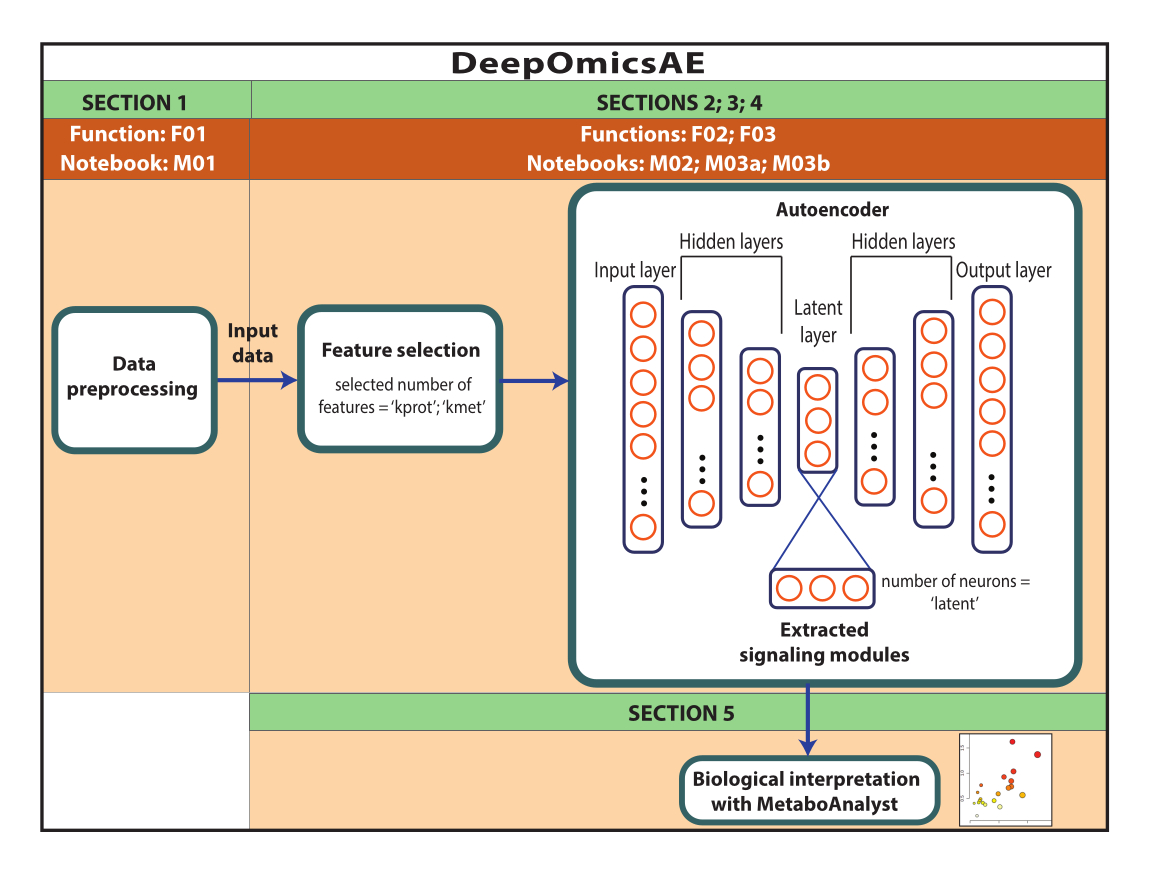
簡単に言うと、ニューラルネットワークは複数の層で構成されており、それぞれに複数の計算単位または「ニューロン」が含まれています。最初と最後の層は、それぞれ入力層と出力層と呼ばれます。オートエンコーダーは、砂時計構造を持つニューラルネットワークで、入力層、それに続く1〜3つの隠れ層、および通常は2〜6個のニューロンを含む小さな「潜在」層で構成されています。この構造の前半はエンコーダーと呼ばれ、エンコーダーをミラーリングするデコーダーと組み合わされています。復号化器は、入力層と同じ数のニューロンを含む出力層で終了します。オートエンコーダーは、ボトルネックを介して入力を取得し、元の情報を可能な限り反映した出力を生成することを目的として、出力層で再構築します。これは、「再構成損失」と呼ばれるパラメータを数学的に最小化することによって実現されます。インプットは、一組の特徴から成り、本明細書に紹介するアプリケーションでは、タンパク質および代謝物の存在量、ならびに臨床的特徴(すなわち、性別、教育、および死亡時年齢)となるであろう。潜在層は、入力の圧縮された情報に富んだ表現を含み、これは、予測モデル17、18などの後続のアプリケーションに使用することができる。
このプロトコルは、1)プロテオミクス、メタボロミクス、および臨床データの前処理(すなわち、正規化、スケーリング、外れ値除去)を含むワークフローDeepOmicsAEを提示し、機械学習分析のための一貫したスケールのデータを取得します。2)特徴の過負荷は関連する疾患パターンを不明瞭にする可能性があるため、適切な自己符号化器入力特徴を選択する。3)選択するタンパク質と代謝物の最適な数、および潜伏層のニューロンの数を決定することを含む、オートエンコーダーの最適化とトレーニング。4)潜伏層から特徴を抽出する。5)分子シグナル伝達モジュールと臨床的特徴との関係を特定することにより、抽出された特徴を生物学的解釈に利用します。
このプロトコルは、Pythonでのプログラミングの基本的な理解を持っている、計算経験が限られている生物学者がシンプルで適用できることを目的としています。このプロトコルは、プロテオミクス、メタボロミクス、臨床的特徴などのマルチオミクスデータの解析に重点を置いていますが、トランスクリプトミクスを含む他のタイプの分子発現データにもその用途を拡張できます。このプロトコルによって導入された重要な新規アプリケーションの1つは、潜在層の個々のニューロンに元の特徴の重要性スコアをマッピングすることです。その結果、潜在層の各ニューロンはシグナル伝達モジュールを表し、特定の分子変化と患者の臨床的特徴との間の相互作用を詳述します。分子シグナル伝達モジュールの生物学的解釈は、遺伝子/タンパク質および代謝物データを統合して、濃縮された代謝および細胞シグナル伝達経路を導き出す公開ツールであるMetaboAnalystを使用することによって得られる17。
プロトコル
注:ここで使用したデータは、AD Knowledge PortalからダウンロードしたROSMAPデータです。データをダウンロードして再利用するためにインフォームドコンセントは必要ありません。本明細書に提示するプロトコールは、深層学習を利用してマルチオミクスデータを解析し、例えば、診断に基づいて特定の患者またはサンプルグループを区別するシグナル伝達モジュールを同定する。また、このプロトコルは、元の大規模データを要約した抽出された特徴量の小さなセットを提供し、機械学習アルゴリズムを使用した予測モデルのトレーニングなどのさらなる分析に使用できます(図1)。プロトコルを実行する前に、コードへのアクセスと計算環境の設定に関する情報については、 補足ファイル1 および 材料表 を参照してください。メソッドは、以下に指定されている順序に従って実行する必要があります。

図1:DeepOmicsAEワークフローの概略図。 ワークフローを使用してマルチオミクスデータを解析するためのワークフローの概略図。自己符号化器の描写では、四角形はニューラル ネットワークの層を表し、円は層内のニューロンを表します。 この図の拡大版をご覧になるには、ここをクリックしてください。
{kind=link}
1. データの前処理
注: このセクションの目的は、欠落しているデータの処理を含め、データを前処理することです。プロテオミクス、メタボロミクス発現、および臨床データの正規化とスケーリング。外れ値の削除。このプロトコルは、log2(ratio)で表されるプロテオミクスデータを含むデータセット用に設計されています。倍率変化として表されるメタボロミクスデータ。連続的およびカテゴリー的特徴を含む臨床的特徴。患者またはサンプルは、診断またはその他の同様のパラメーターに基づいてグループ化する必要があります。サンプルまたは患者は行にまたがり、特徴は列にまたがっている必要があります。
- ブラウザーで Jupyter Notebook の新しいインスタンスを起動するには、新しいターミナル ウィンドウを開き、次のように入力して Enter キーを押します。
Jupyter Notebook (英語) - ブラウザーの Jupyter ホーム ページで、ノートブック M01 - expression data pre-processing.ipynb をクリックして新しいタブで開きます (補足ファイル 2、手順 1.1)。
- ノートブックの 2 番目のセル に、 your_dataset_name.csvの代わりにデータセット ファイルの名前を入力します。
- ノートブック の最後のセル に、 M01_output_data.csvの代わりに出力データ ファイルの名前を入力します。
- ノートブックの 5 番目のセル で、プロテオミクス データ (cols_prot)、メタボロミクス データ (cols_met)、連続臨床データ (年齢など) (cols_clin_con)、バイナリ臨床データ (性別など) (cols_clin_bin) のように、各データ型の列の位置を指定します。 col_start の代わりに各データ型の最初の列インデックスを入力し、 col_endの代わりに最後の列インデックスを入力します。たとえば、 cols_prot = slice(0, 8817) のようになります。スライスオブジェクトで指定された値が、各データ型に対応する最初と最後の列インデックスに対応していることを確認します。同じノートブックの 4 番目のセル (df.iloc[:, :]) のコマンドを使用して、各データ型の開始位置と終了位置を決定します (補足ファイル 2、手順 1.2)。
- セルを選択 |Jupyter のメニュー バーから all を実行して、指定したフォルダーに出力データ ファイルを作成します (補足ファイル 2、手順 1.3)。
注: これらのデータは、セクション 2、3、または 4 で説明するプロトコルの入力として使用されます。
2.ワークフローのカスタム最適化(オプション)
注: セクション 2 は、コンピュータを集中的に使用するため、オプションです。ユーザーは、セクション 2 を実行しないことにした場合、セクション 4 に直接スキップする必要があります。このプロトコルは、自動化された方法でワークフローを最適化することでユーザーをガイドします。具体的には、この手法は、サンプルグループを適切に分離する抽出された特徴を生成するという点で、自己符号化器の最高のパフォーマンスを提供するパラメーターを特定します。出力として生成される最適化されたパラメーターには、特徴選択に使用する特徴の数 (k_prot と k_met) と自己符号化器潜在層のニューロンの数 (潜在) が含まれます。これらのパラメータは、セクション3で説明したプロトコルでモデルを生成するために使用できます。
- ブラウザーの Jupyter ホーム ページで、ノートブック M02 - DeepOmicsAE model optimization.ipynb をクリックして、新しいタブで開きます (補足ファイル 2、手順 2.1)。
- ノートブックの 2 番目のセルに、M01_output_data.csv の代わりに入力ファイルの名前を入力します。この関数への入力は、セクション 1 からの出力データです。
- ノートブックの 5 番目のセルで、プロテオミクス データ (cols_X_prot)、メタボロミクス データ (cols_X_met)、臨床データ (cols_clin、すべての臨床データを含む)、プロテオミクスおよびメタボロミクス データ (cols_X_expr) を含むすべての分子発現データ。各データ型の最初の列インデックスを col_start の代わりに入力し、最後の列インデックスを col_end の代わりに入力します。たとえば、cols_prot = slice(0, 8817) です。スライス オブジェクトで指定された値が、各データ型に対応する最初と最後の列インデックスに対応していることを確認し、ノートブックの 3 番目と 4 番目のセルのコマンドを使用してデータを探索し、各データ型の開始位置と終了位置を決定します。y_column_nameの代わりにターゲット変数を含む列の名前をy_labelとして指定します(補足ファイル2、ステップ2.2)。
注: cols_X_prot、 cols_X_met、 cols_clin、 および cols_X_expr で指定されたインデックスの値は、データの前処理中にデータフレームの再形成が行われるため、セクション 1 で使用されたものとは異なります。 - ノートブックの 6 番目の セルで、 n_comb に値を割り当てて、実行する最適化ラウンドの数を指定します。処理時間は、10ラウンドで約4〜5分です。50ラウンドで20分、100ラウンドで40分(補足ファイル2、ステップ2.3)。
- セルを選択 | Jupyter のメニュー バーからすべて実行します。
注: 出力変数 kprot、 kmet、 latent は保存され、他のノートブックからアクセスでき、分析ワークフローを続行するために使用されます。プロット AE_optimization_plot.pdf が生成され、ローカルフォルダに保存されます(図2)。
3. カスタム最適化パラメータによるワークフロー実装
注:このプロトコルは、分析法の最適化(セクション 2)に従ってのみ実行してください。メソッド最適化を実行しないことを選択した場合は、セクション 4 に直接進んでください。このプロトコルは、セクション2から導出されたカスタム最適化パラメータを使用してモデルを生成する方法をユーザーにガイドします。オートエンコーダーは、1)元のデータを再現する抽出された特徴のセットを生成し、2)潜在層の各ニューロンを駆動する重要な特徴を特定し、固有のシグナル伝達モジュールを効果的に表現します。シグナリングモジュールは、セクション 5 で提供されるプロトコルを使用して解釈されます。
- ブラウザーの Jupyter ホーム ページで、ノートブック M03a - DeepOmicsAE implementation with custom-optimized parameters.ipynb をクリックして、新しいタブで開きます (補足ファイル 2、手順 3.1)。
- ノートブックの 2 番目の セルに、 M01_output_data.csvの代わりに入力ファイルの名前を入力します。この関数への入力は、セクション 1 からの出力データです。
- ノートブックの 5 番目のセルで、プロテオミクス データ (cols_prot)、メタボロミクス データ (cols_met)、臨床データ (cols_clin、すべての臨床データを含む) のように、各データ型の列の位置を指定します。col_startの代わりに各データ型の最初の列インデックスを入力し、col_endの代わりに最後の列インデックスを入力します。たとえば、cols_prot = slice(0, 8817) のようになります。スライス オブジェクトで指定された値が、各データ型に対応する最初と最後の列インデックスに対応していることを確認し、ノートブックの 3 番目と 4 番目のセルのコマンドを使用してデータを探索し、各データ型の開始位置と終了位置を決定します。y_column_nameの代わりに、ターゲット変数を含む列の名前(例:0または1、健康または病気に対応)をy_labelとして指定します。
注: cols_X_prot、 cols_X_met、 cols_clin、 cols_X_expr で指定されたインデックスの値は、データの前処理中にデータフレームの形状が変更されるため、セクション 1 で使用されたものとは異なります。 - セルを選択 | Jupyter のメニュー バーから all を実行して、プロット PCA_initial_data.pdf、PCA_extracted_features.pdf、distribution_important_feature_scores.pdf を生成してローカル フォルダーに保存します (図 3 および補足図 S1)。さらに、識別された各シグナリングモジュールの重要な機能のリストは、module_n.txtという名前のローカルフォルダ内のテキストファイルに保存されます(nはモジュール番号に置き換えられます)。
4. プリセットパラメータによるワークフロー実装
- このメソッドの実行方法の詳細については、セクション3を参照してください(補足ファイル2、ステップ4.1)。これら 2 つのプロトコルの唯一の違いは、パラメーター kprot、 kmet、 および latent (ノートブックの 7 番目の セル内) が、 図 2 に示すように実行された最適化の結果に基づいて数学的に導出されることです。
注: セクション 4 でサンプル グループの分離が不十分で、モデルのパフォーマンスが最適でないことを示す場合は、 少なくとも 15 回、可能であれば 最大 50 回の反復を使用してモデルの最適化 (セクション 2) を実行することをお勧めします。
5. MetaboAnalystを用いた生物学的解釈
- ブラウザを開き、MetaboAnalyst WebサイトのJoint Pathway Analysis機能にアクセスするには、以下のリンクに移動します https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml。
- 方法3または方法4の出力ファイルが保存されたフォルダにアクセスし、方法3または方法4で生成された各シグナリングモジュールnのテキストファイル module_n.txt を開きます。
- テキストファイル内のタンパク質を見つけてコピーします。
- MetaboAnalyst ウェブページの Genes/proteins ウィンドウにタンパク質のリストを貼り付け、必要に応じて折り目を変更します。
- 代謝物について上記の手順を繰り返し、同じWebページ上の オプションの折り目変更を含むウィンドウ に貼り付けます。
- 適切な 生物 と ID タイプを選択し、ページ下部の [送信 ] をクリックします (補足ファイル 2、ステップ 5.1)。
注記: 識別子が MetaboAnalyst によって認識されることを確認してください。認識される識別子には、Entrez ID、公式の遺伝子シンボル、およびタンパク質のUniprot IDが含まれます。代謝物の化合物名、HMDB ID、およびKEGG ID。識別子がこれらの型以外の場合は、分析の前に適切な変換が必要です。 - 次のページで、[ 続行 ] をクリックする前に ID マッピングを確認し、識別子が認識されていることを確認します。
- [パラメーター設定]ページで、[代謝経路(統合)]または[すべての経路(統合)]を選択して、代謝経路のみまたはすべてのシグナル伝達経路への入力の寄与をそれぞれ視覚化します(補足ファイル2、ステップ5.2)。[アルゴリズムの選択] パネルで、[エンリッチメント解析: 超幾何学的検定]、[トポロジー メジャー: 次数中心性]、および [積分方法] で [p 値の結合 (経路レベル)] を選択します。ページ下部の [送信] をクリックします。
- 最後のページは [結果ビュー] で、エンリッチメント解析の結果が表示されます。エンリッチされた経路は、その影響と重要性に基づいてプロットされ、経路のリストも表形式で提供されます。
結果
このプロトコルを紹介するために、健康な人またはアルツハイマー病と診断された142人の死後脳から得られたプロテオーム、メタボローム、および臨床情報を含むデータセットを分析しました。
プロトコールセクション1を実行してデータを前処理した後、データセットには6,497のタンパク質、443の代謝物、および3つの臨床的特徴(性別、死亡時年齢、および教育)が含ま?...
ディスカッション
データセットの構造はプロトコルの成功に不可欠であり、慎重にチェックする必要があります。データは、プロトコル セクション 1 に示されているようにフォーマットする必要があります。列の位置を正しく割り当てることも、分析法の成功に不可欠です。プロテオミクスとメタボロミクスのデータは異なる方法で前処理され、データの性質が異なるため、特徴量の選択は別々に行われます...
開示事項
著者は、利益相反がないことを宣言します。
謝辞
この研究は、NIHの助成金CA201402とCornell Center for Vertebrate Genomics(CVG)のDistinguished Scholar Awardの支援を受けました。ここで公開している結果の全部または一部は、AD Knowledge Portal(https://adknowledgeportal.org)から入手したデータに基づいています。研究データは、シカゴのラッシュ大学医療センターのラッシュアルツハイマー病センターから提供されたサンプルに基づいて、ADの加速医療パートナーシップ(U01AG046161およびU01AG061357)を通じて提供されました。データ収集は、NIAの助成金P30AG10161、R01AG15819、R01AG17917、R01AG30146、R01AG36836、U01AG32984、U01AG46152、イリノイ州公衆衛生局、およびトランスレーショナルゲノミクス研究所からの資金提供によってサポートされました。メタボロミクスデータセットはMetabolonで生成され、ADMCによって前処理されました。
資料
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
参考文献
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved