
Method Article
通过透明化、免疫染色、共聚焦和光片荧光显微镜对 HIV 感染组织中免疫细胞群进行 3D 可视化
摘要
组织透明化与免疫荧光显微镜相结合,可以对完整组织内的免疫细胞群和病毒蛋白进行空间可视化和定量。使用共聚焦和光片荧光显微镜对透明化组织进行光学切片可以生成复杂组织环境的 3D 模型,并揭示 HIV 感染过程中表现出的空间异质性。
摘要
人类免疫缺陷病毒 (HIV) 是获得性免疫缺陷综合症 (AIDS) 的病原体,是一个主要的全球健康问题,全球有近 4000 万人感染,并且没有广泛可用的治疗方法。尽管付出了巨大的努力,但对感染期间和治疗反应中组织中病毒和宿主细胞相互作用的详细了解仍然不完整。为了解决这些限制,应用水基组织透明化技术 CUBIC(清晰、无阻碍的脑/身体成像混合物和计算分析)和 CLARITY(透明脂质交换丙烯酰胺杂交刚性成像/免疫染色/原位杂交兼容的组织 hYdrogel)来可视化来自动物模型和人类的 HIV 感染组织中的复杂病毒宿主细胞相互作用使用共聚焦和光片荧光显微镜。完整组织的光学切片和图像分析可以快速重建整个组织中包含的空间信息,并在感染过程中量化免疫细胞群。这些方法适用于大多数组织来源和各种生物学问题,包括传染病和癌症。
引言
近年来,生物研究中对定量空间组织成像的需求不断增长,这导致了组织透明化技术的出现,该技术用于生成具有单细胞分辨率的完整组织的更大体积 (mm3-cm 3) 图像。组织包括具有独特定义结构、组成和功能的生物分子的复杂组织。不幸的是,组织中存在的许多生物分子(例如脂质和发色团)在通过光学显微镜成像时会散射、吸收或发射光,这使得大体积成像变得困难。此外,组织通常表现出与标准成像解决方案和光学镜头不匹配的折射率,从而导致成像过程中的光学失真。使用光学显微镜对大量组织进行成像的最佳方法应包括匹配组织的折射率、成像溶液和物镜,同时允许光线深入组织,而不会在处理过程中破坏完整组织的生物学特征。德国解剖学家 Werner Spalteholz 在 1800 年代后期首次尝试通过清除不透明组织样本来减少组织和成像溶液之间的折射率差异1。这种组织透明化技术涉及刺激性的化学溶剂,这些溶剂会损坏组织样本,但尽管如此,它还是首次报道了完整组织的大体积成像。现代光学显微镜方法与图像捕获和分析的计算能力相结合,最近使组织透明化作为一种以单细胞分辨率对大型完整组织样本进行成像的方法再次流行起来。在过去的二十年里,出现了数十种先进的组织透明化技术,包括有机基和水基技术,每一种技术在特定应用中都有其优缺点。
3D 组织成像可以探测无法在细胞培养中重现的更复杂的生物相互作用。例如,细胞信号模式2(cell signaling patterns)、不同细胞类型的空间分布3(spatial distribution of different cell types)和脑连接4(brain connectivity) 以前使用全组织/器官成像方法以定量方式绘制。这里描述的是水基组织透明化方案的应用,用于在活动性感染期间清除、免疫染色和可视化完整 HIV 感染淋巴组织内的不同 HIV 靶细胞群。在体内,HIV 主要感染 CD4+ T 细胞,并将其基因组拷贝整合到受感染宿主细胞的基因组中。该病毒随后劫持受感染的宿主细胞机制进行自我复制,导致病毒传播、宿主细胞杀伤、免疫功能障碍和长期发展为 AIDS。值得注意的是,受感染的 T 细胞在组织和细胞培养物中的行为明显不同。与 HIV 一起孵育的培养的 CD4 + T 细胞可以产生大量 HIV 诱导的合胞体,其中可能包括数十个细胞核5,而在 3D 细胞外基质 (ECM) 水凝胶或 HIV 感染的人源化小鼠(胡 小鼠)的组织样本中培养的原代 CD4 + T 细胞的类似实验通常产生具有 2-5 个细胞核的合胞体6。了解 HIV 感染个体中的局部细胞间传播和病毒的全身传播可能更加复杂,涉及多种感染细胞类型将病毒从组织运输到血管再到新组织,其中游离病毒粒子和产生病毒的细胞可以进入大量易感淋巴细胞7.这些场景目前无法在细胞培养系统中重现,来自动物模型和人类的组织仍然是在具有功能免疫系统的复杂生物体背景下了解病毒发病机制的重要资源。
目前的抗逆转录病毒疗法 (ART) 通过抑制 HIV 复制和阻止疾病向 AIDS 发展,大大提高了 HIV 感染者 (PWH) 的预期寿命和生活质量。不幸的是,ART 并不能消除含有逆转录病毒基因组插入物的潜伏感染免疫细胞,这些免疫细胞是静止的且不主动产生病毒。尽管在大多数接受 ART 治疗的个体的血液中检测不到病毒,但在 ART 中断后病毒载量迅速反弹,疾病进展继续8。由受感染细胞的潜在储存库引起的 HIV 感染的持续性是建立 HIV 治愈方法的巨大障碍。HIV 的组织储存库仍然知之甚少,在 ART 之前、期间和之后更深入地了解淋巴组织中的这些储存库至关重要,以完整表征病毒发病机制并评估有效消除不活跃产生病毒的潜伏感染细胞的新疗法。
在这里,CUBIC3 和 CLARITY9 是两种先前适应的水基组织透明化方案,用于对具有人源化免疫系统的 HIV 感染小鼠(胡小鼠)、SIV/SHIV 感染的非人灵长类动物 (NHP) 和 HIV 感染的人类的众多完整淋巴组织内的免疫细胞群进行成像。这些方案适用于共聚焦和光片荧光显微镜检查,具体取决于成像目标(更高的分辨率与更大的体积)和可用的仪器。虽然光学显微镜无法分辨单个病毒粒子,但使用免疫荧光可以识别含有病毒的组织区域和产生病毒的细胞,这些区域可以用更高分辨率的方法进一步分析。这里介绍的方法可以适应以单细胞分辨率可视化体内的几乎任何组织,以量化感染期间不同条件下特定细胞类型之间的空间关系,并且很容易转化为高度相关的人类患者样本,用于传染病或癌症的研究。
研究方案
所有动物实验均根据经批准的机构动物护理方案进行。所有人体组织均根据批准的机构人类研究伦理指南获得。
1. 组织收获和固定(CUBIC 和 CLARITY 相同)
- 如前所述识别和解剖淋巴组织10.
- 在安全的情况下,在死后几分钟内用解剖剪刀和镊子切除淋巴组织。
- 将组织样品放入新鲜制备的冰冷固定缓冲液中,该缓冲液含有 8% 多聚甲醛 (PFA)、5% 蔗糖和 0.1 M 二甲胂酸钠三水合物,以充分保存组织样品用于光学显微镜 (LM)、电子显微镜 (EM) 或免疫 EM。或者,在 0.1 M PBS 中用 4% PFA 固定 LM 样品。在开始清除过程之前,将样品固定过夜,以确保病毒完全灭活。
注意:多聚甲醛对皮肤接触和吸入有毒,也是一种易燃固体;小心处理并存放在易燃储物柜中。二甲胂酸钠三水合物吞咽或吸入有毒。 - 在开始清理过程之前,拍摄组织的参考图像。
注:在这些条件下,LM 样品可以储存至少 1 年。要处理表达内源性荧光蛋白的样品,请在后续步骤中始终将样品置于避光处。
2. CUBIC 组织透明化
- 在无菌 0.1 M PBS 中冲洗淋巴组织样品 3 次,在室温下摇动 15 分钟,以确保在每次更换缓冲液期间去除 PFA。
注意:根据机构指南处理含有 PFA 的液体。 - 将淋巴组织样品在 37 °C 的 CUBIC Reagent-1(参见 材料表)中浸泡 3 天,轻轻摇动。定期拍摄参考图像以监测脱色过程随时间的变化。
- 将 Reagent-1 更换为额外的浸泡 3-4 天,或直到组织脱色完成。清除所需的时间取决于组织的体积和类型。为了加快组织脱色过程,请每天刷新 CUBIC Reagent-1 并使用更大的体积。
- 在室温下用 0.1 M PBS 洗涤淋巴组织样品 3 次,每次 30 分钟,轻轻摇动。
- 将淋巴组织样品浸入 37 °C 的 CUBIC Reagent-2(参见 材料表)中,轻轻摇动 2-7 天或直到达到完全透明。如果样品未达到完全透明,请重复步骤 2.2-2.5,直到清除不再进行。定期拍摄参考图像以监控一段时间内的清除过程。
- 在室温下用 0.1 M PBS 洗涤淋巴组织样品 3 次,每次 30 分钟,轻轻摇动。
- 将样品储存在含有 0.01% 体积/体积 (V/V) 叠氮化钠的 CUBIC Reagent-2 中,在黑暗中保存(参见 材料表)。
注意:使用此方法,样品可以储存至少 6 个月。
注意:叠氮化钠具有剧毒,具有严重的吸入危险。建议购买 5% 或更低叠氮化钠的稀释溶液。
3. 立方体样品的封闭和免疫染色
- 在室温下用 0.1 M PBS 洗涤淋巴组织样品 3 次,每次 30 分钟,轻轻摇动。
- 要使用共聚焦显微镜成像,请使用组织切片机基质将组织切成 ~0.5-1 mm 厚的切片。要进行光片荧光显微镜 (LSFM),请封闭整个组织区域。
- 用 5 mL CUBIC 封闭液在 4 °C 下摇动封闭样品过夜(参见 材料表)。处理 NHP 或人类样品时,请使用抗人 FcR。处理小鼠样品时,请在封闭溶液中使用抗小鼠 FcR。
- 在封闭溶液(不含物种特异性 FcR)中用 5 mL 一抗(参见 材料表)在室温下摇动对样品染色 3 天(可选:使用前将浓缩抗体原液以 2,300 x g 离心 5 分钟,以减少聚集抗体的添加)。
- 在室温下摇动洗涤染色样品至少总共 5 小时,至少更换 5 次洗涤液缓冲液(参见 材料表)。
- 在封闭溶液(无物种特异性 FcR)中用二抗(参见 材料表)在室温下摇动对样品染色 3 天(可选:使用前将抗体以 2,300 x g 离心 5 分钟,以尽量减少抗体聚集)。
- 在室温下用洗涤液洗涤染色样品 5 次,总共摇动至少 5 小时。
- 用 5 mL DAPI 染色溶液对每个组织样品进行染色(参见 材料表),并在室温下孵育 10 分钟。让样品在 4 °C 的黑暗中保留在 DAPI 染色溶液中,以便稍后成像。
- 用洗涤液在室温下洗涤淋巴组织样品 3 次,每次振荡 30 分钟。
- 在样品封片前,将染色样品在室温下避光浸入 CUBIC Reagent-2 中过夜。
4. CLARITY 组织透明化
- 在无菌 0.1 M PBS 中冲洗淋巴组织样品 3 次,在室温下摇动,每次 15 分钟以去除 PFA。
- 将组织样品放入 15 mL 新鲜制备的丙烯酰胺溶液中,并在 4 °C 下轻轻搅拌孵育过夜(参见 材料表)。
注意:未聚合的丙烯酰胺是一种强效神经毒素,很容易通过皮肤吸收。避免与皮肤接触,如果发生接触,请立即冲洗。 - 让组织样品升温至室温。
- 可选:将氮气鼓泡到丙烯酰胺溶液中 1 分钟,对组织样品进行脱气。注意使用低流速,以避免有毒的未聚合丙烯酰胺飞溅(~1-2 个气泡/秒)。
- 将组织样品置于 37 °C 水浴中 1-3 小时聚合,每 15 分钟倒置一次。一旦检测到明显的聚合,如粘性液体、混合时出现 Schleren 线或在组织周围形成透明胶囊所示,请立即取出样品。
注:如果丙烯酰胺溶液发生完全聚合,请从样品中去除多余的水凝胶并继续该方案。 - 在室温下用无菌 0.1 M PBS 洗涤组织样品 3 次,每次 30 分钟,轻轻摇动以去除丙烯酰胺溶液。
- 将组织样品放入 15 mL 8% SDS 中,在 37 °C 下加入 0.1 M PBS 中,轻轻摇动 2-5+ 天以使其透明化。如有必要,定期刷新 8% SDS 溶液,并使用最多 50 mL 的溶液以加快透明化速度。当样品在视觉上透明或不再进行时,停止清除过程。定期拍摄参考图像以监控一段时间内的清除过程。
- 在室温下,用无菌 0.1 M PBS 洗涤组织样品 5 次,在 1 天内轻轻摇动。
- 将样品暂时保存在 0.1 M PBS(加上 0.01% 体积/体积 (v/v) NaN3 以长期储存)中),直到准备好对内源性荧光进行成像。
- 将组织放入 5 mL 成像介质 RI-2 中(参见 材料表)。在室温下避光孵育过夜,以验证免疫染色前透明化过程的完整性。拍摄参考图像以监测组织透明度。
5. CLARITY 样品的封闭和免疫染色
注:这些步骤类似于 CUBIC 透明化组织的封闭和免疫染色,但使用不同的配方进行封闭、洗涤和染色溶液。
- 在室温下用 0.1 M PBS 洗涤淋巴组织样品 3 次,每次 30 分钟,轻轻摇动。
- 要使用共聚焦显微镜成像,请使用 0.5 mm 组织切片机和基质将组织切成 ~0.5-1 mm 厚的切片。要进行 LSFM,请封闭整个组织样本。
- 用 5 mL CLARITY 封闭液(参见 材料表)在 4 °C 下振荡封闭样品过夜。
- 在封闭溶液(不含物种特异性 FcR)中用 5 mL 一抗(参见 材料表)在室温下振荡对样品染色 3 天(可选:使用前将抗体以 2,300 x g 离心 5 分钟,以尽量减少抗体聚集)。
- 在室温下用洗涤液洗涤染色样品 5 次,总共摇动至少 5 小时(参见 材料表)。
- 在封闭溶液(不含物种特异性 FcR)中用 5 mL 二抗(参见 材料表)在室温下振荡对样品染色 3 天(可选:使用前将抗体以 2,300 x g 离心 5 分钟,以尽量减少抗体聚集)。为了缩短总体方案长度,请使用与荧光基团偶联的一抗,以消除与二抗孵育的需要。
- 在室温下用洗涤液洗涤染色样品 5 次,总共摇动至少 5 小时。
- 用 5 mL DAPI 染色溶液对每个组织样品进行染色(参见 材料表),并在室温下孵育 10 分钟。让样品在 DAPI染色溶液中避光保持在 4°C,以便稍后成像。
- 用洗涤液在室温下洗涤淋巴组织样品 3 次,每次振荡 30 分钟。
- 将组织放入 5 mL 成像培养基 RI-2 (RI = 1.46) 中,并在样品安装前在室温下避光孵育过夜(参见方案步骤 6 和 7)。
6. 用于共聚焦显微镜的透明化组织样品的封片和成像
- 撕下粘性硅胶隔离器保护层的一侧。
- 将显微镜盖玻片(22 mm x 40 mm,0.25 mm 厚)贴在硅胶隔离器的剥离侧,为样品形成防液空间。
- 撕下粘性硅胶隔离器保护层的另一侧。
- 将用于成像的样品放在硅胶隔离器的中心,然后根据需要添加 CUBIC Reagent-2 或成像介质 RI-2,直到液体表面与隔离器边缘一样高。
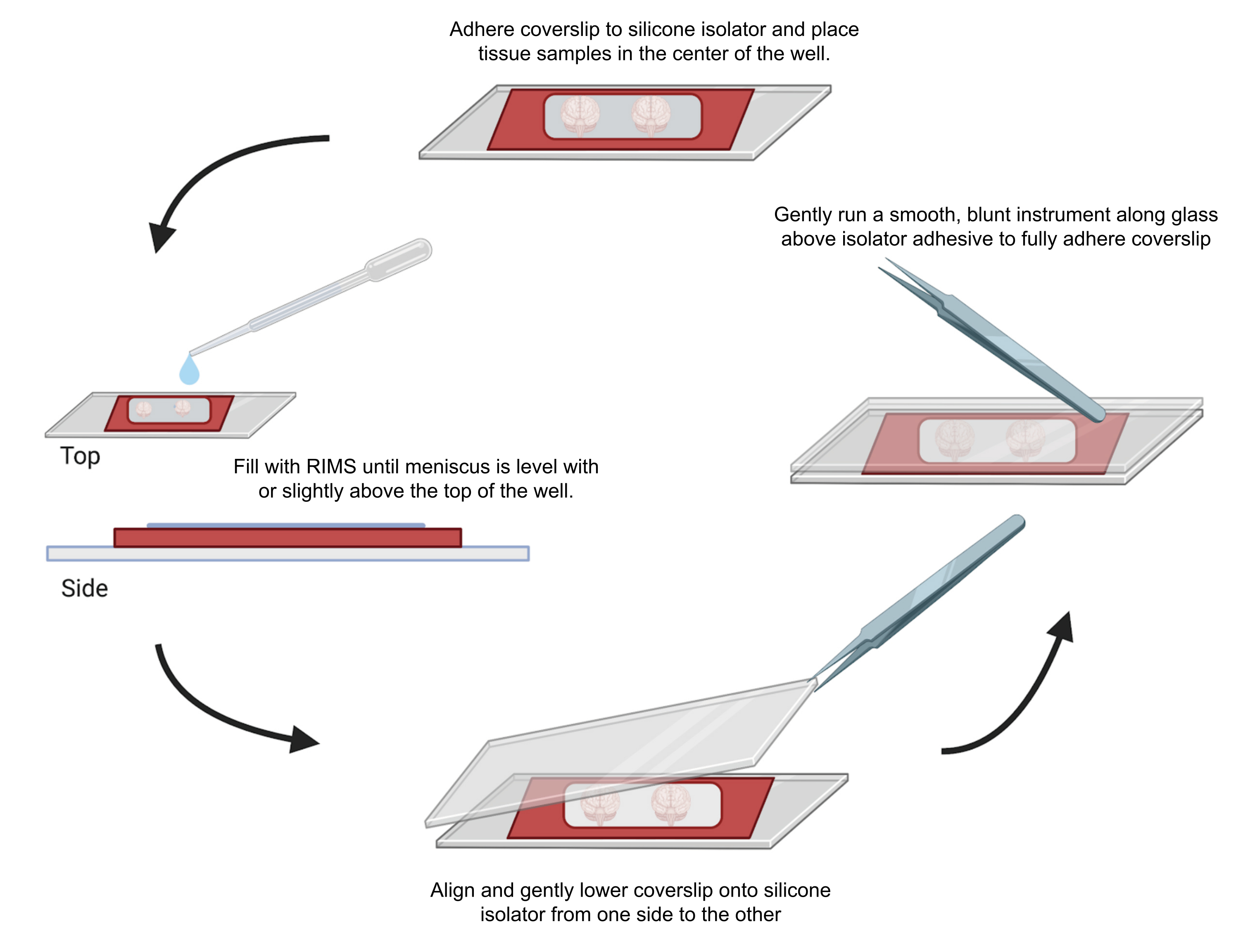
- 为了最大限度地减少硅胶隔离器内气泡的滞留,请使用 EM 镊子从一侧对齐并轻轻地将第二个盖玻片向下分层。擦去多余的液体。使用镊子背面轻轻按压样品孔周围的盖玻片以密封粘合剂。将封固的样品水平存放在黑暗中。
注意:样品可以在封片后数周至数月进行成像;然而,成像质量通常会随着时间的推移而下降。 - 将安装的载玻片放在显微镜载物台上,并使用白光和较低放大倍率的物镜 (2-10x) 定位样品。
- 根据所选的单个荧光基团设置荧光采集曲线。
注:建议单独获取单独的荧光基团通道。这导致采集时间更长,但减少了光谱重叠和非特异性荧光信号的采集。常见的荧光基团谱图可以包括 DAPI (450 nm)、Alexa488、Alexa594 和 Alexa647(或相关组合),以最大限度地减少图像采集过程中的光谱重叠。 - 选择合适的放大物镜来对感兴趣区域进行成像。使用较低放大倍率的物镜 (2-10x) 进行具有单细胞分辨率的大体积或全组织成像,使用较高放大倍率的物镜 (20-63x) 对透明化组织中的亚细胞细节进行更高分辨率的可视化。物镜、成像介质和组织的折射率尽可能匹配,以最大限度地减少图像采集过程中引入的光学失真。
- 选择 Z 堆栈采集的步长。对于低放大倍率物镜 (2-10x),选择 ~3-5 μm 的步长,以检测多个连续 Z 切片中单个细胞的荧光,以进行 3D 建模,同时减少总采集时间和整体文件大小。对于更高放大倍率的物镜 (20-63x),请选择 ~1 μm 或更小的步长,以最大限度地减少单个 Z 切片之间亚细胞信息的损失。
- 缩放视野以在 X 和 Y 维度上可视化要成像的整个组织区域,并尽可能减少未占用区域。设置包含整个感兴趣区域(要成像)的上部和下部 Z 载物台采集坐标。
- 获取 Z 堆栈图像。保存并导出文件,以便使用任何图像分析软件进行后处理。对于某些软件套件,请将文件转换为特定的文件类型(例如,.tiff、.ome-tiff、.jpeg 等)。使用任何显微镜图像采集软件或图像分析免费软件(例如 ImageJ/Fiji)完成转换。
7. 在 LSFM 腔室或比色皿中封片和成像样品
- 根据所使用的特定方案,用 CUBIC Reagent-2 或 RI-2 填充成像室。转移液体时避免形成气泡。用移液管去除多余的气泡。
- 将样品浸入成像室中并限制样品移动。
注:根据所使用的特定显微镜,这可能包括将样品包埋在琼脂糖中、将样品悬浮在钩子或豪猪适配器上、3D 打印样品架或用粘合剂将样品连接到塑料培养皿上。 - 将物镜置于成像溶液中,聚焦于样品。将封固的样品在成像室中放置数小时或过夜,以使比色皿中的溶液和组织完全平衡。
- 获取感兴趣区域的 Z 堆栈(图像采集参见步骤 6.7-6.11)。
注:这种方法可以允许以单细胞分辨率对大于 1 cm3 的组织体积进行成像。
8. 使用 Imaris 图像分析软件进行表面重建和细胞定量
注意:这些步骤特定于 Imaris 图像分析软件,但可以使用其他软件套件(例如 ImageJ/Fiji、Aivia、Arivis、Amira 等)进行类似的图像处理步骤。
- 使用 Imaris 文件转换器将 Z 堆栈图像文件转换为本机 Imaris 格式 .ims。这将有助于更快速地转换文件,同时最大限度地减少打开后的转换错误和潜在的软件问题。
注意:一些较新的 LSFM 允许用户将文件直接保存为 .ims 格式。 - 将要分析的 .ims 文件拖到 Imaris 软件的 Arena 区域中。通过 “显示调整 ”面板调整每个颜色通道的对比度或强度。单击左上角的 Add New Surfaces 图标。
- 单击 Next: Source Channel (带有指向右侧的箭头的蓝色图标)。选择要构建的表面的源通道。请勿更改其他参数。
- 单击 Next: Threshold (带有指向右侧的箭头的蓝色图标)。
- 要调整阈值(绝对强度),请向左或向右拖动阈值线。启用 Split Touching Objects 并输入以微米为单位的平均单元直径作为系统的分割标准,以产生许多点作为每个单独表面的原点。
- 不要包含太小或太亮的荧光信号,因为它们可能代表潜在的染色或显微镜伪影。通过相应地改变平均细胞直径,仅包括具有可接受大小和荧光强度的点。
注:平均细胞直径会因特定组织或细胞类型而异,但通常介于 5-15 μm 之间。
- 不要包含太小或太亮的荧光信号,因为它们可能代表潜在的染色或显微镜伪影。通过相应地改变平均细胞直径,仅包括具有可接受大小和荧光强度的点。
- 单击 Next: Classify Surfaces (蓝色图标,带有指向右侧的箭头)。通过向左或向右拖动阈值线来调整要包含的曲面。确保表面完全接近原始荧光信号,同时将荧光信号与单个细胞分开。
- 单击 Finish: Execute All Creation Steps 并终止向导 (带有两个指向右侧的箭头的绿色图标)。表面是正式建造的。
- 单击左侧面板上标记为 Statistics 的第六个图标,查看单元格数,在这种情况下,分析的特定颜色通道的表面数。
- 确保四个变量 Number of Disconnected Components per Time Point、Number of Surfaces per Time Point、Total Number of Disconnected Component 和 Total Number of Surfaces 具有相同的数字,即该颜色通道的单元计数。
结果
组织透明化包括用化学混合物处理保存的组织,以从组织中提取不透明的生物分子,同时保持组织结构。这些组织透明化解决方案将组织的折射率与周围的成像介质相匹配,以最大限度地减少光学失真,提高组织深处的信噪比,并最大限度地减少背景自发荧光。在免疫荧光染色和用共聚焦和光片荧光显微镜成像之前,使用两种基于水的光学组织透明化方案 CUBIC3 和 CLARITY9 来清除保存的 HIV/SIV 感染的胡小鼠、非人灵长类动物和人体组织样本。
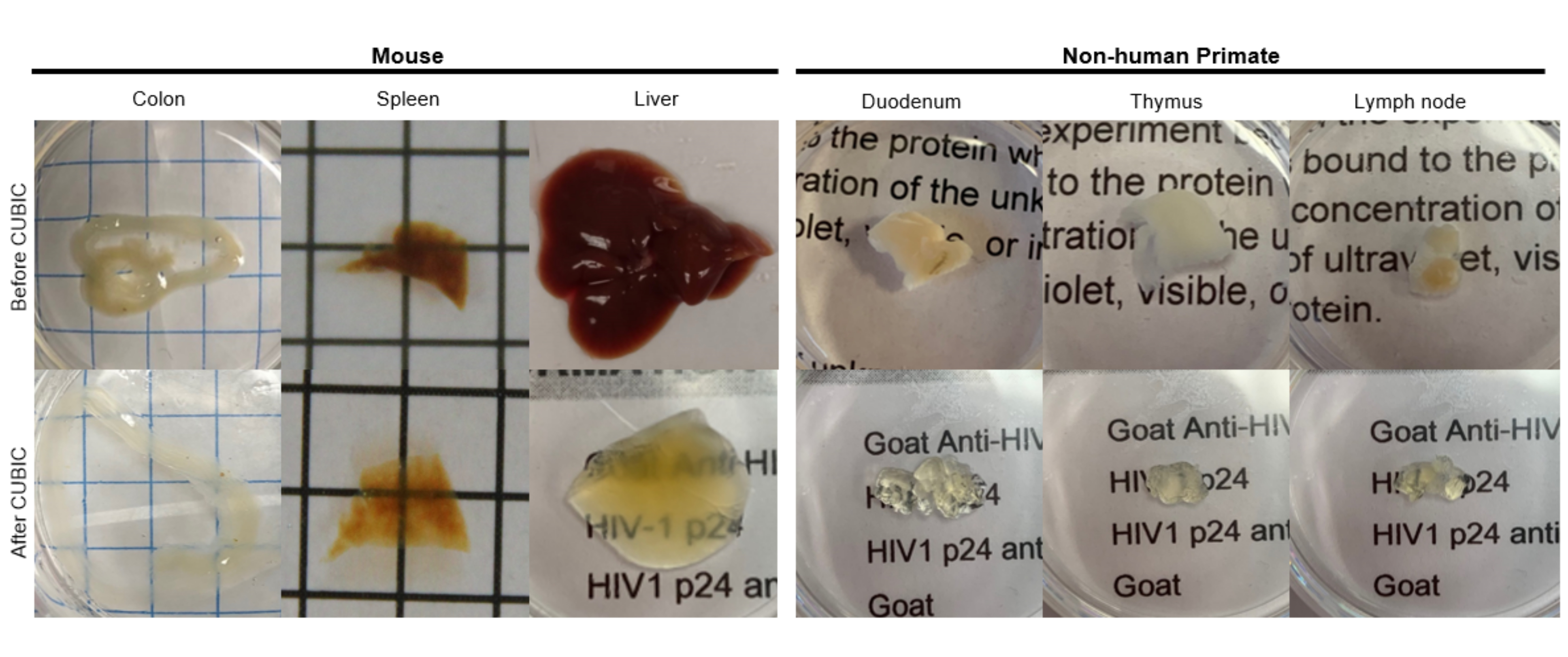
对于 CUBIC 方案,用 PBS 洗涤固定的组织以去除固定剂,并浸入 CUBIC Reagent-1 中,CUBIC Reagent-1 是一种氨基醇的基本缓冲溶液,可洗脱血红素等发色团,导致组织脱色和脱脂(图 1,顶部)。用 CUBIC Reagent-1 处理 3 天后,较小的组织体积 (~mm3) 可以脱色,但较大的组织体积 (~cm3) 或具有大量血红素的组织(如肝脏、脾脏或心脏)需要更长的孵育时间和溶液体积(>1 个月和 ~50 mL),以及每 2-3 天频繁更换一次溶液。脱色后,洗涤组织并置于 CUBIC Reagent-2 中,这是一种含蔗糖的溶液,折射率约为 1.48-1.49,与组织的折射率相匹配,并增加了光的透射率。对透明化的组织进行免疫染色,并在使用共聚焦或光片显微镜成像之前,将其封片在 CUBIC Reagent-2 溶液中。对不同大小和浓度的发色团的几种胡小鼠和非灵长组织进行 CUBIC 透明化程序的效果成像(图 2)。光学透明化使组织在肉眼看得透明,从而可以“透过”组织看到纸上的网格线和文本。富含发色团的组织,如脾脏、肝脏、骨髓和心脏,可能不会完全脱色,但仍适合免疫染色和成像(图 2 和 图 5)。
对于 CLARITY 方案,用 PBS 洗涤固定的组织以去除固定剂,然后在 4°C 下在 40% 丙烯酰胺溶液中与热引发剂一起孵育过夜,以在样品中的蛋白质和丙烯酰胺单体之间形成共价键(图 1,底部)。第二天,将组织平衡至室温,然后在 37 °C 水浴中加热后,开始丙烯酰胺聚合并将样品迅速包裹在水凝胶中。在 2-5 天的过程中用 8% SDS 溶液处理样品,以去除不透明的脂质。在荧光染色之前,将样品浸入含有 90% 非离子密度梯度培养基的 CLARITY 示差折光匹配溶液 (RIMS)(成像介质 RI-2)中。对于含有大量血红素的组织,可以在脱脂步骤9、11、12 结束时添加脱色步骤。比较同一人脾脏样本不同切片的 CUBIC 和 CLARITY 清除进展(图 3)。CLARITY 透明可产生可见的聚丙烯酰胺凝胶,包裹溶液,与 CUBIC 透明相比,除非添加额外的脱色步骤,否则通常脱色减少 9,12。
随后在两种方案中,对透明化的完整组织进行免疫染色以检测特异性免疫细胞群。洗涤样品,用含 α-FcR 的试剂封闭以减少非特异性抗体结合,并使用直接与荧光团偶联的一抗染色 3 天。或者,样品用未偶联的一抗染色 3 天,然后用与荧光团偶联的二抗再染色 3 天。再次洗涤组织,然后与 DAPI 染色剂在 4 °C 下孵育过夜以进行细胞核可视化。洗涤样品并在 CUBIC Reagent-2 中孵育 24-36 小时或在成像介质 RI-2 (CLARITY) 中避光孵育过夜。对于共聚焦显微镜,在成像之前将组织安装在适当的 RIMS 中的显微镜载玻片上(图 4)。对于光片荧光显微镜 (LSFM),在成像前,将样品用 RIMS 完全浸没在成像比色皿中过夜。
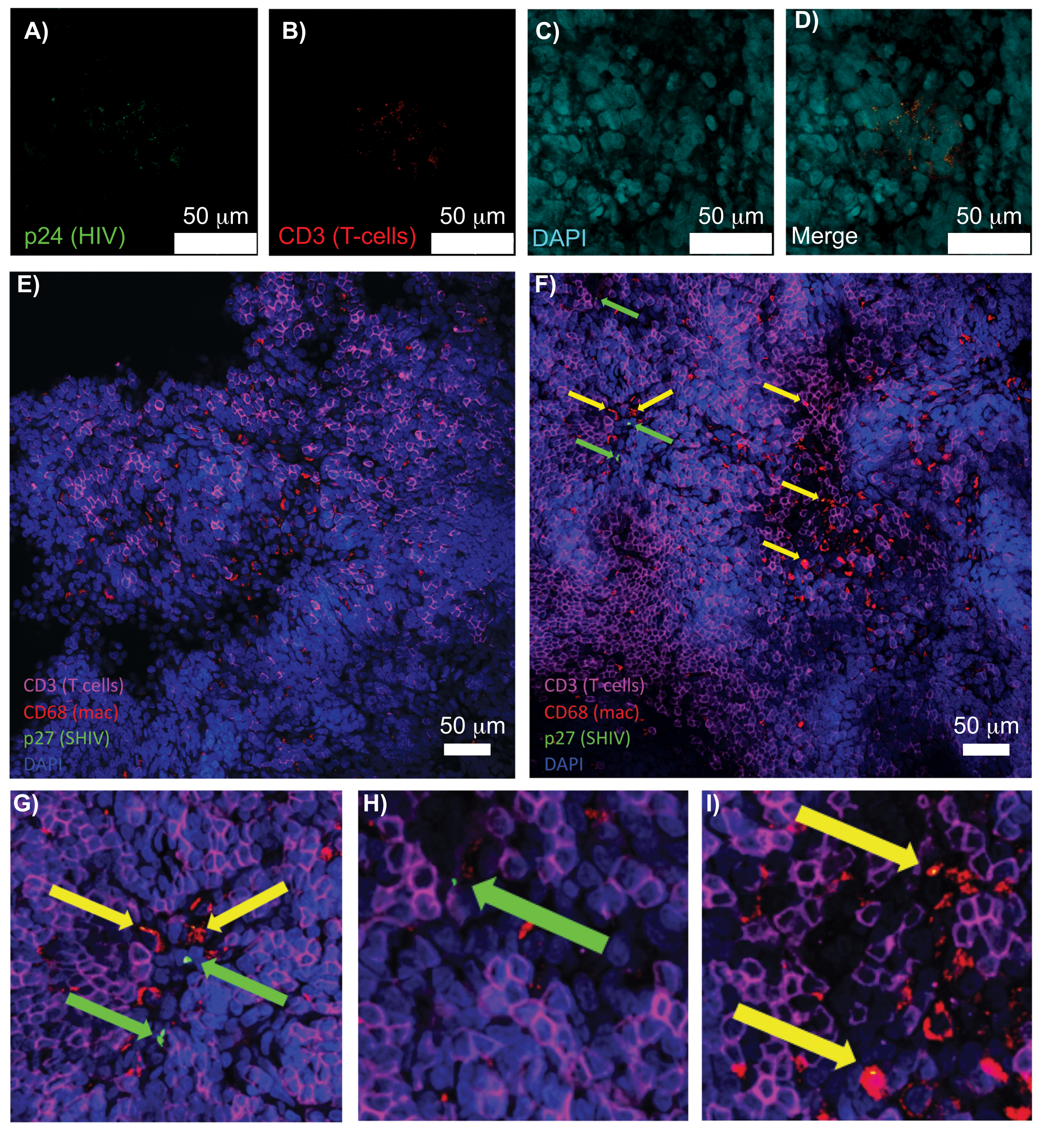
对完整、透明化和免疫染色的淋巴组织进行共聚焦显微镜检查,可以同时观察多种荧光信号,包括细胞核、免疫细胞标志物和 HIV/SIV CA(衣壳)蛋白(图 5)。通过免疫细胞标志物和 HIV 蛋白的荧光共定位来确定产生病毒的细胞。清除和染色的 HIV 感染人脾脏显示多个 CD3 + T 细胞与 HIV p24 共定位,表明在完整组织区域内存在产生病毒的细胞(图 5A-D)。清除和免疫染色的 SHIV 感染的 NHP 淋巴结揭示了 CD3 + T 细胞和 CD68 + 巨噬细胞在未检测到病毒的组织区域(图 5E)以及具有大量病毒产生细胞的区域(图 5F)的分布。这些结果表明,来自不同组织来源的病毒产生细胞与给定视野内的其他细胞是不同的,并允许在复杂的组织环境中检测罕见的生物事件。
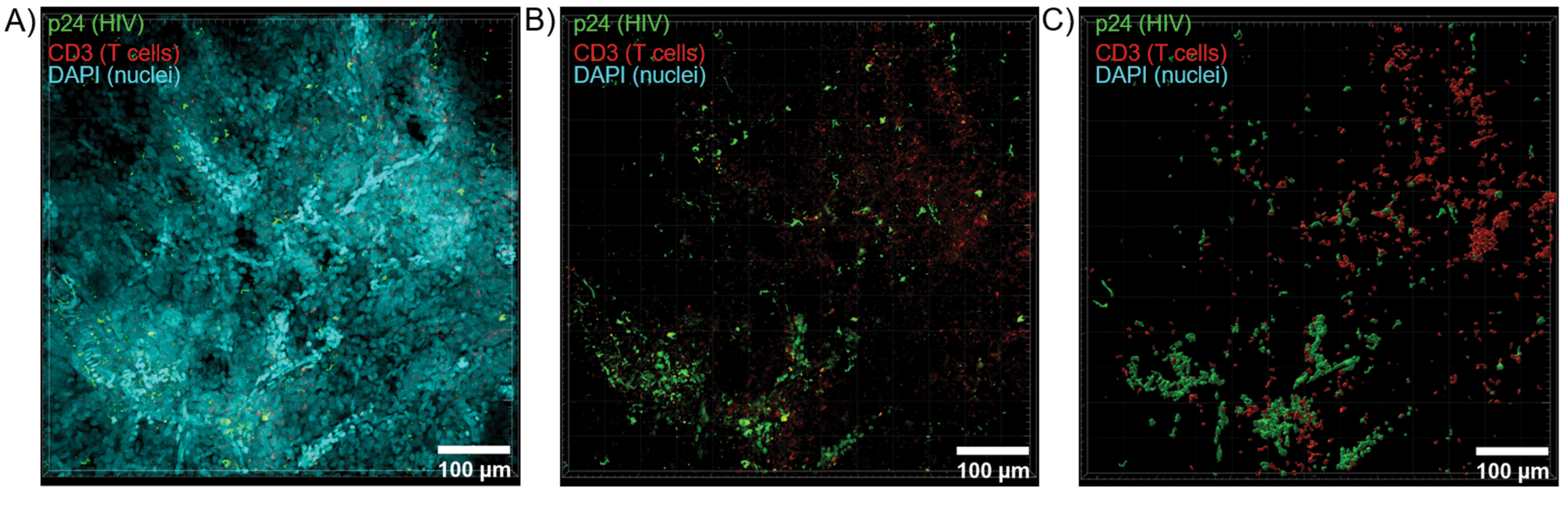
使用共聚焦显微镜对透明化组织进行光学切片,以生成 Z 堆栈和 3D 表面模型,揭示了 HIV 感染过程中表现出的细胞异质性(图 6)。使用 Imaris 软件套件将 Z 堆栈重组为 Z 投影图像(图 6A),并去除 DAPI 核通道,以便在整个组织体积中清晰显示 CD3 + T 细胞和 HIV 衣壳蛋白 (p24) 荧光(图 6B)。使用 Imaris 软件自动分割 Z 投影荧光,以生成重建的 3D 表面模型,用于整个 Z 堆栈中荧光信号的空间可视化和量化(图 6C)。3D 表面模型的分析显示 546 个 CD3+ T 细胞和 218 个产生 HIV p24 的细胞。累积起来,从清除的 HIV 感染淋巴组织中获取免疫荧光的 Z 堆栈可以生成组织内细胞组成的 3D 模型,并自动量化组织体积内的免疫细胞群。
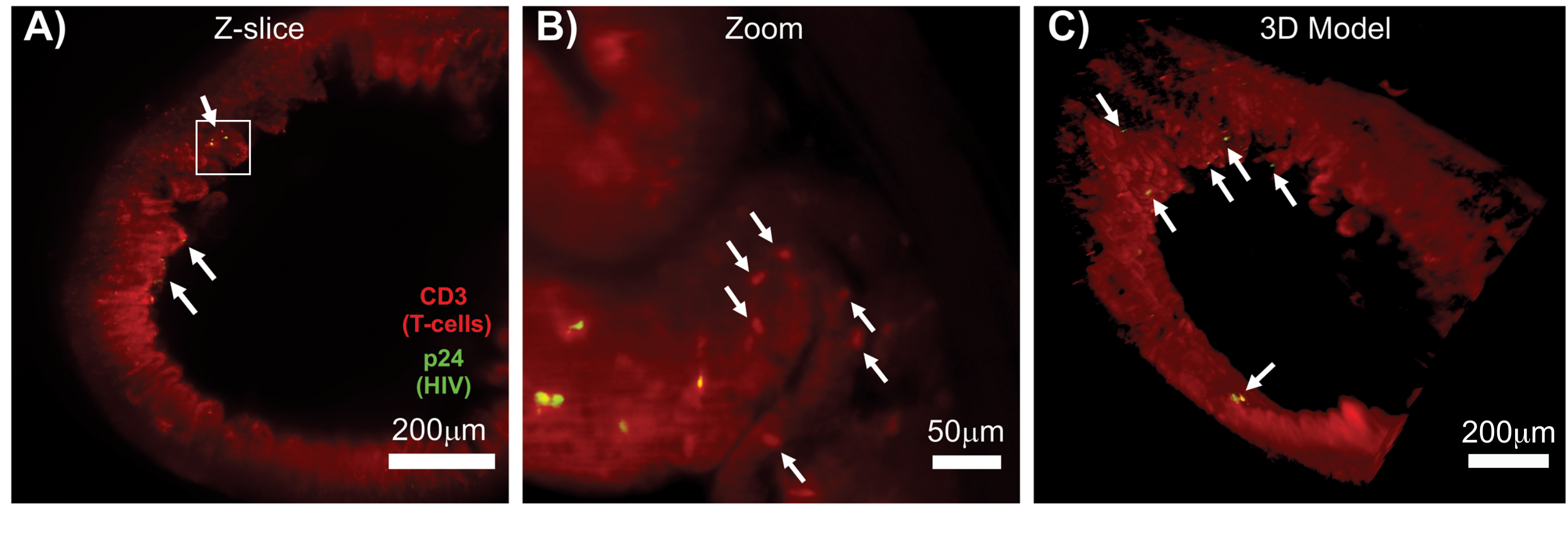
完整、透明化和免疫染色的淋巴组织的 LSFM 允许对淋巴组织中免疫细胞和产生病毒的细胞分布进行更大容量的免疫荧光 (IF) 成像(图 7)。对 HIV 感染的 胡 小鼠的结肠组织进行 hCD3 + T 细胞和 HIV p24 的免疫染色显示,产生病毒的细胞病灶分散在大面积组织中,没有感染的证据(图 7A)。产生病毒的细胞病灶的放大视图显示,多个产生病毒的细胞靠近潜在的靶细胞(图 7B)。组织自发荧光(红色雾霾)用于可视化整个组织结构,同时区分组织内比自发荧光染色更亮的特定免疫细胞群(红色椭圆)。整个 LSFM Z 堆栈体积的 3D 模型显示了完整组织区域内产生病毒的细胞病灶的空间分布,并允许绘制相对于整体组织结构的病毒产生位置(图 7C)。令人惊讶的是,产生病毒的细胞的病灶经常散布在大面积的组织区域之间,没有病毒产生的迹象。这些结果可以量化不同组织内以及感染的不同时间或对不同治疗的反应的病毒分布和感染细胞密度的参数。

图 1:典型的 CUBIC 和 CLARITY 组织透明化、免疫染色和成像的工作流程。 CUBIC(顶部)和 CLARITY(底部)清除时间可能因组织的大小和类型而有很大差异。对于 CLARITY 透明化,在免疫染色之前需要使用折射率匹配的培养基进行额外的孵育步骤,以验证组织是否透明。当一抗与荧光基团偶联时,免疫染色通常需要 3 天,如果需要荧光二抗,则需要 6 天。样品可以使用共聚焦或 LSFM 成像。 请单击此处查看此图的较大版本。
{kind=link}

图 2:胡 小鼠和 NHP 组织样品的 CUBIC 清除。 根据组织样品的血红素和脂质密度的不同,清除每种组织类型所需的时间会有所不同。例如,结肠和十二指肠通常需要相对较短的时间(~7 天),而脾脏和肝脏可能需要更长的时间才能变得透明(~30 天)。 请单击此处查看此图的较大版本。
{kind=link}

图 3:人类样品上组织透明化方法的纵向比较。 CUBIC(上图)和 CLARITY(下图)清除了接受抗逆转录病毒治疗的 HIV 感染个体的脾脏。两种方法在第 32 天时都充分清除了组织以进行免疫染色和成像。CUBIC 方法的脱色步骤明显减少了由脾脏样品中所含血红素引起的自发荧光。 请单击此处查看此图的较大版本。
{kind=link}

图 4:共聚焦显微镜的样品安装。 将样品封片在用 0.5-1 mm 胶粘剂硅胶隔离器隔开的盖玻片之间。将硅胶隔离器粘附在第一个盖玻片上,并将组织放置在孔的中心(顶部)。井中充满 RIMS,直到弯液面与井顶部齐平或略高于井顶部(左)。小心地将第二个盖玻片从一侧降低到另一侧,以避免气泡(底部)。用钝器轻轻地绕着孔的周边运行,将盖玻片完全粘附在硅胶隔离器上(右)。样品在标准共聚焦显微镜中成像。 请单击此处查看此图的较大版本。
{kind=link}

图 5:透明化、完整的人脾脏和 NHP 淋巴结的共聚焦显微镜检查。 (A-D)对清除的 HIV 感染人体组织进行 HIV-1 p24 (绿色)、hCD3+ T 细胞 (红色) 和细胞核 (青色) 染色。(E) 感染后 8 周,来自 SHIV 感染的 NHP 的 CUBIC 清除淋巴结的共聚焦 Z 切片,对 CD3+ T 细胞(品红色)、CD68+ 巨噬细胞(mac/红色)、SHIV p27(绿色)和细胞核(蓝色)进行免疫染色。视野包含 T 细胞、巨噬细胞和其他细胞类型,但没有产生 SHIV 细胞的证据(绿色)。 F) 同一淋巴结相邻区域的共聚焦 Z 切片,显示细胞密度和数量的差异,以及存在产生病毒的 CD3+ T 细胞(绿色箭头)和 CD68+ 巨噬细胞(黄色箭头)。(G-I)(F) 中 p27 染色选定区域的放大视图。比例尺为 50 μm。请单击此处查看此图的较大版本。
{kind=link}

图 6:HIV 感染的人类脾脏的 Z 堆栈体积和 3D 重建表面。 (A) HIV 感染的人类脾脏组织 600 μm x 600 μm x 100 μm Z 堆栈的 Z 投影图像,对 HIV-1 p24(绿色)、hCD3+ T 细胞(红色)和细胞核(青色)进行染色。(B) 没有核 DAPI 染色的相同 Z 投影图像。(C) 从整个 Z 堆栈体积重建的 CD3(红色)和 p24(绿色)荧光的 3D 表面模型。 请单击此处查看此图的较大版本。
{kind=link}

图 7:HIV 感染组织的表面体积的 LSFM 和 3D 重建 (A) 来自 HIV 感染的 胡 小鼠结肠的 Z 切片 (1,000 μm x 1,000 μm),CD3+ T 细胞(红色)和 HIV p24(绿色)。暗红色的雾霾代表组织自发荧光,而明显的红色点表示 T 细胞。在指向中央管腔的外围可见绒毛,有几个活跃病毒产生的焦点(白色箭头)分散在不含病毒的大片区域。(B) 组织的放大区域,显示未感染的 T 细胞(红色)附近产生 hCD3+ T 细胞(黄色)的单个病毒。图像被旋转并更改为附近的 Z 切片,以在一个 Z 平面上显示 p24 阳性细胞的焦点。背景红色自发荧光显示除特异性 hCD3+ T 细胞染色外的一般组织结构(红色点状;白色箭头)。(C) 使用 Imaris 软件生成的完整体积 (1,000 μm x 1,000 μm x 200 μm) 的 3D 表面模型,旋转以显示肠道不同位置的 HIV 感染病灶(黄色)。白色箭头表示体积内的各个焦点。 请单击此处查看此图的较大版本。
{kind=link}
讨论
死后应迅速收集感兴趣的淋巴组织,并立即放入预冷的固定缓冲液中,以避免组织坏死(深色或黑色组织),从而对染色和成像产生负面影响。收获所需组织后,立即将组织浸入冰冷的 4%-8% 多聚甲醛 (PFA) 中过夜进行固定,这也可以灭活与样品相关的潜在病原体。4% PFA 是固定 LM 样品的最佳选择,而 8% PFA 可以充分保存 LM 和 EM 的组织。遵循这些程序并将样品储存在 4 °C 的固定剂中,在黑暗中可以有效地保存组织以进行 LM 成像数年。需要注意的是,在固定剂中长期储存会导致染色伪影的引入,尤其是抗原掩蔽,这是由相邻蛋白质与目标蛋白质交联引起的,这可能会阻碍染色抗体对表位的可及性13,14。如果组织含有内源性表达的荧光蛋白,请在整个方案中采取措施尽可能避免组织暴露在光线下。通常,内源性荧光蛋白在固定后会保留荧光 6-12 个月,但单个组织样品可能会变化较长或较短的时间。如果内源性荧光因蛋白质降解而丢失,通常可以使用对目标蛋白质具有特异性的一抗来检测荧光蛋白。灌注是在清除组织之前快速固定组织的另一种选择12;然而,由于在处理 HIV 等病原体时考虑到问题,选择了组织尸检后浸入冰冷固定剂中的途径,以尽可能安全地制备样品。
所描述的水基清除方案的一个优点是它们通常比有机物方案温和,有机物有时会损害更脆弱的组织,例如肝脏。与有机物分离方案相比,水性透明化方案通常需要更长的时间才能实现完全的样品透明化(数周而不是数天)。CLARITY 和 CUBIC 方案可以更快地进行,使用灌注同时清除啮齿动物内的所有器官11,12;然而,对于 NHP 和人体尸检来说,这不是一个可行的选择。使用 CLARITY 处理的样品往往显示一些体积膨胀,而 CUBIC 显示对样品体积的影响较小9。虽然通常更快,但许多基于有机物的组织透明化方案会导致组织收缩15,这使得在细胞致密组织(如淋巴结和脾脏)中更难观察到单细胞或亚细胞细节的检测。透明化诱导的膨胀可以有效地提高成像的分辨率,从而更容易观察到在组织原始大小下难以观察到的方面。或者,组织收缩可以有效减小样本的整体大小,从而可以在不解剖的情况下进行整个器官成像。CLARITY 和 CUBIC 方案的一个优点是,它们保留了组织中内源性表达的荧光蛋白的荧光,同时保持对免疫荧光染色的适应性11,12。免疫染色可以使用水性或有机组织透明化方法进行;然而,个人经验表明,与有机方案相比,使用水性方案的抗体相容性比例更高。研究人员需要根据成像的组织和解决的生物学问题(例如,整个器官成像与特定感兴趣区域成像)考虑使用哪种组织透明化方法。没有通用的组织透明化技术可以对所有大体积成像问题进行稳健的交钥匙分析,并且根据生物学应用的不同,可用方法表现出明显的优缺点。
进行抗体染色时,需要考虑许多方面。由于 CLARITY 样品包埋在丙烯酰胺水凝胶中,因此它们往往需要更长的孵育时间12。抗体孵育所需的时间还取决于每个样品的体积和厚度。此处描述的大多数样品厚度为 ~2-3 毫米,3 天足以对整个组织进行完全染色。如果目标是对整个小鼠大脑进行成像,则抗体孵育时间可能需要 1 周或更长时间12。选择水性组织透明化方法与有机组织透明化方法进行免疫荧光成像可能取决于抗体的兼容性。一般来说,对于 CUBIC 或 CLARITY,在培养细胞和组织中起作用的抗体的命中率为 ~70%。无论是使用水性组织透明化还是有机组织透明化方法,都需要评估所有抗体与所用特定方法的兼容性和有效性。如本实验步骤部分所示,CUBIC 和 CLARITY 处理的样品的免疫染色在透明化完成后进行。相反,此步骤发生在一些基于有机的方案的清除程序之前,然后是固定后。
将组织完全浸入与其折射率相匹配的成像介质中至关重要。否则,将在成像时引入球面像差,并使图像采集过程中捕获的光线失真。在为共聚焦和 LSFM 封片样品时,必须小心去除成像介质上的所有气泡,因为气泡会破坏进出样品的光路。在最终样品安装之前,可以用移液器手动去除气泡。对于使用共聚焦显微镜对较厚的样品进行成像,可以将多个硅胶垫片相互叠加,以容纳厚度大于 0.5 mm 的组织。一种建议是将 RIMS 中的所有组织平衡数小时至过夜,同时安装在显微镜上,无需额外移动样品。组织和成像介质的完全平衡将防止折射率不匹配的溶液混合,从而在成像过程中产生像差。重要的是要记住,在封片透明化的组织样品时,没有一种单一的交钥匙封片方法可以对所有显微镜中的所有样品进行成像。本实验方案讨论了在一种情况下效果最佳的样品安装选项,但根据所使用的单个显微镜和解决的生物学问题,有多种样品安装方法。这些方法包括但不限于将样品包埋在琼脂糖中、将样品悬浮在钩状或折光率匹配的塑料管线上、使用豪猪适配器、3D 打印样品架或用粘合剂将样品连接到塑料皿上。
共聚焦显微镜可以很好地用于 ~1 mm3-1 cm3 的组织体积成像。对于共聚焦显微镜,使用 2-10 倍物镜初步定位感兴趣区域,并以单细胞分辨率获取更大体积或全组织 Z 堆栈。切换到 20-63 倍物镜,以采集具有亚细胞信息的特定感兴趣区域的更高分辨率图像。对 CUBIC 和 CLARITY 透明化组织进行成像的理想物镜是 CLARITY/Scale 特定物镜,它与组织和成像溶液的折射率精确匹配。如果这种类型的物镜不可用,则最好使用甘油或油浸物镜(例如,LD LCI Plan-Apochromat 25 x 0.8 NA Imm Corr DIC M27 多重浸没物镜:工作距离 = 0.57 mm)而不是空气物镜对样品进行成像。这将最大限度地减少图像捕获过程中由于折射率不匹配而导致的光学失真。20-25 倍物镜可以在复杂组织环境中平衡大体积图像采集与从单个细胞获取染色细节。重要的是,大多数共聚焦显微镜都包含允许对成像体积进行 3D 平铺的模块。这种类型的图像采集可以理想地生成包含亚细胞信息的更大体积的 Z 堆栈。
LSFM 成像可以在大量组织 (>1 cm3) 甚至整个器官的背景下对特定细胞群进行 3D 可视化。在过去的 10 年中,组织透明化与 LSFM 相结合主要侧重于了解啮齿动物体内的大脑连接;然而,最近的应用包括可视化肿瘤转移景观16、解剖区室内的细胞分布 9,17 和病原体分散18。与培养的细胞相比,组织中的大多数生物事件是不均匀的,LSFM 特别擅长可视化和量化这些事件的空间组织异质性(例如,病毒复制、免疫信号传导、细胞分布等)。
通过共聚焦或 LSFM 获取的 3D 数据集可以使用众多图像分析平台进行后处理。Imaris 软件套件可用于表面构建、生成 3D 动画和细胞量化;但是,有许多图像分析系统可用于实现高效的图像后处理和分析。ImageJ/Fiji 免费软件19 是大多数实验室都可以使用的有吸引力的替代图像处理平台,但没有一种万能的分析软件可以擅长所有形式的图像分析和可视化。如果无法通过共享使用设施获得,许多图像分析软件套件可能会非常昂贵。最后,LSFM 或大型平铺共聚焦 3D 数据集的一个关键方面是数据管理。这些成像平台可以生成大量文件 (>1 Tb),这些文件需要更高端的计算机工作站进行数据可视化和量化。最终,这种成像工作流程可以简化整个组织内空间上不同的细胞群的采集和定量,并广泛适用于大多数组织来源和生物系统。
披露声明
作者没有需要披露的利益冲突。
致谢
感谢伊利诺伊大学厄巴纳-香槟分校基因组生物学研究所核心设施使用共聚焦和光片荧光显微镜。感谢“最后的礼物”队列中的杰出个人,他们提供人体组织样本,该队列由以下赠款资助:I147821、DA051915、AI131385 和 P30 AI036214。感谢 Nancy Haigwood 和 Ann Hessell 提供 SHIV 感染的 NHP 组织样本。
材料
| Name | Company | Catalog Number | Comments |
| Acrylamide Solution (in 0.1 M PBS, 40 mL in total) | |||
| 40% Acrylamide: 4 mL | Bio-Rad | 1610144 | |
| VA-044 Thermal Initiator: 0.1g | Fujifilm | 011-19365 | |
| CLARITY Blocking solution (in 0.1 M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Tween-20: 5 µL | Fisher Scientific | BP337-500 | |
| CLARITY wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Tween-20: 50 µL | Fisher Scientific | BP337-500 | |
| CUBIC Blocking solution (in 0.1M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Triton X-100: 5 µL | VWR | M143-1L | |
| CUBIC Reagent-1 (in 0.1M PBS, 50 mL in total) | |||
| N, N, N’, N’-tetrakis (2-hydroxypropyl) ethylenediamine: 12.5 g | Aldrich | 122262 | |
| Triton X-100: 7.5 g | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC Reagent-2 (in 0.1M PBS, 50 mL in total) | |||
| Sucrose: 25 g | Sigma | S1888-500G | |
| Sodium azide (in powder form): 10 g | Sigma | 71289-50G | |
| Sodium azide stock solution (in DI H2O, 50 mL in total) | Sigma | 71289-50G | |
| Triethanolamine: 5 g | Sigma | 90270-500mL | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| DAPI staining solution (0.5 µg/mL) | |||
| DAPI stock solution: 1 µL | |||
| Wash solution: 10 mL | |||
| DAPI stock solution (5 mg/mL) | |||
| DAPI powder: 5 mg | Sigma-Aldrich | D9542-1MG | |
| DMSO (100%): 1 mL | ThermoFisher | D12345 | |
| Imaging Media RI-2 (in dH2O) | |||
| 90% Histodenz | Sigma | D2158-100G | |
| 0.01% Sodium azide | Sigma | 71289-50G | |
| 0.02 Sodium Phosphate Buffer, pH 7.5 | Sigma-Aldrich | S9638-250G | |
| 0.1% Tween-20 | Fisher Scientific | BP337-500 | |
| Primary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Goat anti-HIV p24: 10 µL (1:200) | Creative Diagnostics | DPATB-H81692 | |
| Mouse anti-human CD68: 10 µL(1:200) | Dako | M0876 | |
| Rabbit anti-human CD3: 10 µL (1:200) | Dako | A0452 | |
| 8% SDS Solution (in 0.1 M PBS, 50 mL in total) | |||
| SDS powder: 4 g | Sigma-Aldrich | L3771-500G | |
| Secondary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Donkey anti-goat conjugated with AlexaFluor647: 2 µL | Invitrogen | A21447 | |
| Donkey anti-mouse conjugated with AlexaFluor594: 2 µL | Invitrogen | A21203 | |
| Donkey anti-rabbit conjugated with AlexaFluor488: 2 µL | Invitrogen | A21206 |
参考文献
- Spalteholz, W., Barker, L. F., Mall, F. P. . Hand-Atlas of Human Anatomy. , (1907).
- Jacob, T., Gray, J. W., Troxell, M., Vu, T. Q. Multiplexed imaging reveals heterogeneity of PI3K/MAPK network signaling in breast lesions of known PIK3CA genotype. Breast Cancer Research and Treatment. 159 (3), 575-583 (2016).
- Kieffer, C., Ladinsky, M. S., Ninh, A., Galimidi, R. P., Bjorkman, P. J. Longitudinal imaging of HIV-1 spread in humanized mice with parallel 3d immunofluorescence and electron tomography. eLife. 6, 23282 (2017).
- Chung, K., Deisseroth, K. CLARITY for mapping the nervous system. Nature Methods. 10 (6), 508-513 (2013).
- Compton, A. A., Schwartz, O. They might be giants: Does syncytium formation sink or spread HIV infection. PLoS Pathogens. 13 (2), 2-8 (2017).
- Symeonides, M., et al. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses. 7 (12), 6590-6603 (2015).
- Sharova, N., Swingler, C., Sharkey, M., Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. The EMBO Journal. 24 (13), 2481-2489 (2005).
- Colby, D. J., et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nature Medicine. 24 (7), 923-926 (2018).
- Ladinsky, M. S., et al. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. eLife. 8, 46916 (2019).
- Buettner, M., Bode, U. Lymph node dissection--understanding the immunological function of lymph nodes. Clinical and Experimental Immunology. 169 (3), 205-212 (2012).
- Treweek, J. B., et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nature Protocols. 10, 1860-1896 (2015).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159, 911-924 (2014).
- Sompuram, S. R., Vani, K., Bogen, S. A. A molecular model of antigen retrieval using a peptide array. American Journal of Clinical Pathology. 125 (1), 91-98 (2006).
- Scalia, C. R., et al. Antigen masking during fixation and embedding, dissected. The journal of histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 65 (1), 5-20 (2017).
- Jing, D., et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Research. 28 (8), 803-818 (2018).
- Guldner, I. H., et al. An Integrative platform for three-dimensional quantitative analysis of spatially heterogeneous metastasis landscapes. Scientific Reports. 6, 24201 (2016).
- Muntifering, M., et al. Clearing for deep tissue imaging. Current Protocols in Cytometry. 86 (1), 38 (2018).
- DePas, W. H., et al. Exposing the three-dimensional biogeography and metabolic states of pathogens in cystic fibrosis sputum via hydrogel embedding, clearing, and rRNA labeling. mBio. 7 (5), 00796 (2018).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image. Nature Methods. 9 (7), 676-682 (2012).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。