
Method Article
Visualizzazione 3D delle popolazioni di cellule immunitarie nei tessuti infetti da HIV tramite microscopia a fluorescenza a clearing, immunocolorazione, confocale e a foglio luminoso
In questo articolo
Riepilogo
La pulizia dei tessuti, combinata con la microscopia a immunofluorescenza, consente la visualizzazione spaziale e la quantificazione delle popolazioni di cellule immunitarie e delle proteine virali all'interno dei tessuti intatti. Il sezionamento ottico di tessuti chiariti con microscopia a fluorescenza confocale e a foglio di luce può generare modelli 3D di ambienti tissutali complessi e rivelare l'eterogeneità spaziale esibita durante l'infezione da HIV.
Abstract
Il virus dell'immunodeficienza umana (HIV), l'agente eziologico della sindrome da immunodeficienza acquisita (AIDS), è un grave problema di salute globale con quasi 40 milioni di individui infetti in tutto il mondo e nessuna cura ampiamente accessibile. Nonostante gli intensi sforzi, una comprensione dettagliata delle interazioni tra virus e cellule ospiti nei tessuti durante l'infezione e in risposta alla terapia rimane incompleta. Per affrontare queste limitazioni, le tecniche di pulizia dei tessuti a base d'acqua CUBIC (Clear, Unobstructed Brain/Body Imaging Cocktails and Computational analysis) e CLARITY (Clear Lipid-exchanged Acrylamide-hybridized Rigid Imaging/Immunostaining/in situ-hybridization-compatible Tissue hYdrogel) vengono applicate per visualizzare complesse interazioni virus-cellula ospite in tessuti infetti da HIV da modelli animali e dall'uomo utilizzando la microscopia a fluorescenza confocale e a foglio di luce. Il sezionamento ottico di tessuti intatti e l'analisi delle immagini consentono una rapida ricostruzione delle informazioni spaziali contenute all'interno di interi tessuti e la quantificazione delle popolazioni di cellule immunitarie durante l'infezione. Questi metodi sono applicabili alla maggior parte delle fonti di tessuto e a diverse questioni biologiche, tra cui le malattie infettive e il cancro.
Introduzione
La crescente necessità di imaging quantitativo dei tessuti spaziali nella ricerca biologica ha recentemente portato all'emergere di tecniche di pulizia dei tessuti per generare immagini di volume maggiore (mm3-cm 3) di tessuti intatti con risoluzione di una singola cellula. I tessuti includono organizzazioni complesse di biomolecole con strutture, composizioni e funzioni definite in modo univoco. Sfortunatamente, molte biomolecole presenti nei tessuti (ad esempio, lipidi e cromofori) si disperdono, assorbono o emettono luce quando vengono visualizzate al microscopio ottico, rendendo difficile l'imaging di grandi volumi. Inoltre, i tessuti spesso mostrano un indice di rifrazione non corrispondente alle soluzioni di imaging standard e alle lenti ottiche, con conseguenti distorsioni ottiche durante l'imaging. Un approccio ottimale per l'imaging di grandi volumi di tessuto con un microscopio ottico dovrebbe comportare la corrispondenza dell'indice di rifrazione dei tessuti, delle soluzioni di imaging e degli obiettivi, consentendo al contempo la penetrazione della luce in profondità nel tessuto senza interrompere le caratteristiche biologiche dei tessuti intatti durante l'elaborazione. I primi tentativi di ridurre le differenze nell'indice di rifrazione tra i tessuti e le soluzioni di imaging attraverso la pulizia di campioni di tessuto opaco furono effettuati dall'anatomista tedesco Werner Spalteholz alla fine del 18001. Questa tecnica di pulizia dei tessuti ha coinvolto solventi chimici aggressivi, che possono danneggiare i campioni di tessuto, ma ha comunque rappresentato il primo imaging di grandi volumi di tessuti intatti. I moderni metodi di microscopia ottica, combinati con la potenza di calcolo per l'acquisizione e l'analisi delle immagini, hanno recentemente riportato in auge la pulizia dei tessuti come metodo per l'imaging di campioni di tessuto intatti di grandi dimensioni con risoluzione di una singola cellula. Negli ultimi due decenni, sono emerse dozzine di tecniche avanzate di pulizia dei tessuti, sia a base organica che a base d'acqua, ognuna con punti di forza e di debolezza per applicazioni specifiche.
L'imaging 3D dei tessuti può sondare interazioni biologiche più complesse che non possono essere riprodotte in coltura cellulare. Ad esempio, i modelli di segnalazione cellulare2, le distribuzioni spaziali di tipi cellulari distinti3 e la connettività cerebrale4 sono stati precedentemente mappati in modo quantitativo utilizzando metodi di imaging di interi tessuti/organi. Qui viene descritta un'applicazione di protocolli di pulizia dei tessuti a base d'acqua per eliminare, immunocolorare e visualizzare popolazioni distinte di cellule bersaglio dell'HIV all'interno di tessuti linfoidi intatti infettati da HIV durante l'infezione attiva. All'interno del corpo, l'HIV infetta prevalentemente le cellule T CD4+ e integra una copia del suo genoma nei genomi delle cellule ospiti infette. Il virus successivamente dirotta il macchinario della cellula ospite infetta per replicarsi, con conseguente disseminazione del virus, uccisione della cellula ospite, disfunzione immunitaria e progressione a lungo termine verso l'AIDS. È importante notare che i comportamenti delle cellule T infette nei tessuti e nelle colture cellulari sono notevolmente discrepanti. Le cellule T CD4+ in coltura incubate con l'HIV possono produrre sincizi massicci indotti dall'HIV che possono includere dozzine di nuclei5, mentre esperimenti simili con cellule T CD4+ primarie coltivate in idrogel di matrice extracellulare 3D (ECM) o campioni di tessuto di topi umanizzati infetti da HIV (topi hu) generalmente producono sincizi con 2-5 nuclei6. Comprendere la trasmissione locale da cellula a cellula e la disseminazione sistemica del virus all'interno di individui infetti da HIV è probabilmente ancora più complicato, poiché coinvolge il trasporto del virus da parte di più tipi di cellule infette dai tessuti ai vasi sanguigni a nuovi tessuti, dove i virioni liberi e le cellule produttrici di virus possono accedere a un gran numero di linfociti suscettibili7. Questi scenari non sono attualmente ricapitolabili nei sistemi di coltura cellulare e i tessuti provenienti da modelli animali e dall'uomo rimangono una risorsa importante per comprendere la patogenesi dei virus nel contesto di un organismo complesso con un sistema immunitario funzionante.
Le attuali terapie antiretrovirali (ART) aumentano notevolmente l'aspettativa di vita e la qualità delle persone con HIV (PWH) inibendo la replicazione dell'HIV e arrestando la progressione della malattia verso l'AIDS. Sfortunatamente, l'ART non elimina le cellule immunitarie infette latenti contenenti un'inserzione del genoma retrovirale che sono quiescenti e non producono attivamente il virus. Sebbene il virus non sia rilevabile nel sangue della maggior parte degli individui in terapia antiretrovirale, le cariche virali rimbalzano rapidamente dopo l'interruzione della terapia antiretrovirale e la progressione della malattia continua8. La natura persistente dell'infezione da HIV causata dal serbatoio latente di cellule infette rappresenta un enorme ostacolo alla creazione di una cura per l'HIV. I serbatoi tissutali dell'HIV rimangono poco conosciuti ed è fondamentale stabilire una comprensione più profonda di questi serbatoi nei tessuti linfoidi prima, durante e dopo l'ART, per caratterizzare completamente la patogenesi del virus e valutare nuovi trattamenti che eliminino efficacemente le cellule infettate latenti che non producono attivamente il virus.
Qui, CUBIC3 e CLARITY9, due protocolli di pulizia dei tessuti a base d'acqua precedentemente adattati, sono stati applicati per visualizzare popolazioni di cellule immunitarie all'interno di numerosi tessuti linfoidi intatti da topi infetti da HIV con sistemi immunitari umanizzati (hu-mice), primati non umani infetti da SIV/SHIV (NHP) e esseri umani infetti da HIV. Questi protocolli sono adattabili sia alla microscopia a fluorescenza confocale che a quella a foglio leggero, a seconda degli obiettivi dell'imaging (risoluzione più elevata rispetto a volume maggiore) e della strumentazione disponibile. Sebbene la microscopia ottica non sia in grado di risolvere singoli virioni, l'uso dell'immunofluorescenza può identificare regioni di tessuto contenenti virus e cellule produttrici di virus che possono essere ulteriormente analizzate con metodi a risoluzione più elevata. I metodi qui presentati possono essere adattati per visualizzare quasi tutti i tessuti del corpo con risoluzione a singola cellula al fine di quantificare le relazioni spaziali tra specifici tipi di cellule in diverse condizioni durante l'infezione e sono facilmente traducibili in campioni di pazienti umani altamente rilevanti per lo studio di malattie infettive o cancro.
Protocollo
Tutti gli esperimenti sugli animali sono stati condotti secondo i protocolli istituzionali approvati per la cura degli animali. Tutti i tessuti umani sono stati acquisiti secondo le linee guida istituzionali approvate per l'etica della ricerca umana.
1. Prelievo e fissazione dei tessuti (lo stesso per CUBIC e CLARITY)
- Identificare e sezionare i tessuti linfoidi come descritto in precedenza10.
- Asportare i tessuti linfoidi con forbici da dissezione e pinzette entro pochi minuti dall'autopsia, quando possibile in sicurezza.
- Posizionare i campioni di tessuto in un tampone fissativo ghiacciato appena fatto contenente l'8% di paraformaldeide (PFA), il 5% di saccarosio in 0,1 M di sodio cacodilato triidrato per conservare adeguatamente i campioni di tessuto per la microscopia ottica (LM), la microscopia elettronica (EM) o l'immuno-EM. In alternativa, fissare i campioni per LM con il 4% di PFA in 0,1 M PBS. Fissare i campioni durante la notte prima di iniziare il processo di eliminazione per garantire la completa disattivazione del virus.
ATTENZIONE: La paraformaldeide è tossica per contatto con la pelle e per inalazione ed è anche un solido infiammabile; Maneggiare con cura e conservare in un armadietto infiammabile. Il sodio cacodilato triidrato è tossico se ingerito o inalato. - Scatta un'immagine di riferimento del tessuto prima di iniziare il processo di pulizia.
NOTA: I campioni LM possono essere conservati per almeno 1 anno in queste condizioni. Per lavorare con campioni che esprimono proteine fluorescenti endogene, tenere sempre i campioni al buio nelle fasi successive.
2. Pulizia dei tessuti CUBICI
- Sciacquare i campioni di tessuto linfoide in PBS sterile da 0,1 M tre volte agitando a temperatura ambiente per 15 minuti per garantire la rimozione del PFA durante ogni cambio di tampone.
NOTA: Smaltire i liquidi contenenti PFA secondo le linee guida istituzionali. - Immergere il campione di tessuto linfoide nel reagente-1 CUBIC (vedere la tabella dei materiali) a 37 °C per 3 giorni agitando delicatamente. Scatta immagini di riferimento regolari per monitorare il processo di decolorazione nel tempo.
- Sostituire il Reagente-1 per altri 3-4 giorni di immersione o fino al completamento della decolorazione dei tessuti. Il tempo necessario per la pulizia dipende sia dal volume che dal tipo di tessuto. Per accelerare il processo di decolorazione dei tessuti, aggiornare quotidianamente il reagente-1 CUBIC e utilizzare volumi maggiori.
- Lavare i campioni di tessuto linfoide tre volte con 0,1 M PBS per 30 minuti a temperatura ambiente agitando delicatamente.
- Immergere i campioni di tessuto linfoide nel reagente-2 CUBIC (vedere la tabella dei materiali) a 37 °C agitando delicatamente per 2-7 giorni o fino a raggiungere la completa trasparenza. Se i campioni non raggiungono la completa trasparenza, ripetere i passaggi 2.2-2.5 fino a quando la pulizia non procede più. Scatta immagini di riferimento regolari per monitorare il processo di sgombero nel tempo.
- Lavare i campioni di tessuto linfoide tre volte con 0,1 M PBS per 30 minuti a temperatura ambiente agitando delicatamente.
- Conservare i campioni nel reagente-2 CUBIC con azoturo di sodio allo 0,01% in volume/volume (V/V) al buio (vedere la tabella dei materiali).
NOTA: I campioni possono essere conservati per almeno 6 mesi utilizzando questo metodo.
ATTENZIONE: L'azoturo di sodio è altamente tossico e rappresenta un grave rischio di inalazione. Si consiglia l'acquisto di soluzioni diluite con sodio azide al 5% o meno.
3. Blocco e immunocolorazione di campioni cubici
- Lavare i campioni di tessuto linfoide tre volte con 0,1 M PBS per 30 minuti ciascuno a temperatura ambiente agitando delicatamente.
- Per l'imaging con un microscopio confocale, tagliare il tessuto in fette spesse ~0,5-1 mm utilizzando una matrice affetta tessuti. Per eseguire la microscopia a fluorescenza a foglio ottico (LSFM), bloccare l'intera regione del tessuto.
- Bloccare i campioni con 5 mL di soluzione bloccante CUBIC per una notte a 4 °C con agitazione (vedere la Tabella dei materiali). Quando si lavora con NHP o campioni umani, utilizzare FcR anti-umano. Quando si utilizzano campioni di topo, utilizzare FcR anti-topo nella soluzione di blocco.
- Colorare i campioni con 5 mL di anticorpi primari (vedere la Tabella dei materiali) in soluzione bloccante (senza FcR specie-specifica) per 3 giorni a temperatura ambiente con agitazione (opzionale: centrifugare lo stock di anticorpi concentrati a 2.300 x g per 5 minuti prima dell'uso, per ridurre l'aggiunta di anticorpi aggregati).
- Lavare il campione colorato a temperatura ambiente agitando per un periodo di tempo minimo di 5 ore in totale con almeno cinque sostituzioni di tampone soluzione di lavaggio (vedere la tabella dei materiali).
- Colorare i campioni con anticorpi secondari (vedi Tabella dei materiali) in soluzione bloccante (senza FcR specie-specifica) per 3 giorni a temperatura ambiente con agitazione (opzionale: centrifugare gli anticorpi a 2.300 x g per 5 minuti prima dell'uso per ridurre al minimo l'aggregazione degli anticorpi).
- Lavare il campione colorato cinque volte con soluzione di lavaggio a temperatura ambiente agitando per almeno 5 ore in totale.
- Colorare i campioni con 5 mL di soluzione colorante DAPI (vedere la Tabella dei materiali) su ciascun campione di tessuto e incubare per 10 minuti a temperatura ambiente. Lasciare che i campioni rimangano nella soluzione di colorante DAPI al buio a 4 °C per l'imaging successivo.
- Lavare i campioni di tessuto linfoide con soluzione di lavaggio tre volte a temperatura ambiente agitando per 30 minuti ciascuna.
- Immergere il campione colorato nel reagente-2 CUBIC per una notte a temperatura ambiente al buio prima di montare il campione.
4. Pulizia dei tessuti CLARITY
- Sciacquare i campioni di tessuto linfoide in PBS sterile 0,1 M tre volte agitando a temperatura ambiente per 15 minuti ciascuno per rimuovere il PFA.
- Immergere i campioni di tessuto in 15 mL di soluzione di acrilammide appena prodotta e incubare a 4 °C per una notte con una leggera agitazione (vedere la Tabella dei materiali).
ATTENZIONE: L'acrilammide non polimerizzata è una potente neurotossina e viene facilmente assorbita attraverso la pelle. Evitare qualsiasi contatto con la pelle e sciacquare immediatamente in caso di contatto. - Lasciare che i campioni di tessuto si riscaldino a temperatura ambiente.
- OPZIONALE: Degassare i campioni di tessuto facendo gorgogliare l'azoto nella soluzione di acrilammide per 1 minuto. Fare attenzione a utilizzare una bassa portata che eviti schizzi di acrilammide tossico non polimerizzato (~1-2 bolle/s).
- Porre i campioni di tessuto in un bagno d'acqua a 37 °C per 1-3 ore per polimerizzare, capovolgendoli ogni 15 minuti. Rimuovere i campioni non appena viene rilevata una polimerizzazione evidente, come indicato da un liquido viscoso, dalla comparsa di linee di Schleren durante la miscelazione o dalla formazione di una capsula trasparente attorno al tessuto.
NOTA: Se si verifica la polimerizzazione completa della soluzione di acrilammide, eliminare l'idrogel in eccesso dal campione e continuare il protocollo. - Lavare i campioni di tessuto con PBS sterile 0,1 M tre volte per 30 minuti ciascuna a temperatura ambiente agitando delicatamente per rimuovere la soluzione di acrilammide.
- Posizionare i campioni di tessuto in 15 mL di SDS all'8% in 0,1 M PBS a 37 °C con un leggero oscillazione per 2-5+ giorni per consentire la cancellazione. Aggiornare periodicamente la soluzione SDS all'8% e utilizzare fino a 50 ml di soluzione per accelerare la pulizia, se necessario. Interrompere il processo di schiaritura quando i campioni sono visivamente trasparenti o non progrediscono più. Scatta immagini di riferimento regolari per monitorare il processo di sgombero nel tempo.
- Lavare i campioni di tessuto con PBS sterile 0,1 M cinque volte al giorno a temperatura ambiente agitando delicatamente.
- Conservare temporaneamente i campioni in 0,1 M PBS (più 0,01% volume/volume (v/v) NaN3 per la conservazione a lungo termine) al buio fino al momento dell'imaging della fluorescenza endogena.
- Posizionare il tessuto in 5 mL di Terreno di Imaging RI-2 (vedere Tabella dei Materiali). Incubare per una notte a temperatura ambiente al buio per verificare la completezza del processo di chiarificazione prima dell'immunocolorazione. Acquisisci immagini di riferimento per monitorare la trasparenza dei tessuti.
5. Blocco e immunocolorazione dei campioni CLARITY
NOTA: Questi passaggi sono simili al blocco e all'immunocolorazione dei tessuti puliti CUBIC, ma utilizzano formulazioni diverse per il blocco, il lavaggio e la colorazione delle soluzioni.
- Lavare i campioni di tessuto linfoide tre volte con 0,1 M PBS per 30 minuti ogni volta a temperatura ambiente agitando delicatamente.
- Per l'imaging con un microscopio confocale, tagliare il tessuto in fette spesse ~0,5-1 mm utilizzando un'affettatrice e una matrice da 0,5 mm. Per eseguire LSFM, bloccare l'intero campione di tessuto.
- Bloccare i campioni con 5 mL di soluzione bloccante CLARITY (vedere la Tabella dei materiali) per una notte a 4 °C con agitazione.
- Colorare i campioni con 5 mL di anticorpi primari (vedere la Tabella dei materiali) in soluzione bloccante (senza FcR specie-specifica) per 3 giorni a temperatura ambiente con agitazione (opzionale: centrifugare gli anticorpi a 2.300 x g per 5 minuti prima dell'uso per ridurre al minimo l'aggregazione degli anticorpi).
- Lavare il campione colorato cinque volte con la soluzione di lavaggio a temperatura ambiente agitando per almeno 5 ore in totale (vedere la tabella dei materiali).
- Colorare i campioni con 5 mL di anticorpi secondari (vedere la Tabella dei materiali) in soluzione bloccante (senza FcR specie-specifica) per 3 giorni a temperatura ambiente con agitazione (opzionale: centrifugare gli anticorpi a 2.300 x g per 5 minuti prima dell'uso per ridurre al minimo l'aggregazione degli anticorpi). Per ridurre la lunghezza complessiva del protocollo, utilizzare anticorpi primari coniugati con fluorofori per eliminare la necessità di incubazione con anticorpi secondari.
- Lavare il campione colorato cinque volte con la soluzione di lavaggio a temperatura ambiente agitando per almeno 5 ore in totale.
- Colorare i campioni con 5 mL di soluzione colorante DAPI (vedere la Tabella dei materiali) su ciascun campione di tessuto e incubare per 10 minuti a temperatura ambiente. Lasciare che i campioni rimangano a 4°C al buio nella soluzione colorante DAPI per l'imaging successivo.
- Lavare i campioni di tessuto linfoide con la soluzione di lavaggio tre volte a temperatura ambiente agitando per 30 minuti ogni volta.
- Posizionare il tessuto in 5 mL di Imaging Media RI-2 (R.I. = 1,46) e incubare per una notte a temperatura ambiente al buio prima del montaggio del campione (vedere le fasi 6 e 7 del protocollo).
6. Montaggio e imaging di campioni di tessuto chiarito per microscopia confocale
- Staccare un lato dello strato protettivo di un isolatore in silicone adesivo.
- Attaccare un vetro di copertura del microscopio (22 mm x 40 mm, 0,25 mm di spessore) sul lato sbucciato dell'isolatore in silicone per formare uno spazio a prova di liquidi per il campione.
- Staccare l'altro lato dello strato protettivo dell'isolatore in silicone adesivo.
- Posizionare il campione per l'imaging al centro dell'isolatore in silicone, quindi aggiungere il reagente-2 CUBIC o il supporto di imaging RI-2 a seconda dei casi fino a quando la superficie del liquido non raggiunge l'altezza del bordo dell'isolatore.
- Per ridurre al minimo l'intrappolamento di bolle d'aria all'interno dell'isolatore in silicone, allineare e stratificare delicatamente il secondo vetro di copertura verso il basso da un lato utilizzando una pinza EM. Eliminare il liquido in eccesso. Premere delicatamente il vetro di copertura attorno al pozzetto del campione utilizzando la parte posteriore della pinza per sigillare l'adesivo. Conservare i campioni montati orizzontalmente al buio.
NOTA: I campioni possono essere ripresi settimane o mesi dopo essere stati montati; Tuttavia, la qualità dell'imaging generalmente diminuisce nel tempo. - Posizionare il vetrino montato sul tavolino del microscopio e localizzare il campione utilizzando la luce bianca e un obiettivo con ingrandimento inferiore (2-10x).
- Impostare il profilo di acquisizione della fluorescenza in base ai singoli fluorofori scelti.
NOTA: Si consiglia di acquisire individualmente canali fluorofori separati. Ciò si traduce in un tempo di acquisizione più lungo, ma riduce la sovrapposizione spettrale e l'acquisizione di un segnale di fluorescenza non specifico. Un profilo fluoroforo comune può includere DAPI (450 nm), Alexa488, Alexa594 e Alexa647 (o combinazioni correlate) per ridurre al minimo la sovrapposizione spettrale durante l'acquisizione dell'immagine. - Scegliere un obiettivo di ingrandimento appropriato per visualizzare le regioni di interesse. Utilizzare obiettivi con ingrandimento inferiore (2-10x) per l'imaging di volumi maggiori o di interi tessuti con risoluzione di una singola cellula e utilizzare obiettivi con ingrandimento più elevato (20-63x) per la visualizzazione a risoluzione più elevata di dettagli subcellulari in tessuto pulito. Abbina il più possibile l'indice di rifrazione degli obiettivi, dei mezzi di imaging e dei tessuti per ridurre al minimo l'introduzione di distorsioni ottiche durante l'acquisizione dell'immagine.
- Scegliere una dimensione del passo per l'acquisizione Z-stack. Per obiettivi con ingrandimento inferiore (2-10x), selezionare una dimensione del passo di ~3-5 μm per rilevare la fluorescenza da una singola cella in più sezioni Z continue per la modellazione 3D, riducendo al contempo il tempo di acquisizione totale e le dimensioni complessive del file. Per obiettivi con ingrandimento più elevato (20-63x), selezionare una dimensione del passo di ~1 μm o inferiore per ridurre al minimo la perdita di informazioni subcellulari tra le singole sezioni Z.
- Ingrandisci il campo visivo per visualizzare l'intera regione del tessuto da visualizzare nelle dimensioni X e Y con la minor area libera possibile. Imposta le coordinate di acquisizione dello stadio Z superiore e inferiore che comprendono l'intera regione di interesse da riprendere.
- Acquisisci le immagini Z-stack. Salva ed esporta il file per la post-elaborazione utilizzando qualsiasi software di analisi delle immagini. Per alcune suite software, converti i file in tipi di file specifici (ad esempio, .tiff, .ome-tiff, .jpeg, ecc.). Esegui la conversione utilizzando qualsiasi software di acquisizione di immagini per microscopio o freeware per l'analisi delle immagini (ad esempio, ImageJ/Fiji).
7. Montaggio e imaging dei campioni in camera LSFM o cuvetta
- Riempire la camera di imaging con CUBIC Reagent-2 o RI-2 a seconda del protocollo specifico utilizzato. Evitare la formazione di bolle durante il trasferimento del liquido. Rimuovere le bolle in eccesso con una pipetta.
- Immergere il campione nella camera di imaging e limitare il movimento del campione.
NOTA: A seconda del microscopio specifico utilizzato, ciò può includere l'inclusione del campione nell'agarosio, la sospensione del campione a un gancio o a un adattatore per porcospino, la stampa 3D di un supporto per campioni o il fissaggio del campione con adesivo a un piatto di plastica. - Posizionare l'obiettivo nella soluzione di imaging, focalizzato sul campione. Lasciare il campione montato nella camera di imaging per diverse ore o durante la notte per consentire il pieno equilibrio delle soluzioni e dei tessuti nella cuvetta.
- Acquisire lo Z-stack della regione di interesse (vedere i passaggi 6.7-6.11 per l'acquisizione dell'immagine).
NOTA: Questo approccio può consentire l'imaging di volumi di tessuto superiori a 1 cm3 con risoluzione a singola cellula.
8. Ricostruzione della superficie e quantificazione cellulare con il software di analisi delle immagini Imaris
NOTA: questi passaggi sono specifici per il software di analisi delle immagini Imaris, ma passaggi simili per l'elaborazione delle immagini possono essere eseguiti utilizzando altre suite software (ad esempio, ImageJ/Fiji, Aivia, Arivis, Amira, ecc.).
- Usa Imaris File Converter per convertire il file immagine Z-stack nel formato nativo Imaris .ims. Ciò faciliterà una conversione più rapida dei file, riducendo al minimo gli errori di conversione e i potenziali problemi del software una volta aperti.
NOTA: Alcuni LSFM più recenti consentono all'utente di salvare i file direttamente nel formato .ims. - Trascinare il file .ims da analizzare nell'area Arena del software Imaris. Regolate il contrasto o l'intensità di ciascun canale di colore tramite il pannello Regolazione schermo . Fare clic sull'icona Aggiungi nuove superfici in alto a sinistra.
- Fare clic su Avanti: Canale sorgente (l'icona blu con una freccia che punta a destra). Scegliere il canale di origine della superficie da costruire. Non modificare gli altri parametri.
- Fare clic su Avanti: Soglia (l'icona blu con una freccia che punta a destra).
- Per regolare la soglia (intensità assoluta), trascinare la linea di soglia a sinistra o a destra. Abilita Dividi oggetti a contatto e inserisci il diametro medio della cella in micron come standard di divisione per il sistema per produrre molti punti come origine per ogni singola superficie.
- Non includere segnali fluorescenti troppo piccoli o troppo luminosi in quanto possono rappresentare potenziali macchie o artefatti del microscopio. Includi solo i punti che hanno dimensioni e intensità di fluorescenza accettabili modificando di conseguenza il diametro medio della cella.
NOTA: Il diametro medio delle cellule varia per tessuti o tipi di cellule specifici, ma generalmente risiede tra 5-15 μm.
- Non includere segnali fluorescenti troppo piccoli o troppo luminosi in quanto possono rappresentare potenziali macchie o artefatti del microscopio. Includi solo i punti che hanno dimensioni e intensità di fluorescenza accettabili modificando di conseguenza il diametro medio della cella.
- Fare clic su Avanti: Classifica superfici (l'icona blu con una freccia che punta a destra con l'etichetta). Regolare le superfici da includere trascinando la linea di soglia a sinistra o a destra. Assicurarsi che le superfici si avvicinino esattamente al segnale di fluorescenza grezzo, separando il segnale di fluorescenza dalle singole cellule.
- Fare clic su Fine: Esegui tutti i passaggi di creazione e termina la procedura guidata (l'icona verde con due frecce che puntano a destra etichettata). La superficie è ufficialmente costruita.
- Fare clic sulla sesta icona denominata Statistiche nel pannello di sinistra per vedere il numero di celle, in questa circostanza, il numero di superfici per il canale di colore specifico analizzato.
- Assicurarsi che le quattro variabili Numero di componenti disconnessi per punto temporale, Numero di superfici per punto temporale, Numero totale di componenti disconnessi e Numero totale di superfici abbiano lo stesso numero, ovvero il numero di celle di quel canale di colore.
Risultati
La pulizia dei tessuti comporta il trattamento dei tessuti conservati con cocktail chimici per estrarre biomolecole opache dal tessuto mantenendo l'architettura dei tessuti. Queste soluzioni per la pulizia dei tessuti abbinano l'indice di rifrazione del tessuto con il mezzo di imaging circostante per ridurre al minimo le distorsioni ottiche, migliorare il rapporto segnale/rumore in profondità all'interno dei tessuti e ridurre al minimo l'autofluorescenza di fondo. Due protocolli a base d'acqua per la chiarificazione ottica dei tessuti, CUBIC3 e CLARITY9, sono stati utilizzati per eliminare campioni di hu-topo, primati non umani e tessuti umani conservati infetti da HIV/SIV prima della colorazione in immunofluorescenza e dell'imaging con microscopia a fluorescenza confocale e a foglio di luce.
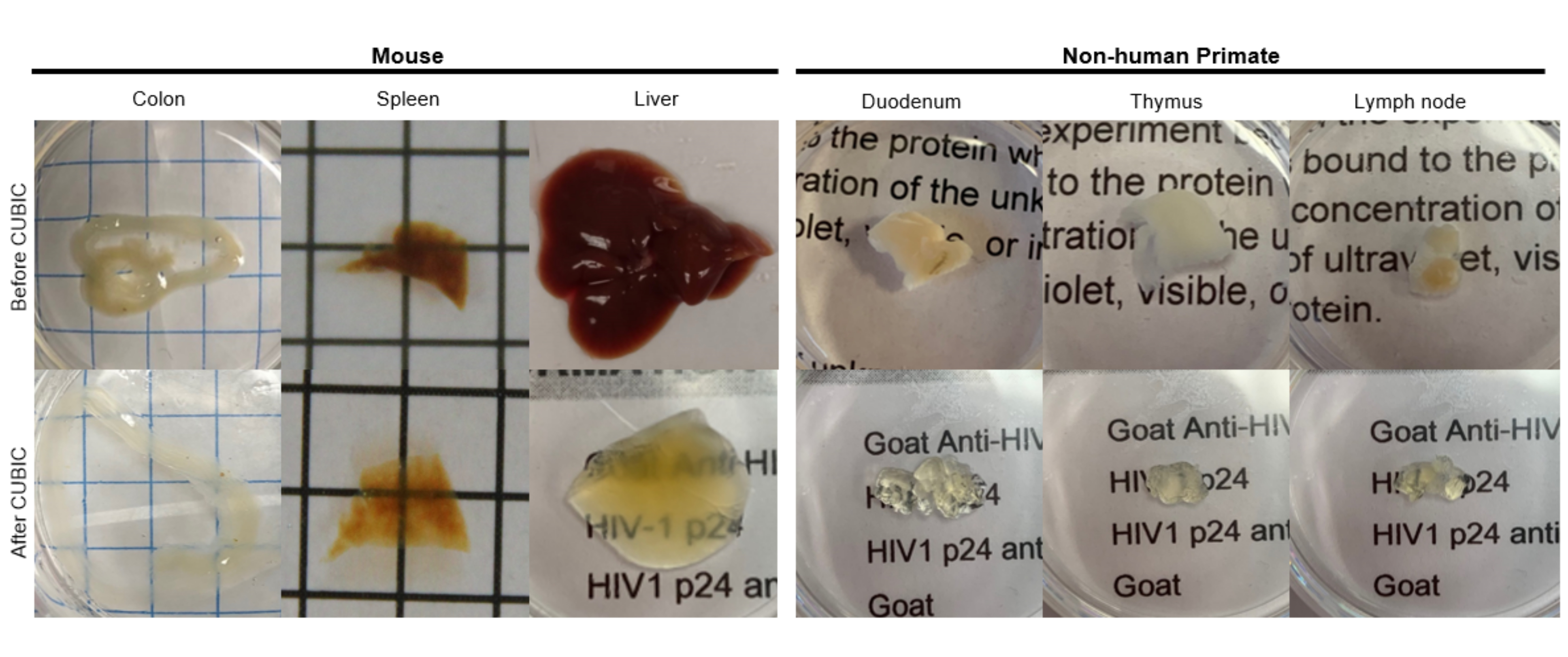
Per il protocollo CUBIC, i tessuti fissati sono stati lavati con PBS per rimuovere i fissativi e immersi in CUBIC Reagent-1, una soluzione basica tamponata di aminoacidi che eluisce cromofori come l'eme, con conseguente decolorazione e delipidazione del tessuto (Figura 1, in alto). I volumi di tessuto più piccoli (~ mm3) possono essere decolorati dopo 3 giorni di trattamento con il reagente-1 CUBIC, ma volumi di tessuto più grandi (~ cm3) o tessuti con una grande quantità di eme (come fegato, milza o cuore) richiedono tempi di incubazione e volumi di soluzione più lunghi (>1 mese e ~ 50 mL), nonché un frequente scambio della soluzione ogni 2-3 giorni. Dopo la decolorazione, i tessuti sono stati lavati e posti in CUBIC Reagent-2, una soluzione contenente saccarosio con un indice di rifrazione di circa 1,48-1,49, che corrisponde all'indice di rifrazione del tessuto e aumenta la trasmittanza della luce. I tessuti chiariti sono stati immunocolorati e montati in una soluzione di CUBIC Reagent-2 prima dell'imaging con un microscopio confocale o a foglio ottico. Gli effetti della procedura di chiarificazione CUBIC sono stati ripresi per diversi tessuti hu-mouse e NHP di varie dimensioni e concentrazioni di cromofori (Figura 2). La pulizia ottica ha reso i tessuti visibilmente trasparenti ad occhio nudo, consentendo di vedere le linee della griglia e il testo su fogli di carta "attraverso" il tessuto. I tessuti ricchi di cromofori come la milza, il fegato, il midollo osseo e il cuore potrebbero non decolorarsi completamente, ma rimanere adatti per l'immunocolorazione e l'imaging (Figura 2 e Figura 5).
Per il protocollo CLARITY, i tessuti fissati sono stati lavati con PBS per rimuovere i fissativi e poi incubati per una notte a 4 °C in una soluzione di acrilammide al 40% con un iniziatore termico per formare legami covalenti tra le proteine nel campione e i monomeri di acrilammide (Figura 1, in basso). Il giorno seguente, dopo che il tessuto è stato equilibrato a temperatura ambiente e poi riscaldato in un bagno d'acqua a 37 °C, è stata avviata la polimerizzazione dell'acrilammide che ha rapidamente racchiuso il campione in un idrogel. Il campione è stato trattato con una soluzione di SDS all'8% in un corso di 2-5 giorni per rimuovere i lipidi opachi. Immediatamente prima della colorazione fluorescente, il campione è stato immerso in una soluzione di corrispondenza dell'indice di rifrazione (RIMS) per CLARITY (Imaging Media RI-2) contenente il 90% di terreno a gradiente di densità non ionica. Per i tessuti contenenti grandi quantità di eme, è possibile aggiungere una fase di decolorazione alla fine della fase di delipidazione 9,11,12. La progressione della pulizia CUBIC e CLARITY è stata confrontata su diverse sezioni dello stesso campione di milza umana (Figura 3). La chiarificazione CLARITY produce un gel di poliacrilammide visibile che avvolge la soluzione e in genere mostra una decolorazione ridotta rispetto alla chiarificazione CUBIC, a meno che non venga aggiunta un'ulteriore fase di decolorazione 9,12.
Successivamente, in entrambi i protocolli, i tessuti puliti e intatti sono stati immunocolorati per rilevare specifiche popolazioni di cellule immunitarie. I campioni sono stati lavati, bloccati con un reagente contenente α-FcR per ridurre il legame con gli anticorpi non specifici e colorati per 3 giorni utilizzando un anticorpo primario direttamente coniugato a un fluoroforo. In alternativa, i campioni sono stati colorati per 3 giorni con un anticorpo primario non coniugato seguito da altri 3 giorni con un anticorpo secondario coniugato a un fluoroforo. I tessuti sono stati nuovamente lavati e quindi incubati con colorante DAPI per una notte a 4 °C per la visualizzazione nucleare. I campioni sono stati lavati e incubati nel reagente-2 CUBIC per 24-36 ore o nel terreno di imaging RI-2 (CLARITY) durante la notte al buio. Per la microscopia confocale, i tessuti sono stati montati su un vetrino da microscopio nel RIMS appropriato prima dell'imaging (Figura 4). Per la microscopia a fluorescenza a foglio di luce (LSFM), i campioni sono stati completamente immersi con RIMS in una cuvetta di imaging durante la notte prima dell'imaging.
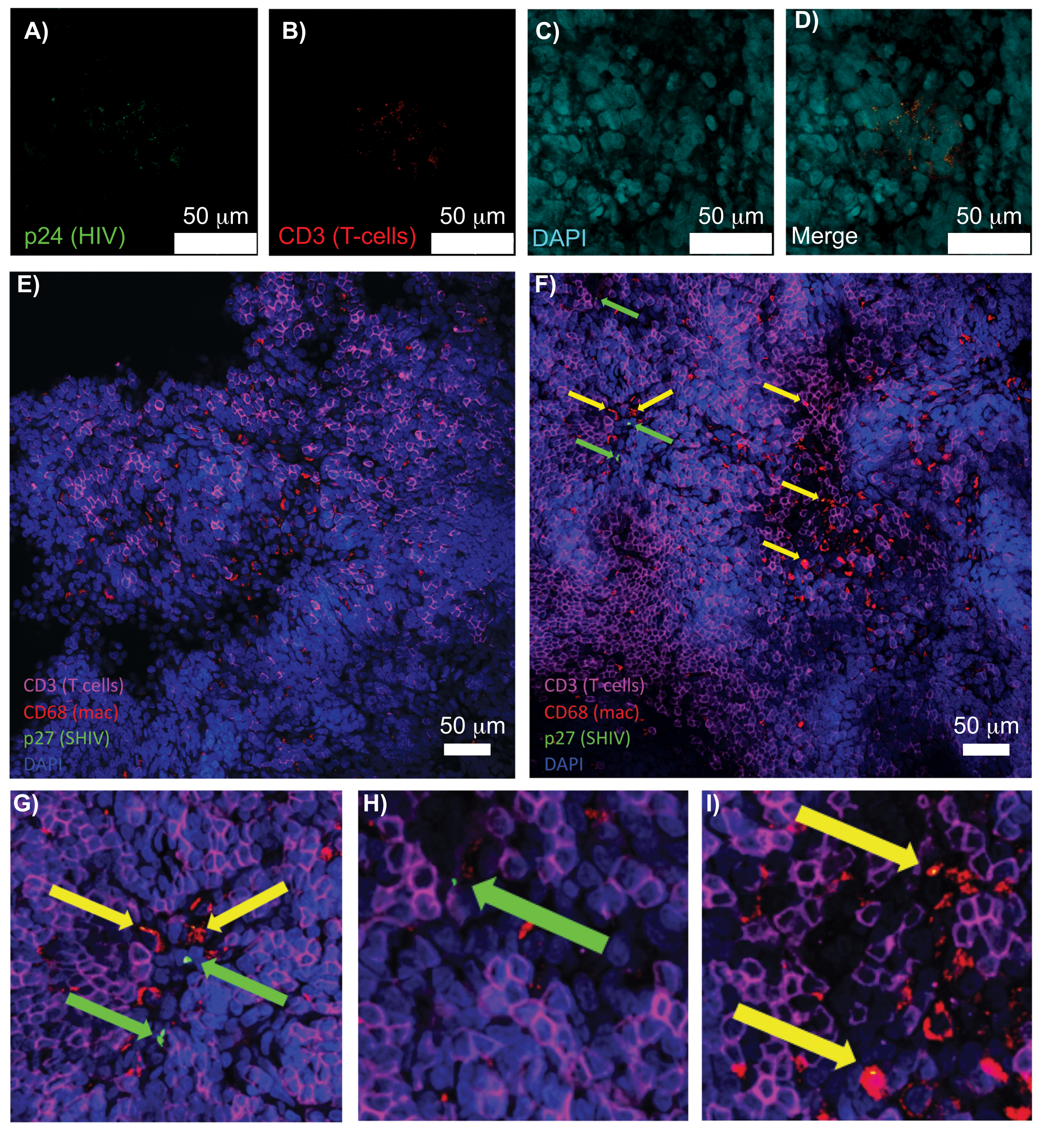
La microscopia confocale di tessuti linfoidi intatti, chiariti e immunocolorati ha permesso la visualizzazione simultanea di più segnali fluorescenti, inclusi nuclei, marcatori di cellule immunitarie e proteine HIV/SIV CA (capside) (Figura 5). Le cellule produttrici di virus sono state determinate mediante colocalizzazione a fluorescenza di marcatori cellulari immunitari e proteine dell'HIV. La milza umana infetta da HIV ripulita e colorata ha rivelato più cellule T CD3+ co-localizzate con HIV p24, indicando la presenza di cellule produttrici di virus all'interno di una regione di tessuto intatto (Figura 5A-D). I linfonodi NHP infetti da SHIV eliminati e immunocolorati hanno rivelato la distribuzione delle cellule T CD3+ e dei macrofagi CD68+ nelle regioni tissutali senza virus rilevato (Figura 5E), oltre alle regioni con numerose cellule produttrici di virus (Figura 5F). Questi risultati hanno mostrato che le cellule produttrici di virus provenienti da diverse fonti di tessuto erano distinguibili da altre cellule all'interno di un determinato campo visivo e consentivano il rilevamento di eventi biologici rari all'interno di un ambiente tissutale complesso.
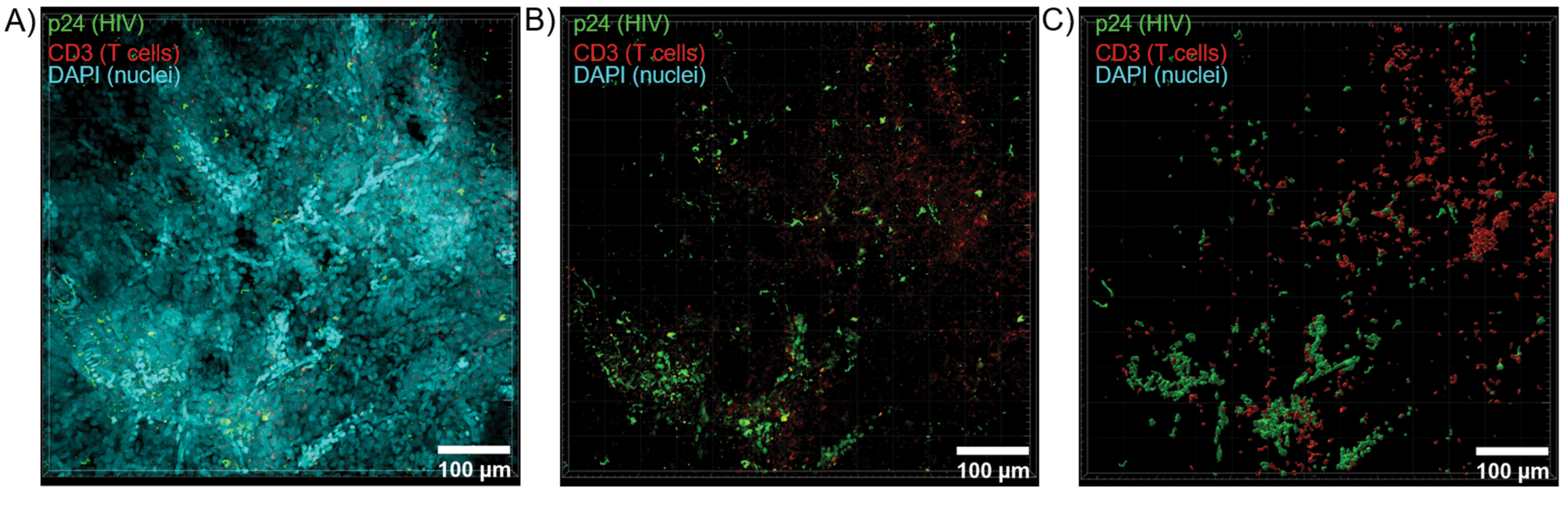
Il sezionamento ottico di tessuti chiariti con un microscopio confocale è stato applicato per generare Z-stack e modelli di superficie 3D, che hanno rivelato l'eterogeneità cellulare mostrata durante l'infezione da HIV (Figura 6). Gli Z-stack sono stati ricombinati in un'immagine di proiezione Z utilizzando la suite software Imaris (Figura 6A) e il canale nucleare DAPI è stato rimosso per una chiara visualizzazione della fluorescenza delle cellule T CD3+ e della proteina del capside dell'HIV (p24) in interi volumi di tessuto (Figura 6B). La fluorescenza a proiezione Z è stata segmentata in modo automatizzato con il software Imaris per generare un modello di superficie 3D ricostruito per la visualizzazione spaziale e la quantificazione del segnale di fluorescenza nell'intero stack Z (Figura 6C). L'analisi del modello 3D di superficie ha rivelato 546 cellule T CD3+ e 218 cellule che producono HIV p24. Cumulativamente, l'acquisizione Z-stack dell'immunofluorescenza da tessuti linfoidi puliti e infetti da HIV ha permesso la generazione di modelli 3D della composizione cellulare all'interno del tessuto e la quantificazione automatizzata delle popolazioni di cellule immunitarie all'interno dei volumi dei tessuti.
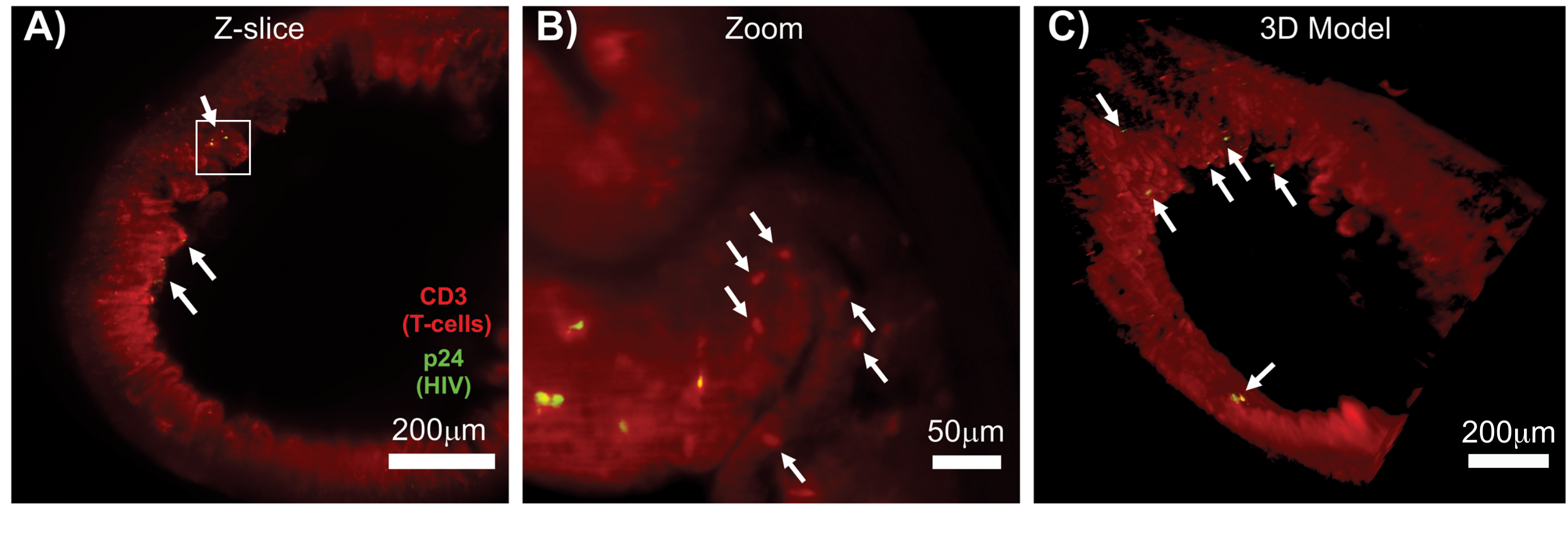
L'LSFM di tessuti linfoidi intatti, chiariti e immunocolorati ha consentito l'imaging in immunofluorescenza (IF) a volume maggiore della distribuzione delle cellule immunitarie e delle cellule produttrici di virus nei tessuti linfoidi (Figura 7). L'immunocolorazione del tessuto del colon di un topo hu infetto da HIV per le cellule T hCD3+ e HIV p24 ha rivelato focolai di cellule produttrici di virus disperse in ampie regioni di tessuto senza evidenza di infezione (Figura 7A). Una vista ingrandita di un focolaio di cellule produttrici di virus ha rivelato più cellule produttrici di virus in prossimità di potenziali cellule bersaglio (Figura 7B). L'autofluorescenza tissutale (foschia rossa) è stata utilizzata per visualizzare l'intera architettura del tessuto, distinguendo specifiche popolazioni di cellule immunitarie all'interno del tessuto che si coloravano più intensamente rispetto all'autofluorescenza (ovali rossi). Un modello 3D dell'intero volume Z-stack di LSFM ha mostrato la distribuzione spaziale dei focolai di cellule produttrici di virus all'interno di una regione di tessuto intatto e ha permesso di mappare le posizioni di produzione del virus rispetto all'architettura tissutale complessiva (Figura 7C). Sorprendentemente, i focolai di cellule produttrici di virus erano spesso sparsi tra grandi regioni di tessuto senza prove di produzione di virus. Questi risultati possono consentire la quantificazione dei parametri di distribuzione del virus e della densità delle cellule infette all'interno di diversi tessuti e in diversi momenti di infezione o risposta a diversi trattamenti.

Figura 1: Flusso di lavoro tipico della pulizia dei tessuti, dell'immunocolorazione e dell'imaging tipici di CUBIC e CLARITY. I tempi di schiaritura CUBIC (in alto) e CLARITY (in basso) possono variare notevolmente a seconda delle dimensioni e del tipo di tessuto. Per la chiarificazione con CLARITY, è necessaria un'ulteriore fase di incubazione con terreno di rifrazione corrispondente all'indice prima dell'immunocolorazione per verificare che il tessuto sia pulito. L'immunocolorazione richiede in genere 3 giorni quando gli anticorpi primari sono coniugati con fluorofori e 6 giorni se sono necessari anticorpi secondari fluorescenti. I campioni possono essere visualizzati con un confocale o LSFM. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Clearing CUBIC di campioni di tessuto hu-mouse e NHP. A seconda delle diverse densità di eme e lipidi dei campioni di tessuto, il tempo necessario per l'eliminazione di ciascun tipo di tessuto varia. Ad esempio, il colon e il duodeno richiedono in genere periodi relativamente brevi (~7 giorni), mentre la milza e il fegato possono impiegare più tempo per diventare trasparenti (~30 giorni). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Confronto longitudinale dei metodi di chiarificazione dei tessuti su campioni umani. CUBIC (pannelli superiori) e CLARITY (pannelli inferiori) hanno eliminato la milza da un individuo infetto da HIV in terapia antiretrovirale. Entrambi i metodi hanno eliminato adeguatamente il tessuto entro il giorno 32 per l'immunocolorazione e l'imaging. La fase di decolorazione per il metodo CUBIC riduce visibilmente l'autofluorescenza causata dalla presenza di eme contenuto nei campioni di milza. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

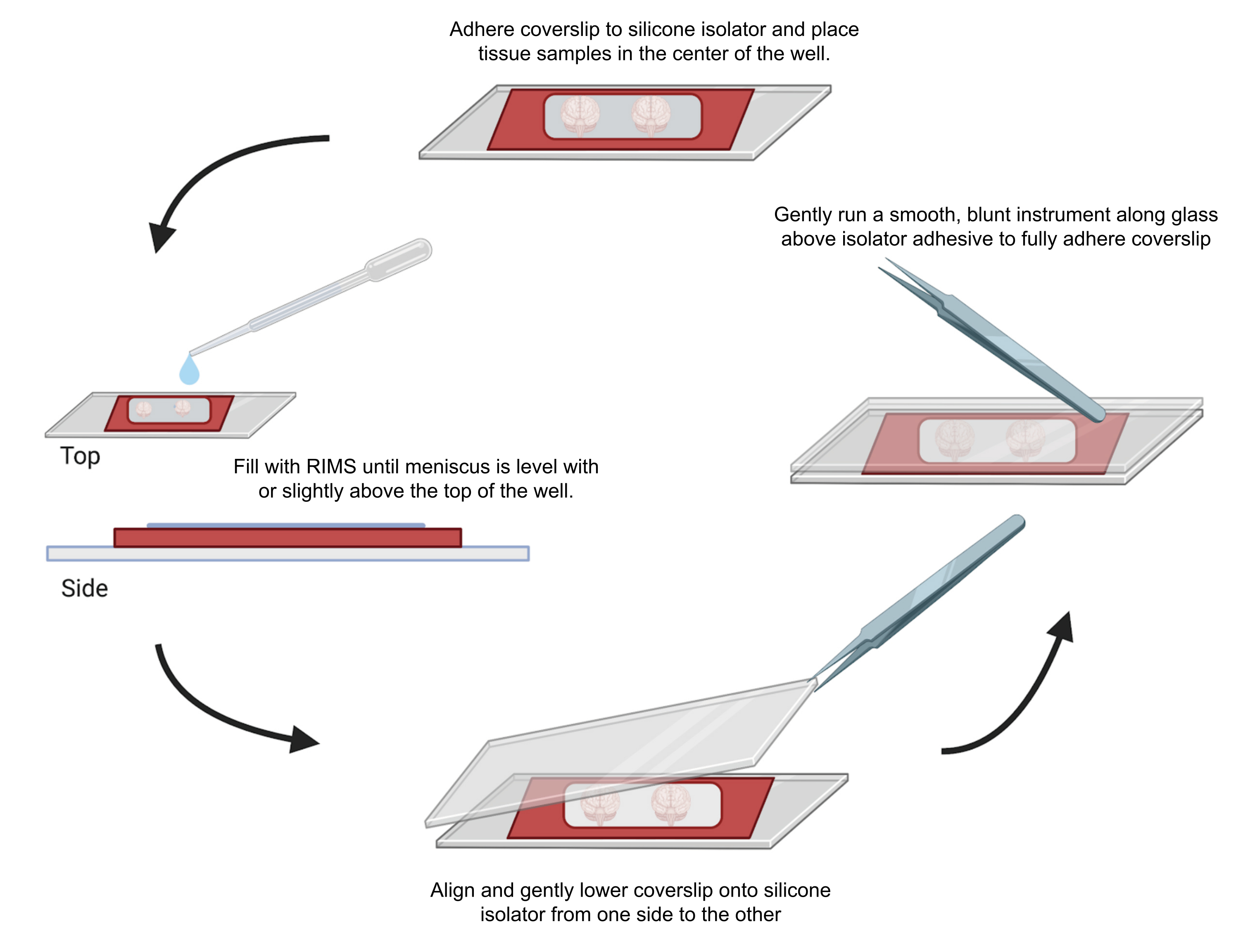
Figura 4: Montaggio del campione per microscopia confocale. I campioni sono stati montati tra vetrini coprioggetti separati con isolatori siliconici adesivi da 0,5-1 mm. Gli isolatori in silicone sono stati fatti aderire al primo vetrino coprioggetti e il tessuto è stato posizionato al centro del pozzetto (in alto). Il pozzo è stato riempito con RIMS fino a quando il menisco non è stato a livello o leggermente al di sopra della parte superiore del pozzo (a sinistra). Il secondo vetrino coprioggetti è stato accuratamente calato in posizione da un lato all'altro, evitando bolle (in basso). I vetrini coprioggetti sono stati completamente aderiti all'isolatore in silicone facendo scorrere delicatamente uno strumento smussato attorno al perimetro del pozzetto (a destra). I campioni sono stati ripresi in un microscopio confocale standard. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Microscopia confocale di milza umana pulita e intatta e linfonodi NHP. (A-D) Il tessuto umano infetto da HIV eliminato è stato colorato per HIV-1 p24 (verde), cellule T hCD3+ (rosso) e nuclei (ciano). (E) Fetta Z confocale di linfonodo CUBIC cancellato da un NHP infetto da SHIV 8 settimane dopo l'infezione immunocolorato per cellule T CD3+ (magenta), macrofagi CD68+ (mac/rosso), SHIV p27 (verde) e nuclei (blu). Il campo visivo contiene cellule T, macrofagi e altri tipi di cellule, ma nessuna evidenza di cellule produttrici di SHIV (verde). F) F) Flice Z confocale di una regione adiacente dello stesso linfonodo che mostra differenze nella densità e nel numero cellulare insieme alla presenza di cellule T CD3+ produttrici di virus (frecce verdi) e macrofagi CD68+ (frecce gialle). (G-I) Vista ingrandita delle regioni selezionate della colorazione p27 da (F). Le barre della scala sono di 50 μm. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Volume Z-stack e superficie ricostruita in 3D da milza umana infetta da HIV. (A) Immagine di proiezione Z da una pila Z di 600 μm x 600 μm x 100 μm di tessuto di milza umana infetta da HIV colorato per HIV-1 p24 (verde), cellule T hCD3+ (rosso) e nuclei (ciano). (B) La stessa immagine a proiezione Z senza colorazione DAPI nucleare. (C) Modello 3D ricostruito della fluorescenza CD3 (rosso) e p24 (verde) dall'intero volume Z-stack. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 7: LSFM e ricostruzione 3D dei volumi superficiali di tessuti infetti da HIV (A) Z-slice (1.000 μm x 1.000 μm) di colon da un topo hu infetto da HIV immunocolorato per le cellule T CD3+ (rosso) e HIV p24 (verde). La foschia rossa opaca rappresenta l'autofluorescenza dei tessuti, mentre i distinti punti rossi indicano le cellule T. I villi sono visibili intorno alla periferia e puntano verso il lume centrale con diversi focolai di produzione attiva di virus (frecce bianche) sparsi in vaste aree che non contengono virus. La casella indica la regione approssimativa di interesse per il pannello B. (B) Regione ingrandita del tessuto che mostra le singole cellule T produttrici di virus hCD3+ (giallo) in prossimità di cellule T non infette (rosso). L'immagine è stata ruotata e cambiata in una fetta Z vicina per mostrare una messa a fuoco di celle positive p24 in un singolo piano Z. L'autofluorescenza rossa di sfondo mostra l'architettura generale del tessuto oltre alla colorazione specifica delle cellule T hCD3+ (punti rossi; frecce bianche). (C) Modello 3D della superficie del volume completo (1.000 μm x 1.000 μm x 200 μm) generato con il software Imaris ruotato per mostrare i focolai di infezione da HIV (giallo) in posizioni distinte dell'intestino. Le frecce bianche indicano i singoli focolai all'interno del volume. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
I tessuti linfoidi di interesse devono essere raccolti rapidamente post-mortem e immediatamente inseriti in tamponi fissativi pre-raffreddati per evitare la necrosi dei tessuti (tessuto scuro o nero) che può influire negativamente sulla colorazione e sull'imaging. Dopo aver raccolto i tessuti desiderati, immergere immediatamente i tessuti in paraformaldeide (PFA) ghiacciata al 4%-8% durante la notte per il fissaggio, che inattiva anche potenziali agenti patogeni associati ai campioni. Il 4% di PFA è ottimale per il fissaggio dei campioni LM, mentre l'8% di PFA può conservare adeguatamente i tessuti sia per LM che per EM. Seguendo queste procedure e conservando i campioni in fissativo a 4 °C al buio, è possibile preservare efficacemente i tessuti per l'imaging LM per diversi anni. Un avvertimento è che la conservazione a lungo termine nel fissativo può portare all'introduzione di artefatti di colorazione, in particolare il mascheramento dell'antigene che è causato dalla reticolazione di proteine adiacenti alla proteina di interesse, che può occludere l'accessibilità degli anticorpi di colorazione all'epitopo13,14. Se i tessuti contengono proteine fluorescenti espresse endogenamente, adottare misure per evitare di esporre i tessuti alla luce quando possibile durante l'intero protocollo. In genere, le proteine fluorescenti endogene mantengono la fluorescenza per 6-12 mesi dopo il fissaggio, ma i singoli campioni di tessuto possono variare per periodi di tempo più o meno lunghi. Se la fluorescenza endogena viene persa a causa della degradazione delle proteine, le proteine fluorescenti possono spesso essere rilevate utilizzando un anticorpo primario specifico per la proteina di interesse. La perfusione è un'altra opzione per fissare rapidamente i tessuti prima di eliminare12; tuttavia, a causa delle preoccupazioni quando si lavora con agenti patogeni come l'HIV, è stata scelta la via della necroscopia tissutale seguita dall'immersione in fissativo ghiacciato per preparare i campioni nel modo più sicuro possibile.
Un vantaggio dei protocolli di pulizia a base d'acqua descritti è che sono generalmente più lievi dei protocolli a base organica, che a volte possono danneggiare i tessuti più fragili, come il fegato. I protocolli di chiarificazione a base d'acqua richiedono generalmente un tempo più lungo per ottenere la pulizia completa del campione (settimane anziché giorni) rispetto ai protocolli di chiarificazione a base organica. I protocolli CLARITY e CUBIC possono essere condotti più rapidamente utilizzando la perfusione per eliminare simultaneamente tutti gli organi all'interno di un roditore11,12; tuttavia, questa non era un'opzione fattibile per NHP e autopsie umane. I campioni processati con CLARITY tendono a mostrare una certa espansione del volume, mentre CUBIC ha rivelato una ridotta influenza sul volume del campione9. Sebbene generalmente più rapidi, molti protocolli di pulizia dei tessuti su base organica causano un restringimentodei tessuti 15, che può rendere più difficile il rilevamento di dettagli di singole cellule o subcellulari all'interno di tessuti densi di cellule come linfonodi e milza. L'espansione indotta dalla pulizia può aumentare efficacemente la risoluzione dell'imaging, rendendo più facile osservare aspetti che sarebbero difficili da osservare nelle dimensioni originali del tessuto. In alternativa, il restringimento dei tessuti può ridurre efficacemente le dimensioni complessive del campione, il che può rendere possibile l'imaging dell'intero organo senza dissezione. Un vantaggio di entrambi i protocolli CLARITY e CUBIC è che preservano la fluorescenza dalle proteine fluorescenti espresse endogenamente nei tessuti, pur rimanendo suscettibili di colorazione in immunofluorescenza11,12. L'immunocolorazione può essere condotta utilizzando metodi acquosi o organici di pulizia dei tessuti; Tuttavia, l'esperienza personale ha mostrato una percentuale più elevata di compatibilità anticorpale utilizzando protocolli a base d'acqua rispetto ai protocolli a base organica. I ricercatori devono considerare quale metodo di pulizia dei tessuti utilizzare in base ai tessuti ripresi e alle questioni biologiche affrontate (ad esempio, imaging dell'intero organo rispetto all'imaging di una regione specifica di interesse). Non esiste una tecnica universale di pulizia dei tessuti che consenta un'analisi robusta chiavi in mano per tutte le domande di imaging di grandi volumi e i metodi disponibili mostrano vantaggi e svantaggi distinti a seconda dell'applicazione biologica.
Quando si esegue la colorazione degli anticorpi, è necessario considerare numerosi aspetti. Poiché i campioni CLARITY sono incorporati in idrogel di acrilammide, tendono a richiedere tempi più lunghi per l'incubazione12. Il tempo necessario per l'incubazione degli anticorpi dipende anche dal volume e dallo spessore di ciascun campione. La maggior parte dei campioni qui descritti aveva uno spessore di ~2-3 millimetri e 3 giorni erano sufficienti per la colorazione completa in tutto il tessuto. Se l'obiettivo è quello di visualizzare un intero cervello di topo, il tempo di incubazione degli anticorpi può richiedere 1 settimana o più12. La scelta di un metodo di pulizia dei tessuti acquosi rispetto a quelli organici per l'imaging in immunofluorescenza può dipendere dalla compatibilità degli anticorpi. In generale, per CUBIC o CLARITY, il tasso di successo per gli anticorpi che funzionano nelle cellule e nei tessuti in coltura è ~70%. Indipendentemente dal fatto che si utilizzi un metodo di chiarificazione dei tessuti acquoso o organico, è necessario valutare la compatibilità e l'efficacia di tutti gli anticorpi con il metodo specifico utilizzato. Come mostrato in questa sezione del protocollo, l'immunocolorazione per i campioni trattati con CUBIC e CLARITY avviene al termine dello sgombero. Al contrario, questa fase avviene prima della procedura di clearing per alcuni protocolli a base organica, seguita dal post-fixing.
È di fondamentale importanza che i tessuti siano completamente immersi in un mezzo di imaging che corrisponda al loro indice di rifrazione. In caso contrario, si introdurranno aberrazioni sferiche durante l'imaging e si distorcerà la luce catturata durante l'acquisizione dell'immagine. È necessario prestare attenzione a rimuovere tutte le bolle d'aria dai supporti di imaging durante il montaggio di campioni sia per confocali che per LSFM, poiché le bolle possono interrompere il percorso della luce verso o lontano dal campione. Le bolle possono essere rimosse manualmente con una pipetta prima del montaggio finale del campione. Per l'imaging di campioni più spessi con un microscopio confocale, è possibile sovrapporre più distanziatori in silicone per ospitare tessuti di spessore superiore a 0,5 mm. Una raccomandazione è quella di equilibrare tutti i tessuti in RIMS per diverse ore o durante la notte mentre sono montati sul microscopio senza ulteriori movimenti del campione. Il pieno equilibrio del tessuto e dei mezzi di imaging impedirà la miscelazione di soluzioni con indici di rifrazione non corrispondenti che possono generare aberrazioni durante l'imaging. È importante ricordare che quando si montano campioni di tessuto chiarito, non esiste un unico metodo di montaggio chiavi in mano per visualizzare tutti i campioni in tutti i microscopi. Questo protocollo illustra le opzioni di montaggio dei campioni che hanno funzionato in modo ottimale in un unico contesto, ma esistono numerosi approcci per il montaggio dei campioni a seconda del singolo microscopio utilizzato e della questione biologica affrontata. Questi approcci possono includere, a titolo esemplificativo ma non esaustivo, l'inclusione del campione nell'agarosio, la sospensione del campione da un gancio o da una linea di plastica abbinata all'indice di rifrazione, l'utilizzo di un adattatore per porcospino, la stampa 3D di un supporto per campioni o il fissaggio del campione con adesivo a un piatto di plastica.
I microscopi confocali possono funzionare bene per l'imaging di volumi di tessuto ~1 mm3-1 cm3. Per i microscopi confocali, utilizzare un obiettivo 2-10x per localizzare inizialmente le regioni di interesse e acquisire stack Z di volume maggiore o di tessuto intero con risoluzione a singola cellula. Passa a obiettivi 20-63x per acquisire immagini ad alta risoluzione di specifiche regioni di interesse con informazioni subcellulari. L'obiettivo ideale per l'imaging di tessuti chiari CUBIC e CLARITY è un obiettivo specifico CLARITY/Scale che viene accuratamente abbinato all'indice di rifrazione del tessuto e della soluzione di imaging. Se questo tipo di obiettivo non è disponibile, è ottimale visualizzare i campioni con un obiettivo a immersione in glicerolo o in olio (ad esempio, obiettivo multi-immersione LD LCI Plan-Apochromat 25 x 0,8 NA Imm Corr DIC M27: distanza di lavoro = 0,57 mm) piuttosto che un obiettivo ad aria. Ciò ridurrà al minimo l'introduzione di distorsioni ottiche dovute a indici di rifrazione non corrispondenti durante l'acquisizione dell'immagine. Gli obiettivi 20-25x possono bilanciare l'acquisizione di immagini di grandi volumi con l'ottenimento di dettagli di colorazione da singole cellule in un ambiente tissutale complesso. È importante sottolineare che la maggior parte dei microscopi confocali contiene moduli che consentono l'affiancamento 3D dei volumi di imaging. Questo tipo di acquisizione di immagini può idealmente generare Z-stack di volume maggiore che contengono informazioni subcellulari.
L'imaging LSFM può consentire la visualizzazione 3D di specifiche popolazioni cellulari nel contesto di grandi volumi di tessuto (>1cm3) e persino di interi organi. Negli ultimi 10 anni, la pulizia dei tessuti combinata con l'LSFM si è concentrata in gran parte sulla comprensione della connettività cerebrale all'interno dei roditori; Tuttavia, le applicazioni più recenti includono la visualizzazione dei paesaggi metastatici tumorali16, la distribuzione cellulare all'interno dei compartimenti anatomici 9,17 e la dispersione del patogeno18. Rispetto alle cellule in coltura, la maggior parte degli eventi biologici nei tessuti non sono uniformi e l'LSFM può essere particolarmente abile per visualizzare e quantificare l'eterogeneità spaziale dei tessuti di questi eventi (ad esempio, replicazione del virus, segnalazione immunitaria, distribuzione cellulare, ecc.).
I set di dati 3D acquisiti tramite confocale o LSFM possono essere post-elaborati con numerose piattaforme di analisi delle immagini. La suite software Imaris può essere utilizzata per la costruzione di superfici, la generazione di animazioni 3D e la quantificazione di celle; Tuttavia, sono disponibili numerosi sistemi di analisi delle immagini che consentono un'efficiente post-elaborazione e analisi delle immagini. ImageJ/Fiji freeware19 è un'interessante piattaforma alternativa di elaborazione delle immagini accessibile alla maggior parte dei laboratori, ma non esiste un software di analisi unico che eccelle in tutte le forme di analisi e visualizzazione delle immagini. Molte suite di software per l'analisi delle immagini possono essere proibitive se non disponibili tramite strutture ad uso condiviso. Infine, un aspetto critico dei set di dati 3D LSFM o confocali a piastrelle di grandi dimensioni è la gestione dei dati. Queste piattaforme di imaging possono generare file di grandi dimensioni (>1 Tb) che richiedono workstation informatiche di fascia alta per la visualizzazione e la quantificazione dei dati. In definitiva, questo flusso di lavoro di imaging può semplificare l'acquisizione e la quantificazione di popolazioni cellulari spazialmente distinte all'interno di interi tessuti ed è ampiamente applicabile alla maggior parte delle fonti tissutali e dei sistemi biologici.
Divulgazioni
Gli autori non hanno conflitti di interesse da divulgare.
Riconoscimenti
Grazie all'Università dell'Illinois presso l'Urbana-Champaign Institute for Genomic Biology Core Facilities per l'uso di microscopi a fluorescenza confocali e a foglio di luce. Grazie alle straordinarie persone della coorte "The Last Gift" per i campioni di tessuto umano, che è stata finanziata dalle seguenti sovvenzioni: I147821, DA051915, AI131385 e P30 AI036214. Grazie a Nancy Haigwood e Ann Hessell per i campioni di tessuto NHP infetti da SHIV.
Materiali
| Name | Company | Catalog Number | Comments |
| Acrylamide Solution (in 0.1 M PBS, 40 mL in total) | |||
| 40% Acrylamide: 4 mL | Bio-Rad | 1610144 | |
| VA-044 Thermal Initiator: 0.1g | Fujifilm | 011-19365 | |
| CLARITY Blocking solution (in 0.1 M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Tween-20: 5 µL | Fisher Scientific | BP337-500 | |
| CLARITY wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Tween-20: 50 µL | Fisher Scientific | BP337-500 | |
| CUBIC Blocking solution (in 0.1M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Triton X-100: 5 µL | VWR | M143-1L | |
| CUBIC Reagent-1 (in 0.1M PBS, 50 mL in total) | |||
| N, N, N’, N’-tetrakis (2-hydroxypropyl) ethylenediamine: 12.5 g | Aldrich | 122262 | |
| Triton X-100: 7.5 g | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC Reagent-2 (in 0.1M PBS, 50 mL in total) | |||
| Sucrose: 25 g | Sigma | S1888-500G | |
| Sodium azide (in powder form): 10 g | Sigma | 71289-50G | |
| Sodium azide stock solution (in DI H2O, 50 mL in total) | Sigma | 71289-50G | |
| Triethanolamine: 5 g | Sigma | 90270-500mL | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| DAPI staining solution (0.5 µg/mL) | |||
| DAPI stock solution: 1 µL | |||
| Wash solution: 10 mL | |||
| DAPI stock solution (5 mg/mL) | |||
| DAPI powder: 5 mg | Sigma-Aldrich | D9542-1MG | |
| DMSO (100%): 1 mL | ThermoFisher | D12345 | |
| Imaging Media RI-2 (in dH2O) | |||
| 90% Histodenz | Sigma | D2158-100G | |
| 0.01% Sodium azide | Sigma | 71289-50G | |
| 0.02 Sodium Phosphate Buffer, pH 7.5 | Sigma-Aldrich | S9638-250G | |
| 0.1% Tween-20 | Fisher Scientific | BP337-500 | |
| Primary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Goat anti-HIV p24: 10 µL (1:200) | Creative Diagnostics | DPATB-H81692 | |
| Mouse anti-human CD68: 10 µL(1:200) | Dako | M0876 | |
| Rabbit anti-human CD3: 10 µL (1:200) | Dako | A0452 | |
| 8% SDS Solution (in 0.1 M PBS, 50 mL in total) | |||
| SDS powder: 4 g | Sigma-Aldrich | L3771-500G | |
| Secondary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Donkey anti-goat conjugated with AlexaFluor647: 2 µL | Invitrogen | A21447 | |
| Donkey anti-mouse conjugated with AlexaFluor594: 2 µL | Invitrogen | A21203 | |
| Donkey anti-rabbit conjugated with AlexaFluor488: 2 µL | Invitrogen | A21206 |
Riferimenti
- Spalteholz, W., Barker, L. F., Mall, F. P. . Hand-Atlas of Human Anatomy. , (1907).
- Jacob, T., Gray, J. W., Troxell, M., Vu, T. Q. Multiplexed imaging reveals heterogeneity of PI3K/MAPK network signaling in breast lesions of known PIK3CA genotype. Breast Cancer Research and Treatment. 159 (3), 575-583 (2016).
- Kieffer, C., Ladinsky, M. S., Ninh, A., Galimidi, R. P., Bjorkman, P. J. Longitudinal imaging of HIV-1 spread in humanized mice with parallel 3d immunofluorescence and electron tomography. eLife. 6, 23282 (2017).
- Chung, K., Deisseroth, K. CLARITY for mapping the nervous system. Nature Methods. 10 (6), 508-513 (2013).
- Compton, A. A., Schwartz, O. They might be giants: Does syncytium formation sink or spread HIV infection. PLoS Pathogens. 13 (2), 2-8 (2017).
- Symeonides, M., et al. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses. 7 (12), 6590-6603 (2015).
- Sharova, N., Swingler, C., Sharkey, M., Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. The EMBO Journal. 24 (13), 2481-2489 (2005).
- Colby, D. J., et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nature Medicine. 24 (7), 923-926 (2018).
- Ladinsky, M. S., et al. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. eLife. 8, 46916 (2019).
- Buettner, M., Bode, U. Lymph node dissection--understanding the immunological function of lymph nodes. Clinical and Experimental Immunology. 169 (3), 205-212 (2012).
- Treweek, J. B., et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nature Protocols. 10, 1860-1896 (2015).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159, 911-924 (2014).
- Sompuram, S. R., Vani, K., Bogen, S. A. A molecular model of antigen retrieval using a peptide array. American Journal of Clinical Pathology. 125 (1), 91-98 (2006).
- Scalia, C. R., et al. Antigen masking during fixation and embedding, dissected. The journal of histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 65 (1), 5-20 (2017).
- Jing, D., et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Research. 28 (8), 803-818 (2018).
- Guldner, I. H., et al. An Integrative platform for three-dimensional quantitative analysis of spatially heterogeneous metastasis landscapes. Scientific Reports. 6, 24201 (2016).
- Muntifering, M., et al. Clearing for deep tissue imaging. Current Protocols in Cytometry. 86 (1), 38 (2018).
- DePas, W. H., et al. Exposing the three-dimensional biogeography and metabolic states of pathogens in cystic fibrosis sputum via hydrogel embedding, clearing, and rRNA labeling. mBio. 7 (5), 00796 (2018).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image. Nature Methods. 9 (7), 676-682 (2012).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati