
Method Article
Visualisation 3D des populations de cellules immunitaires dans les tissus infectés par le VIH par clarification, immunomarquage, microscopie confocale et microscopie à fluorescence à feuillet de lumière
Dans cet article
Résumé
Le nettoyage des tissus, combiné à la microscopie d’immunofluorescence, permet la visualisation spatiale et la quantification des populations de cellules immunitaires et des protéines virales dans les tissus intacts. La coupe optique des tissus clairsemés à l’aide de la microscopie confocale et de la microscopie à fluorescence à feuillet de lumière peut générer des modèles 3D d’environnements tissulaires complexes et révéler l’hétérogénéité spatiale manifestée lors de l’infection par le VIH.
Résumé
Le virus de l’immunodéficience humaine (VIH), l’agent causal du syndrome d’immunodéficience acquise (sida), est un problème de santé mondial majeur avec près de 40 millions de personnes infectées dans le monde et aucun remède largement accessible. Malgré des efforts intensifs, une compréhension détaillée des interactions entre le virus et les cellules hôtes dans les tissus pendant l’infection et en réponse au traitement reste incomplète. Pour remédier à ces limitations, les techniques de nettoyage des tissus à base d’eau CUBIC (Clear, Unobstructed Brain/Body Imaging Cocktails and Computational analysis) et CLARITY (Clear Lipid-exchange Acrylamide-hybridized Rigid Imaging/Immunostaining/in situ-hybridization-compatible Tissue hYdrogel) sont appliquées pour visualiser les interactions complexes entre l’hôte du virus et les cellules dans des tissus infectés par le VIH à partir de modèles animaux et d’humains à l’aide de la microscopie confocale et à fluorescence à feuillet de lumière. Le sectionnement optique de tissus intacts et l’analyse d’images permettent de reconstituer rapidement les informations spatiales contenues dans des tissus entiers et de quantifier les populations de cellules immunitaires au cours de l’infection. Ces méthodes sont applicables à la plupart des sources tissulaires et à diverses questions biologiques, y compris les maladies infectieuses et le cancer.
Introduction
Le besoin croissant d’imagerie spatiale quantitative des tissus dans la recherche biologique a récemment conduit à l’émergence de techniques de clarification tissulaire permettant de générer des images de plus grand volume (mm3-cm 3) de tissus intacts avec une résolution unicellulaire. Les tissus comprennent des organisations complexes de biomolécules avec des structures, des compositions et des fonctions définies de manière unique. Malheureusement, de nombreuses biomolécules présentes dans les tissus (par exemple, les lipides et les chromophores) diffusent, absorbent ou émettent de la lumière lorsqu’elles sont imagées par microscopie optique, ce qui rend difficile l’imagerie à grand volume. De plus, les tissus présentent souvent un indice de réfraction inadapté aux solutions d’imagerie standard et aux lentilles optiques, ce qui entraîne des distorsions optiques pendant l’imagerie. Une approche optimale pour l’imagerie de grands volumes de tissus à l’aide d’un microscope optique doit consister à faire correspondre l’indice de réfraction des tissus, les solutions d’imagerie et les objectifs, tout en permettant à la lumière de pénétrer profondément dans les tissus sans perturber les caractéristiques biologiques des tissus intacts pendant le traitement. À la fin des années 1800, l’anatomiste allemand Werner Spalteholz a tenté de réduire les différences d’indice de réfraction entre les tissus et les solutions d’imagerie en éclaircissant des échantillons de tissusopaques. Cette technique de nettoyage des tissus impliquait des solvants chimiques agressifs, qui peuvent endommager les échantillons de tissus, mais représentait néanmoins la première imagerie de plus grand volume de tissus intacts. Les méthodes modernes de microscopie optique, combinées à la puissance de calcul pour la capture et l’analyse d’images, ont récemment remis la clarification tissulaire à la mode en tant que méthode d’imagerie de grands échantillons de tissus intacts avec une résolution unicellulaire. Au cours des deux dernières décennies, des dizaines de techniques avancées de nettoyage des tissus ont émergé, y compris à base d’alcool et d’eau, chacune ayant des forces et des faiblesses pour des applications spécifiques.
L’imagerie tissulaire 3D permet de sonder des interactions biologiques plus complexes qui ne peuvent pas être reproduites en culture cellulaire. Par exemple, les modèles de signalisation cellulaire2, les distributions spatiales de types cellulaires distincts3 et la connectivité cérébrale4 ont été précédemment cartographiés de manière quantitative à l’aide de méthodes d’imagerie de tissus et d’organes entiers. Il s’agit d’une application de protocoles d’élimination des tissus à base d’eau pour éliminer, immunocolorer et visualiser des populations distinctes de cellules cibles du VIH dans des tissus lymphoïdes infectés par le VIH intacts pendant l’infection active. Dans le corps, le VIH infecte principalement les lymphocytes T CD4+ et intègre une copie de son génome dans les génomes des cellules hôtes infectées. Par la suite, le virus détourne la machinerie de la cellule hôte infectée pour se répliquer, ce qui entraîne la dissémination du virus, la destruction des cellules hôtes, un dysfonctionnement immunitaire et une progression à long terme vers le sida. Il est important de noter que les comportements des lymphocytes T infectés dans les tissus et la culture cellulaire sont notablement divergents. Des lymphocytes T CD4+ cultivés incubés avec le VIH peuvent produire des syncytia massives induites par le VIH qui peuvent inclure des dizaines de noyaux5, tandis que des expériences similaires avec des lymphocytes T CD4+ primaires cultivés dans des hydrogels de matrice extracellulaire (MEC) 3D ou des échantillons de tissus de souris humanisées infectées par le VIH (hu-souris) produisent généralement des syncytia avec 2 à 5 noyaux6. Il est probablement encore plus compliqué de comprendre la transmission locale de cellule à cellule et la dissémination systémique du virus chez les personnes infectées par le VIH, car il implique le transport du virus par plusieurs types de cellules infectées, des tissus aux vaisseaux sanguins et aux nouveaux tissus, où les virions libres et les cellules productrices de virus peuvent accéder à un grand nombre de lymphocytes sensibles7. Ces scénarios ne sont actuellement pas possibles à récapituler dans les systèmes de culture cellulaire, et les tissus provenant de modèles animaux et d’humains restent une ressource importante pour comprendre la pathogenèse du virus dans le contexte d’un organisme complexe doté d’un système immunitaire fonctionnel.
Les traitements antirétroviraux actuels augmentent considérablement l’espérance de vie et la qualité de vie des personnes vivant avec le VIH en inhibant la réplication du VIH et en arrêtant la progression de la maladie vers le sida. Malheureusement, le TAR n’élimine pas les cellules immunitaires infectées de manière latente contenant une insertion du génome rétroviral qui sont quiescentes et ne produisent pas activement le virus. Bien que le virus ne soit pas détectable dans le sang de la plupart des personnes sous TAR, les charges virales rebondissent rapidement après l’interruption du TAR et la progression de la maladie se poursuit8. La nature persistante de l’infection par le VIH causée par le réservoir latent de cellules infectées représente un obstacle majeur à l’établissement d’un remède contre le VIH. Les réservoirs tissulaires du VIH restent mal compris, et il est crucial d’établir une compréhension plus approfondie de ces réservoirs dans les tissus lymphoïdes avant, pendant et après le TAR, afin de caractériser complètement la pathogenèse du virus et d’évaluer de nouveaux traitements qui éliminent efficacement les cellules infectées de manière latente qui ne produisent pas activement le virus.
Ici, CUBIC3 et CLARITY9, deux protocoles de nettoyage des tissus à base d’eau précédemment adaptés, ont été appliqués pour imager des populations de cellules immunitaires dans de nombreux tissus lymphoïdes intacts de souris infectées par le VIH avec un système immunitaire humanisé (souris hu), de primates non humains (NHP) infectés par le SIV/SHIV et d’humains infectés par le VIH. Ces protocoles sont adaptables à la fois à la microscopie confocale et à la microscopie à fluorescence à feuillet de lumière, en fonction des objectifs de l’imagerie (résolution plus élevée ou volume plus important) et de l’instrumentation disponible. Bien que la microscopie optique ne puisse pas résoudre les virions individuels, l’utilisation de l’immunofluorescence permet d’identifier des régions de tissus contenant des virus et des cellules productrices de virus qui peuvent être analysées plus en détail avec des méthodes à plus haute résolution. Les méthodes présentées ici peuvent être adaptées pour visualiser presque n’importe quel tissu du corps avec une résolution unicellulaire afin de quantifier les relations spatiales entre des types de cellules spécifiques dans différentes conditions pendant l’infection et sont facilement transposables à des échantillons de patients humains très pertinents pour l’étude des maladies infectieuses ou du cancer.
Protocole
Toutes les expériences sur les animaux ont été menées conformément aux protocoles de soins aux animaux approuvés par les établissements. Tous les tissus humains ont été acquis conformément aux lignes directrices institutionnelles approuvées en matière d’éthique de la recherche sur les humains.
1. Prélèvement et fixation des tissus (idem pour CUBIC et CLARITY)
- Identifier et disséquer les tissus lymphoïdes comme décrit précédemment10.
- Exciser les tissus lymphoïdes à l’aide de ciseaux de dissection et d’une pince à épiler dans les minutes qui suivent l’autopsie, lorsque cela est possible en toute sécurité.
- Placez les échantillons de tissus dans un tampon fixateur fraîchement préparé et glacé contenant 8 % de paraformaldéhyde (PFA), 5 % de saccharose dans 0,1 M de cacodylate de sodium trihydraté afin de préserver adéquatement les échantillons de tissus pour la microscopie optique (LM), la microscopie électronique (EM) ou l’immuno-EM. Alternativement, fixez les échantillons pour LM avec 4 % de PFA dans 0,1 M PBS. Réparez les échantillons pendant la nuit avant de commencer le processus d’élimination pour assurer la désactivation complète du virus.
ATTENTION : Le paraformaldéhyde est toxique par contact cutané et inhalation et est également un solide inflammable ; Manipulez-le avec précaution et rangez-le dans une armoire de stockage inflammable. Le cacodylate de sodium trihydraté est toxique en cas d’ingestion ou d’inhalation. - Prenez une image de référence du tissu avant de commencer le processus de nettoyage.
REMARQUE : Les échantillons LM peuvent être stockés pendant au moins 1 an dans ces conditions. Pour travailler avec des échantillons exprimant des protéines fluorescentes endogènes, gardez toujours les échantillons dans l’obscurité lors des étapes suivantes.
2. Nettoyage des tissus CUBIC
- Rincer trois fois les échantillons de tissu lymphoïde dans du PBS stérile de 0,1 M en agitant à température ambiante pendant 15 minutes pour s’assurer de l’élimination du PFA à chaque changement de tampon.
REMARQUE : Éliminez les liquides contenant du PFA conformément aux directives institutionnelles. - Immerger l’échantillon de tissu lymphoïde dans le réactif CUBIC-1 (voir tableau des matériaux) à 37 °C pendant 3 jours en agitant doucement. Prenez régulièrement des images de référence pour surveiller le processus de décoloration au fil du temps.
- Remplacez le réactif-1 par 3 à 4 jours supplémentaires d’immersion, ou jusqu’à ce que la décoloration des tissus soit complète. Le temps nécessaire à l’élimination dépend à la fois du volume et du type de tissu. Pour accélérer le processus de décoloration des tissus, rafraîchissez le réactif CUBIC-1 quotidiennement et utilisez de plus grands volumes.
- Laver les échantillons de tissu lymphoïde trois fois avec 0,1 M de PBS pendant 30 min à température ambiante en secouant doucement.
- Immerger les échantillons de tissu lymphoïde dans le réactif CUBIC-2 (voir tableau des matériaux) à 37 °C en agitant doucement pendant 2 à 7 jours ou jusqu’à ce qu’une transparence complète soit obtenue. Si les échantillons n’atteignent pas une transparence totale, répétez les étapes 2.2 à 2.5 jusqu’à ce que l’effacement ne progresse plus. Prenez régulièrement des images de référence pour surveiller le processus de nettoyage au fil du temps.
- Laver les échantillons de tissu lymphoïde trois fois avec 0,1 M de PBS pendant 30 min à température ambiante en secouant doucement.
- Conservez les échantillons dans le réactif CUBIC-2 avec 0,01 % volume/volume (V/V) d’azoture de sodium dans l’obscurité (voir le tableau des matériaux).
REMARQUE : Les échantillons peuvent être conservés pendant au moins 6 mois en utilisant cette méthode.
ATTENTION : L’azoture de sodium est hautement toxique et présente un grave risque d’inhalation. Il est recommandé d’acheter des solutions diluées d’azoture de sodium à 5 % ou moins.
3. Blocage et immunomarquage d’échantillons cubiques
- Laver les échantillons de tissu lymphoïde trois fois avec 0,1 M de PBS pendant 30 minutes chacune à température ambiante en secouant doucement.
- Pour imager à l’aide d’un microscope confocal, coupez le tissu en tranches de ~0,5 à 1 mm d’épaisseur à l’aide d’une matrice de coupe de tissus. Pour effectuer la microscopie à fluorescence à feuillet de lumière (LSFM), bloquez toute la région tissulaire.
- Bloquer les échantillons avec 5 mL de solution de blocage CUBIC pendant une nuit à 4 °C en agitant (voir le tableau des matériaux). Lorsque vous travaillez avec des échantillons de PHN ou d’échantillons humains, utilisez du FcR anti-humain. Lorsque vous travaillez avec des échantillons de souris, utilisez un FcR anti-souris dans la solution de blocage.
- Colorer les échantillons avec 5 mL d’anticorps primaires (voir tableau des matériaux) dans une solution bloquante (sans FcR spécifique à l’espèce) pendant 3 jours à température ambiante en agitant (facultatif : centrifuger le stock d’anticorps concentré à 2 300 x g pendant 5 min avant utilisation, pour réduire l’ajout d’anticorps agrégés).
- Lavez l’échantillon coloré à température ambiante en agitant pendant une période minimale de 5 h au total avec au moins cinq remplacements de tampon de solution de lavage (voir tableau des matériaux).
- Colorer les échantillons avec des anticorps secondaires (voir tableau des matières) dans une solution bloquante (sans FcR spécifique à l’espèce) pendant 3 jours à température ambiante en agitant (facultatif : centrifuger les anticorps à 2 300 x g pendant 5 min avant utilisation pour minimiser l’agrégation d’anticorps).
- Lavez l’échantillon taché cinq fois avec une solution de lavage à température ambiante en agitant pendant au moins 5 h au total.
- Colorer les échantillons avec 5 mL de solution de coloration DAPI (voir le tableau des matériaux) sur chaque échantillon de tissu et incuber pendant 10 min à température ambiante. Laissez les échantillons rester dans la solution de coloration DAPI dans l’obscurité à 4 °C pour une imagerie ultérieure.
- Lavez les échantillons de tissu lymphoïde avec une solution de lavage trois fois à température ambiante en agitant pendant 30 minutes chacun.
- Immergez l’échantillon coloré dans CUBIC Reagent-2 pendant une nuit à température ambiante dans l’obscurité avant de monter l’échantillon.
4. Clarification des tissus CLARITY
- Rincer les échantillons de tissu lymphoïde dans du PBS stérile de 0,1 M trois fois en agitant à température ambiante pendant 15 minutes chacun pour éliminer le PFA.
- Placez les échantillons de tissus dans 15 mL de solution d’acrylamide fraîchement préparée et incubez à 4 °C pendant la nuit en agitant doucement (voir le tableau des matériaux).
ATTENTION : L’acrylamide non polymérisé est une neurotoxine puissante et facilement absorbée par la peau. Évitez tout contact avec la peau et rincez immédiatement en cas de contact. - Laissez les échantillons de tissu se réchauffer à température ambiante.
- FACULTATIF : Dégazer les échantillons de tissus en faisant bouillonner de l’azote dans la solution d’acrylamide pendant 1 min. Veillez à utiliser un faible débit qui évite les éclaboussures d’acrylamide toxique non polymérisé (~1-2 bulles/s).
- Placez les échantillons de tissu dans un bain-marie à 37 °C pendant 1 à 3 h pour polymériser, en inversant toutes les 15 minutes. Retirez les échantillons dès qu’une polymérisation notable est détectée, comme l’indique un liquide visqueux, l’apparition de lignes de Schleren lors du mélange ou la formation d’une capsule transparente autour du tissu.
REMARQUE : Si la polymérisation complète de la solution d’acrylamide se produit, coupez l’excès d’hydrogel de l’échantillon et continuez le protocole. - Lavez les échantillons de tissus avec du PBS stérile de 0,1 M trois fois pendant 30 minutes chacune à température ambiante en secouant doucement pour éliminer la solution d’acrylamide.
- Placez les échantillons de tissu dans 15 mL de FDS à 8 % dans 0,1 M PBS à 37 °C avec un léger balancement pendant 2 à 5+ jours pour permettre l’élimination. Actualisez périodiquement la solution SDS à 8 % et utilisez jusqu’à 50 ml de la solution pour accélérer le nettoyage, si nécessaire. Arrêtez le processus de nettoyage lorsque les échantillons sont visuellement transparents ou ne progressent plus. Prenez régulièrement des images de référence pour surveiller le processus de nettoyage au fil du temps.
- Lavez les échantillons de tissus avec du PBS stérile 0,1 M cinq fois sur 1 jour à température ambiante en secouant doucement.
- Conservez temporairement les échantillons dans 0,1 M de PBS (plus 0,01 % volume/volume (v/v) NaN3 pour un stockage à plus long terme) dans l’obscurité jusqu’à ce qu’ils soient prêts à imager la fluorescence endogène.
- Placez le tissu dans 5 mL de support d’imagerie RI-2 (voir le tableau des matériaux). Incuber toute la nuit à température ambiante dans l’obscurité pour vérifier l’exhaustivité du processus de clarification avant l’immunomarquage. Prenez des images de référence pour surveiller la transparence des tissus.
5. Blocage et immunocoloration des échantillons CLARITY
REMARQUE : Ces étapes sont similaires au blocage et à l’immunocoloration des tissus éliminés par CUNIC, mais utilisent des formulations différentes pour les solutions de blocage, de lavage et de coloration.
- Laver les échantillons de tissu lymphoïde trois fois avec 0,1 M de PBS pendant 30 minutes, à chaque fois à température ambiante en secouant doucement.
- Pour imager à l’aide d’un microscope confocal, coupez le tissu en tranches de ~0,5 à 1 mm d’épaisseur à l’aide d’un trancheur de tissu et d’une matrice de 0,5 mm. Pour effectuer la LSFM, bloquez l’ensemble de l’échantillon de tissu.
- Bloquer les échantillons avec 5 mL de solution de blocage CLARITY (voir le tableau des matériaux) pendant une nuit à 4 °C en agitant.
- Colorer les échantillons avec 5 mL d’anticorps primaires (voir le tableau des matières) dans une solution bloquante (sans FcR spécifique à l’espèce) pendant 3 jours à température ambiante en agitant (facultatif : centrifuger les anticorps à 2 300 x g pendant 5 min avant utilisation pour minimiser l’agrégation des anticorps).
- Lavez l’échantillon taché cinq fois avec la solution de lavage à température ambiante en agitant pendant au moins 5 h au total (voir tableau des matériaux).
- Colorer les échantillons avec 5 mL d’anticorps secondaires (voir le tableau des matériaux) dans une solution bloquante (sans FcR spécifique à l’espèce) pendant 3 jours à température ambiante en agitant (facultatif : centrifuger les anticorps à 2 300 x g pendant 5 min avant utilisation pour minimiser l’agrégation des anticorps). Pour raccourcir la durée totale du protocole, utilisez des anticorps primaires conjugués à des fluorophores afin d’éliminer la nécessité d’une incubation avec des anticorps secondaires.
- Lavez l’échantillon taché cinq fois avec la solution de lavage à température ambiante en agitant pendant au moins 5 h au total.
- Colorer les échantillons avec 5 mL de solution de coloration DAPI (voir le tableau des matériaux) sur chaque échantillon de tissu et incuber pendant 10 min à température ambiante. Laissez les échantillons rester à 4 °C dans l’obscurité dans une solution de coloration DAPI pour une imagerie ultérieure.
- Lavez les échantillons de tissu lymphoïde avec la solution de lavage trois fois à température ambiante en agitant pendant 30 minutes à chaque fois.
- Placez le tissu dans 5 mL de support d’imagerie RI-2 (R.I. = 1,46) et incubez toute la nuit à température ambiante dans l’obscurité avant de monter l’échantillon (voir les étapes 6 et 7 du protocole).
6. Montage et imagerie d’échantillons de tissus clairsemés pour la microscopie confocale
- Décollez un côté de la couche protectrice d’un isolateur adhésif en silicone.
- Collez un verre de protection de microscope (22 mm x 40 mm, 0,25 mm d’épaisseur) sur le côté pelé de l’isolateur en silicone pour former un espace étanche pour l’échantillon.
- Décollez l’autre côté de la couche protectrice de l’isolateur adhésif en silicone.
- Placez l’échantillon pour l’imagerie au centre de l’isolateur en silicone, puis ajoutez le réactif CUNIC-2 ou le support d’imagerie RI-2 selon le cas jusqu’à ce que la surface du liquide soit aussi haute que le bord de l’isolateur.
- Pour minimiser le piégeage des bulles d’air à l’intérieur de l’isolateur en silicone, alignez et superposez doucement le deuxième verre de couverture d’un côté à l’aide d’une pince EM. Essuyez tout excès de liquide. Appuyez doucement sur le verre de protection autour du ou des puits de l’échantillon à l’aide du dos de la pince pour sceller l’adhésif. Stockez les échantillons montés horizontalement dans l’obscurité.
REMARQUE : Les échantillons peuvent être imagés des semaines à des mois après avoir été montés ; Cependant, la qualité de l’image diminue généralement avec le temps. - Placez la lame montée sur la platine du microscope et localisez l’échantillon à l’aide d’une lumière blanche et d’un objectif à faible grossissement (2-10x).
- Configurez le profil d’acquisition de fluorescence en fonction des fluorophores individuels choisis.
REMARQUE : Il est recommandé d’acquérir individuellement des canaux fluorophores séparés. Cela se traduit par un temps d’acquisition plus long mais réduit le chevauchement spectral et l’acquisition de signaux de fluorescence non spécifiques. Un profil fluorophore courant peut inclure DAPI (450 nm), Alexa488, Alexa594 et Alexa647 (ou des combinaisons associées) pour minimiser le chevauchement spectral lors de l’acquisition d’images. - Choisissez un objectif d’agrandissement approprié pour imager les régions d’intérêt. Utilisez des objectifs à faible grossissement (2-10x) pour l’imagerie de plus grands volumes ou de tissus entiers avec une résolution de cellule unique et utilisez des objectifs à grossissement plus élevé (20-63x) pour une visualisation à plus haute résolution des détails subcellulaires dans les tissus clairs. Adaptez autant que possible l’indice de réfraction des objectifs, des supports d’imagerie et des tissus afin de minimiser l’introduction de distorsions optiques lors de l’acquisition de l’image.
- Choisissez une taille de pas pour l’acquisition de la pile Z. Pour les objectifs à faible grossissement (2-10x), sélectionnez une taille de pas de ~3-5 μm pour détecter la fluorescence d’une cellule individuelle dans plusieurs tranches Z continues pour la modélisation 3D tout en réduisant le temps d’acquisition total et la taille globale du fichier. Pour les objectifs à grossissement plus élevé (20-63x), sélectionnez une taille de pas de ~1 μm ou moins pour minimiser la perte d’information subcellulaire entre les tranches Z individuelles.
- Zoomez sur le champ de vision pour visualiser toute la région du tissu à imager dans les dimensions X et Y avec le moins de surface inoccupée possible. Définissez les coordonnées d’acquisition des étages Z supérieur et inférieur qui englobent toute la région d’intérêt à imager.
- Acquérez les images de la pile Z. Enregistrez et exportez le fichier pour le post-traitement à l’aide d’un logiciel d’analyse d’images. Pour certaines suites logicielles, convertissez les fichiers en types de fichiers spécifiques (par exemple, .tiff, .ome-tiff, .jpeg, etc.). Effectuez la conversion à l’aide d’un logiciel d’acquisition d’images microscope ou d’un logiciel gratuit d’analyse d’images (par exemple, ImageJ/Fiji).
7. Montage et imagerie des échantillons dans la chambre ou la cuvette LSFM
- Remplissez la chambre d’imagerie avec CUBIC Reagent-2 ou RI-2 selon le protocole spécifique utilisé. Évitez la formation de bulles lors du transfert du liquide. Retirez les bulles en excès à l’aide d’une pipette.
- Immergez l’échantillon dans la chambre d’imagerie et limitez le mouvement de l’échantillon.
REMARQUE : Selon le microscope spécifique utilisé, cela peut inclure l’intégration de l’échantillon dans de l’agarose, la suspension de l’échantillon à un crochet ou à un adaptateur de porc-épic, l’impression 3D d’un porte-échantillon ou la fixation de l’échantillon avec de l’adhésif à une boîte en plastique. - Placez l’objectif dans la solution d’imagerie, en vous concentrant sur l’échantillon. Laissez l’échantillon monté dans la chambre d’imagerie pendant plusieurs heures ou toute la nuit pour permettre un équilibre complet des solutions et des tissus dans la cuvette.
- Acquérez l’empilement Z de la région d’intérêt (voir les étapes 6.7-6.11 pour l’acquisition d’images).
REMARQUE : Cette approche peut permettre l’imagerie de volumes tissulaires supérieurs à 1cm3 avec une résolution unicellulaire.
8. Reconstruction de surface et quantification cellulaire avec le logiciel d’analyse d’images Imaris
REMARQUE : Ces étapes sont spécifiques au logiciel d’analyse d’images Imaris, mais des étapes de traitement d’images similaires peuvent être effectuées à l’aide d’autres suites logicielles (par exemple, ImageJ/Fiji, Aivia, Arivis, Amira, etc.).
- Utilisez le convertisseur de fichiers Imaris pour convertir le fichier image Z-stack au format natif Imaris .ims. Cela facilitera une conversion plus rapide des fichiers tout en minimisant les erreurs de conversion et les problèmes logiciels potentiels une fois ouverts.
REMARQUE : Certains LSFM plus récents permettent à l’utilisateur d’enregistrer des fichiers directement au format .ims. - Faites glisser le fichier .ims à analyser dans la zone Arena du logiciel Imaris. Ajustez le contraste ou l’intensité de chaque couche de couleur à l’aide du panneau Réglage de l’affichage . Cliquez sur l’icône Ajouter de nouvelles surfaces en haut à gauche.
- Cliquez sur Suivant : Canal source (l’icône bleue avec une flèche pointant vers la droite). Choisissez le canal source de la surface à construire. Ne modifiez pas les autres paramètres.
- Cliquez sur Suivant : Seuil (l’icône bleue avec une flèche pointant vers la droite).
- Pour régler le seuil (intensité absolue), faites glisser la ligne de seuil vers la gauche ou la droite. Activez l’option Diviser les objets en contact et entrez le diamètre moyen de la cellule en microns comme norme de division pour que le système produise de nombreux points comme origine pour chaque surface individuelle.
- N’incluez pas de signaux fluorescents trop petits ou trop brillants, car ils peuvent représenter des artefacts potentiels de coloration ou de microscope. N’incluez que les points qui ont des tailles et des intensités de fluorescence acceptables en modifiant le diamètre moyen des cellules en conséquence.
REMARQUE : Le diamètre moyen des cellules varie pour des tissus ou des types de cellules spécifiques, mais se situe généralement entre 5 et 15 μm.
- N’incluez pas de signaux fluorescents trop petits ou trop brillants, car ils peuvent représenter des artefacts potentiels de coloration ou de microscope. N’incluez que les points qui ont des tailles et des intensités de fluorescence acceptables en modifiant le diamètre moyen des cellules en conséquence.
- Cliquez sur Suivant : Classifier les surfaces (l’icône bleue avec une flèche pointant vers la droite étiquetée). Ajustez les surfaces à inclure en faisant glisser la ligne de seuil vers la gauche ou la droite. Assurez-vous que les surfaces se rapprochent exactement du signal de fluorescence brut, tout en séparant le signal de fluorescence des cellules individuelles.
- Cliquez sur Terminer : Exécuter toutes les étapes de création et terminer l’assistant (l’icône verte avec deux flèches pointant vers la droite étiquetées). La surface est officiellement construite.
- Cliquez sur la sixième icône intitulée Statistiques dans le panneau de gauche pour voir le nombre de cellules, dans ce cas, le nombre de surfaces pour le canal de couleur spécifique analysé.
- Assurez-vous que les quatre variables Nombre de composants déconnectés par point temporel, Nombre de surfaces par point temporel, Nombre total de composants déconnectés et Nombre total de surfaces ont le même numéro, qui est le nombre de cellules de cette couche de couleur.
Résultats
Le nettoyage des tissus consiste à traiter les tissus préservés avec des cocktails chimiques pour extraire des biomolécules opaques du tissu tout en maintenant l’architecture tissulaire. Ces solutions de nettoyage des tissus font correspondre l’indice de réfraction du tissu avec le support d’imagerie environnant pour minimiser les distorsions optiques, améliorer le rapport signal/bruit en profondeur dans les tissus et minimiser l’autofluorescence de fond. Deux protocoles à base d’eau pour l’élimination optique des tissus, CUBIC3 et CLARITY9, ont été utilisés pour éliminer les échantillons préservés de souris infectées par le VIH/VIS, de primates non humains et de tissus humains avant la coloration par immunofluorescence et l’imagerie par microscopie confocale et microscopie à fluorescence à feuillet de lumière.
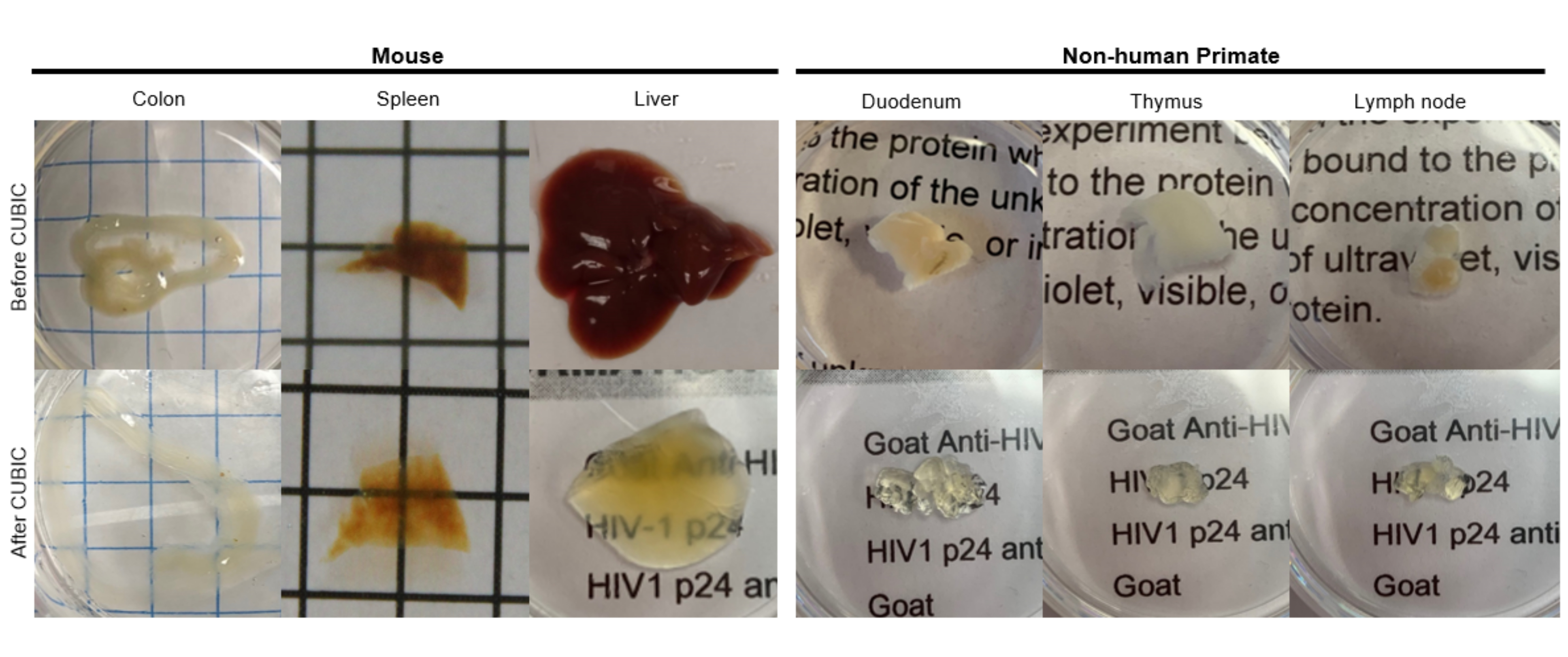
Pour le protocole CUNIC, les tissus fixés ont été lavés avec du PBS pour éliminer les fixateurs et immergés dans le réactif CUBIC-1, une solution tamponnée basique d’aminoalcools qui élue les chromophores tels que l’hème, entraînant une décoloration et une délipidation des tissus (Figure 1, en haut). Les petits volumes de tissus (~ mm3) peuvent être décolorés après 3 jours de traitement avec CUBIC Reagent-1, mais les volumes de tissus plus importants (~ cm3) ou les tissus contenant une grande quantité d’hème (comme le foie, la rate ou le cœur) nécessitent des temps d’incubation et des volumes de solution plus longs (>1 mois et ~50 ml), ainsi qu’un échange fréquent de la solution tous les 2-3 jours. Après la décoloration, les tissus ont été lavés et placés dans CUBIC Reagent-2, une solution contenant du saccharose avec un indice de réfraction d’environ 1,48-1,49, qui correspond à l’indice de réfraction du tissu et augmente la transmission de la lumière. Les tissus éliminés ont été immunomarqués et montés dans une solution de réactif CUBIC-2 avant l’imagerie avec un microscope confocal ou à feuillet de lumière. Les effets de la procédure de clarification CUBIC ont été imagés pour plusieurs tissus de souris et de PNH de différentes tailles et concentrations de chromophores (Figure 2). La clairance optique a rendu les tissus visiblement transparents à l’œil nu, ce qui a permis de voir les lignes de la grille et le texte sur des feuilles de papier « à travers » le tissu. Les tissus riches en chromophores tels que la rate, le foie, la moelle osseuse et le cœur peuvent ne pas se décolorer complètement, mais restent adaptés à l’immunocoloration et à l’imagerie (Figure 2 et Figure 5).
Dans le cadre du protocole CLARITY, les tissus fixés ont été lavés avec du PBS pour éliminer les fixateurs, puis incubés pendant la nuit à 4 °C dans une solution d’acrylamide à 40 % avec un initiateur thermique pour former des liaisons covalentes entre les protéines de l’échantillon et les monomères d’acrylamide (figure 1, en bas). Le lendemain, après que le tissu a été équilibré à la température ambiante, puis réchauffé dans un bain d’eau à 37 °C, la polymérisation de l’acrylamide a été initiée et rapidement enfermée l’échantillon dans un hydrogel. L’échantillon a été traité avec une solution de SDS à 8 % pendant 2 à 5 jours pour éliminer les lipides opaques. Immédiatement avant la coloration fluorescente, l’échantillon a été immergé dans une solution d’appariement de l’indice de réfraction (RIMS) pour CLARITY (Imaging Media RI-2) contenant 90 % de milieu à gradient de densité non ionique. Pour les tissus contenant de grandes quantités d’hème, une étape de décoloration peut être ajoutée à la fin de l’étapede délipidation 9,11,12. La progression de la clairance CUBIC et CLARITY a été comparée sur différentes sections du même échantillon de rate humaine (Figure 3). Le nettoyage CLARITY produit un gel de polyacrylamide visible qui enveloppe la solution et présente généralement une décoloration réduite par rapport au nettoyage CUNIC, à moins qu’une étape de décoloration supplémentaire ne soit ajoutée 9,12.
Par la suite, dans les deux protocoles, des tissus intacts et nettoyés ont été immunomarqués pour détecter des populations spécifiques de cellules immunitaires. Les échantillons ont été lavés, bloqués avec un réactif contenant du α-FcR pour réduire la liaison d’anticorps non spécifiques, et colorés pendant 3 jours à l’aide d’un anticorps primaire directement conjugué à un fluorophore. Alternativement, les échantillons ont été colorés pendant 3 jours avec un anticorps primaire non conjugué, suivis de 3 jours supplémentaires avec un anticorps secondaire conjugué à un fluorophore. Les tissus ont été lavés à nouveau, puis incubés avec un colorant DAPI pendant la nuit à 4 °C pour une visualisation nucléaire. Les échantillons ont été lavés et incubés dans le réactif CUBIC-2 pendant 24 à 36 heures ou dans le milieu d’imagerie RI-2 (CLARITY) pendant la nuit dans l’obscurité. Pour la microscopie confocale, les tissus ont été montés sur une lame de microscope dans le RIMS approprié avant l’imagerie (figure 4). Pour la microscopie à fluorescence à feuillet de lumière (LSFM), les échantillons ont été complètement immergés avec du RIMS dans une cuvette d’imagerie pendant la nuit avant l’imagerie.
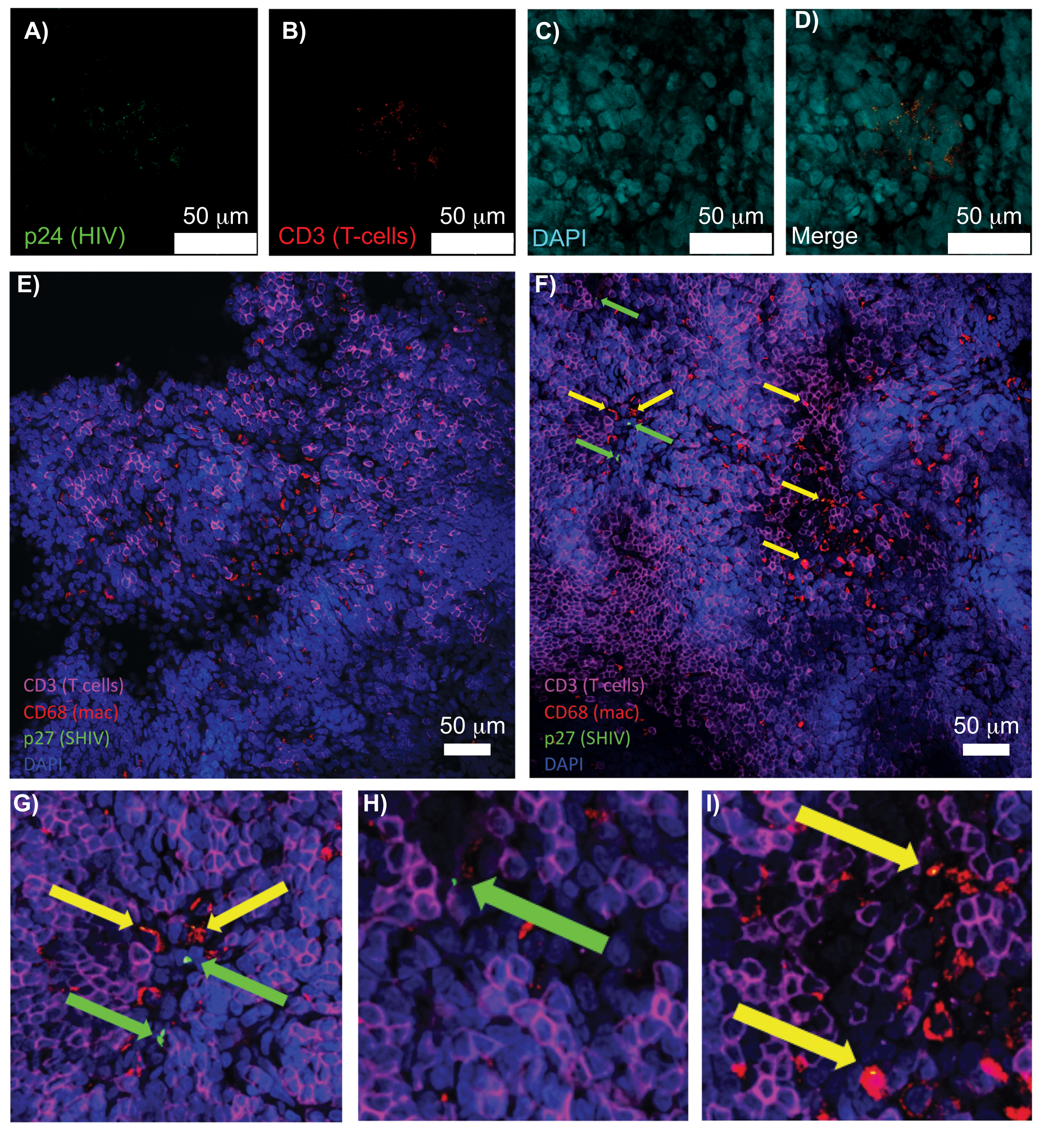
La microscopie confocale de tissus lymphoïdes intacts, clairsemés et immunomarqués a permis de visualiser simultanément plusieurs signaux fluorescents, y compris des noyaux, des marqueurs de cellules immunitaires et des protéines HIV/SIV CA (capside) (Figure 5). Les cellules productrices de virus ont été déterminées par colocalisation en fluorescence des marqueurs cellulaires immunitaires et des protéines du VIH. La rate humaine infectée par le VIH éliminée et colorée a révélé la présence de plusieurs lymphocytes T CD3+ co-localisés avec le HIV p24, indiquant la présence de cellules productrices de virus dans une région de tissu intact (Figure 5A-D). Des ganglions lymphatiques de PNH infectés par le virus SHIV ont révélé la distribution des lymphocytes T CD3+ et des macrophages CD68+ dans les régions tissulaires où aucun virus n’a été détecté (figure 5E) en plus des régions où il y a de nombreuses cellules productrices de virus (figure 5F). Ces résultats ont montré que les cellules productrices de virus provenant de diverses sources tissulaires se distinguaient des autres cellules dans un champ de vision donné et permettaient de détecter des événements biologiques rares dans un environnement tissulaire complexe.
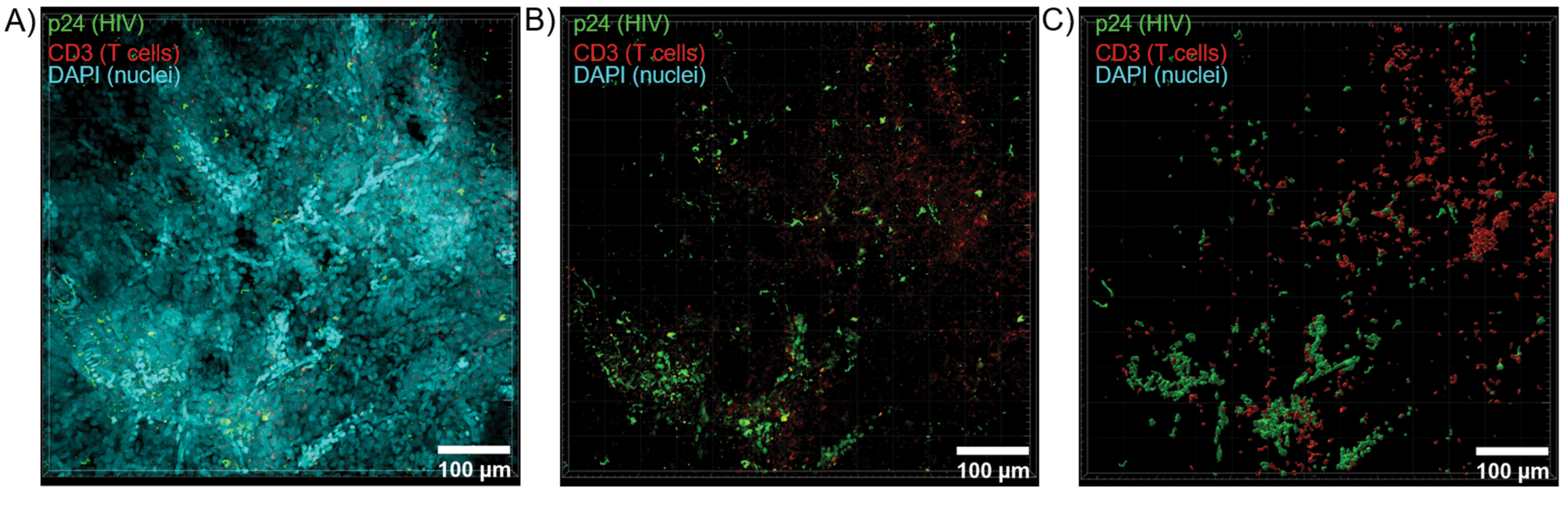
La section optique des tissus clairsemés à l’aide d’un microscope confocal a été appliquée pour générer des empilements Z et des modèles de surface 3D, qui ont révélé l’hétérogénéité cellulaire manifestée pendant l’infection par le VIH (figure 6). Les piles Z ont été recombinées en une image de projection Z à l’aide de la suite logicielle Imaris (Figure 6A) et le canal nucléaire DAPI a été supprimé pour une visualisation claire de la fluorescence des lymphocytes T CD3+ et de la protéine de capside du VIH (p24) dans des volumes entiers de tissu (Figure 6B). La fluorescence de projection Z a été segmentée automatiquement avec le logiciel Imaris pour générer un modèle de surface 3D reconstruit pour la visualisation spatiale et la quantification du signal de fluorescence dans l’ensemble de la pile Z (Figure 6C). L’analyse du modèle de surface 3D a révélé la présence de 546 lymphocytes T CD3+ et de 218 cellules produisant le HIV p24. Cumulativement, l’acquisition de l’immunofluorescence par pile Z à partir de tissus lymphoïdes infectés par le VIH a permis de générer des modèles 3D de la composition cellulaire dans le tissu et de quantifier automatiquement les populations de cellules immunitaires dans les volumes tissulaires.
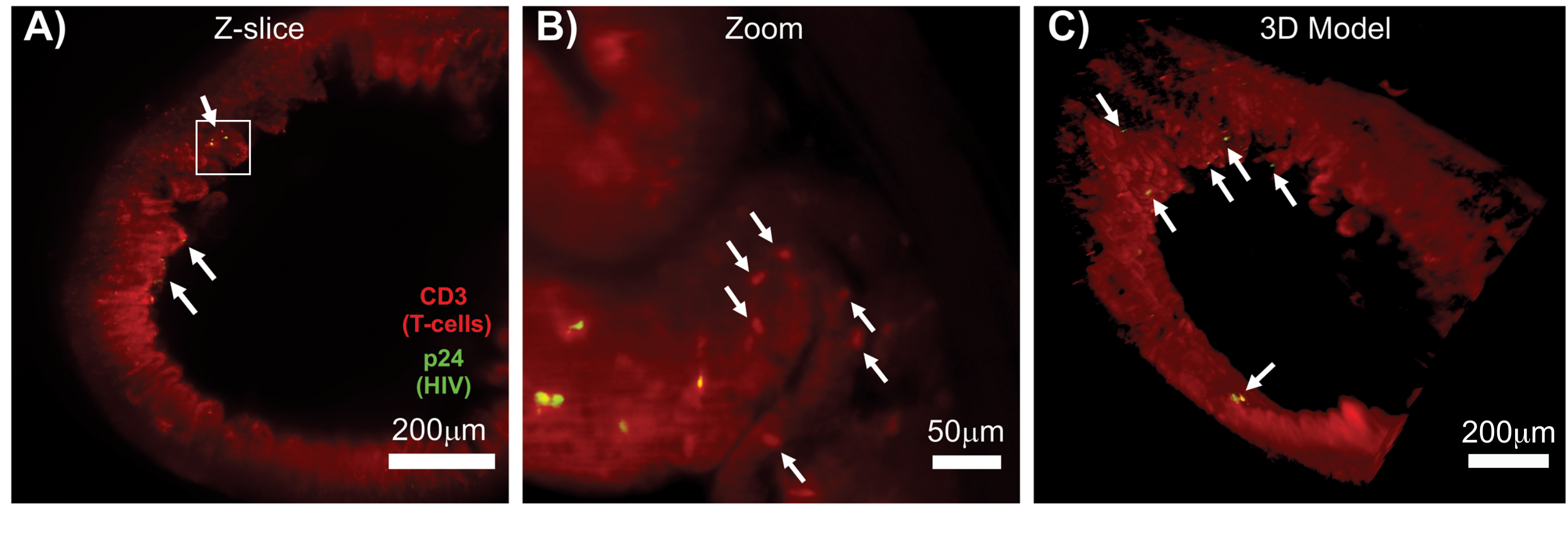
La LSFM des tissus lymphoïdes intacts, clairsemés et immunomarqués a permis l’imagerie par immunofluorescence (IF) de plus grand volume de la distribution des cellules immunitaires et des cellules productrices de virus dans les tissus lymphoïdes (Figure 7). L’immunocoloration du tissu du côlon d’une souris hu-infection par le VIH pour les lymphocytes T hCD3+ et le HIV p24 a révélé des foyers de cellules productrices de virus dispersées dans de grandes régions de tissu sans signe d’infection (figure 7A). Une vue zoomée d’un foyer de cellules productrices de virus a révélé que plusieurs cellules productrices de virus se trouvaient à proximité de cellules cibles potentielles (Figure 7B). L’autofluorescence tissulaire (brume rouge) a été utilisée pour visualiser l’ensemble de l’architecture tissulaire tout en distinguant des populations spécifiques de cellules immunitaires dans le tissu qui se coloraient plus vivement que l’autofluorescence (ovales rouges). Un modèle 3D de l’ensemble du volume de l’empilement Z du LSFM a montré la distribution spatiale des foyers de cellules productrices de virus dans une région de tissu intact et a permis de cartographier les emplacements de production de virus par rapport à l’architecture tissulaire globale (Figure 7C). Étonnamment, les foyers de cellules productrices de virus étaient souvent intercalés dans de grandes régions de tissus sans aucune preuve de production de virus. Ces résultats peuvent permettre de quantifier les paramètres de distribution du virus et de densité cellulaire infectée au sein de différents tissus et à différents moments de l’infection ou de la réponse à différents traitements.

Figure 1 : Flux de travail typique de l’éclaircissement, de l’immunocoloration et de l’imagerie tissulaires CUBIC et CLARITY. Les temps de nettoyage CUBIC (en haut) et CLARITY (en bas) peuvent varier considérablement en fonction de la taille et du type de tissu. Pour l’élimination CLARITY, une étape d’incubation supplémentaire avec un milieu à indice de réfraction apparié est nécessaire avant l’immunocoloration pour vérifier que le tissu est clair. L’immunocoloration prend généralement 3 jours lorsque les anticorps primaires sont conjugués à des fluorophores et 6 jours si des anticorps secondaires fluorescents sont nécessaires. Les échantillons peuvent être imagés avec un confocal ou un LSFM. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Clarification CUBIC d’échantillons de tissus de souris hu et de NHP. En fonction des différentes densités d’hème et de lipides des échantillons de tissus, le temps nécessaire à l’élimination de chaque type de tissu varie. Par exemple, le côlon et le duodénum nécessitent généralement des périodes relativement courtes (~7 jours), tandis que la rate et le foie peuvent prendre plus de temps pour devenir transparents (~30 jours). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Comparaison longitudinale des méthodes d’élimination des tissus sur des échantillons humains. CUBIC (panneaux du haut) et CLARITY (panneaux du bas) ont éliminé la rate d’une personne infectée par le VIH sous traitement antirétroviral. Les deux méthodes ont permis d’éliminer adéquatement les tissus au 32e jour pour l’immunocoloration et l’imagerie. L’étape de décoloration de la méthode CUBIC réduit visiblement l’autofluorescence causée par la présence d’hème contenu dans les échantillons de rate. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

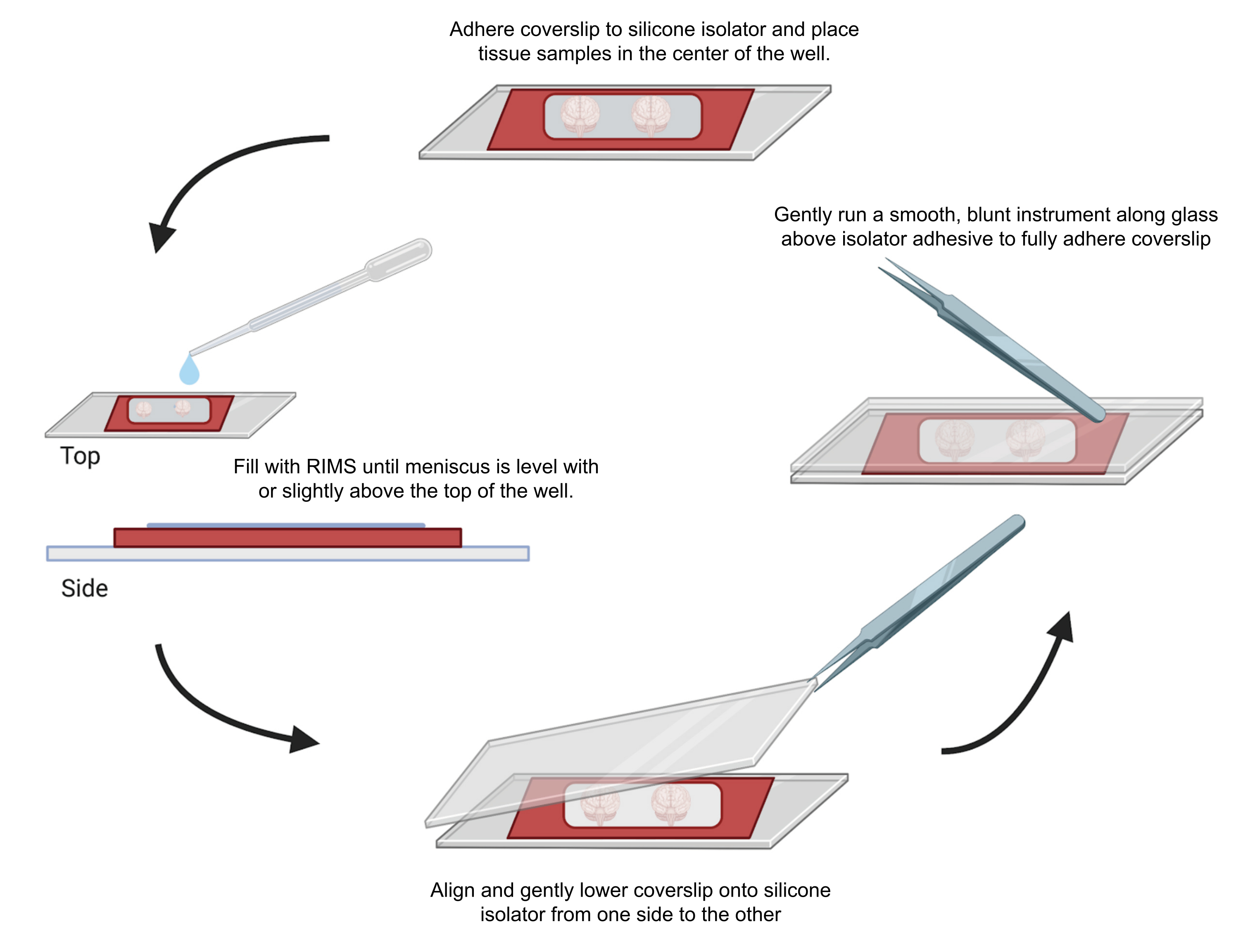
Figure 4 : Montage d’échantillons pour la microscopie confocale. Les échantillons ont été montés entre des lamelles séparées par des isolateurs adhésifs en silicone de 0,5 à 1 mm. Des isolateurs en silicone ont été collés sur la première lamelle et du tissu a été placé au centre du puits (en haut). Le puits a été rempli de RIMS jusqu’à ce que le ménisque soit au niveau ou légèrement au-dessus du sommet du puits (à gauche). La deuxième lamelle a été soigneusement abaissée en place d’un côté à l’autre, en évitant les bulles (en bas). Les lamelles ont été entièrement collées à l’isolateur en silicone en faisant passer doucement un instrument contondant autour du périmètre du puits (à droite). Les échantillons ont été imagés à l’aide d’un microscope confocal standard. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5 : Microscopie confocale de la rate humaine et des ganglions lymphatiques du PNH clairs et intacts. (A-D) Les tissus humains infectés par le VIH éliminés ont été colorés pour les cellules T VIH-1 p24 (vert), les lymphocytes T hCD3+ (rouge) et les noyaux (cyan). (E) Tranche Z confocale de CUBIC ayant éliminé le ganglion lymphatique d’un NHP infecté par le SHIV 8 semaines après l’infection, immunomarquée pour les lymphocytes T CD3+ (magenta), les macrophages CD68+ (mac/rouge), SHIV p27 (vert) et les noyaux (bleu). Le champ de vision contient des lymphocytes T, des macrophages et d’autres types de cellules, mais aucune preuve de cellules productrices de SHIV (vert). F) Tranche Z confocale d’une région adjacente du même ganglion lymphatique montrant des différences de densité et de nombre de cellules ainsi que la présence de lymphocytes T CD3+ producteurs de virus (flèches vertes) et de macrophages CD68+ (flèches jaunes). (G-I) Vue agrandie des régions sélectionnées de la coloration p27 de (F). Les barres d’échelle mesurent 50 μm. Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 6 : Volume de la pile Z et surface reconstruite en 3D à partir de rate humaine infectée par le VIH. (A) Image de projection Z de 600 μm x 600 μm x 100 μm de tissu de rate humaine infecté par le VIH coloré pour les lymphocytes T hCD-1 p24 (vert), les lymphocytes T hCD3+ (rouge) et les noyaux (cyan). (B) La même image de projection Z sans coloration DAPI nucléaire. (C) Modèle de surface 3D reconstruit de la fluorescence CD3 (rouge) et p24 (vert) à partir de l’ensemble du volume Z-stack. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 7 : LSFM et reconstruction 3D des volumes de surface des tissus infectés par le VIH (A) Tranche Z (1 000 μm x 1 000 μm) du côlon d’une souris hu-souris infectée par le VIH immunomarquée pour les lymphocytes T CD3+ (rouge) et le VIH p24 (vert). La brume rouge terne représente l’autofluorescence des tissus, tandis que les ponctuations rouges distinctes indiquent les lymphocytes T. Des villosités sont visibles autour de la périphérie pointant vers la lumière centrale avec plusieurs foyers de production active de virus (flèches blanches) dispersés dans de grandes zones ne contenant aucun virus. La case indique la région d’intérêt approximative pour le panneau B. (B) Région zoomée du tissu montrant des lymphocytes T hCD3+ producteurs de virus individuels (jaune) à proximité de lymphocytes T non infectés (rouge). L’image a été tournée et remplacée par une tranche Z voisine pour montrer un foyer de cellules positives p24 dans un seul plan Z. L’autofluorescence rouge d’arrière-plan montre l’architecture générale des tissus en plus d’une coloration spécifique des lymphocytes T hCD3+ (puncta rouge ; flèches blanches). (C) Modèle de surface 3D du volume complet (1 000 μm x 1 000 μm x 200 μm) généré avec le logiciel Imaris tourné pour montrer les foyers d’infection par le VIH (jaune) à des endroits distincts de l’intestin. Les flèches blanches indiquent les foyers individuels dans le volume. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Discussion
Les tissus lymphoïdes d’intérêt doivent être prélevés rapidement post-mortem et immédiatement placés dans des tampons fixateurs pré-réfrigérés pour éviter la nécrose tissulaire (tissu sombre ou noir) qui peut avoir un impact négatif sur la coloration et l’imagerie. Après avoir récolté les tissus souhaités, immergez immédiatement les tissus dans du paraformaldéhyde (PFA) glacé à 4 % à 8 % pendant la nuit pour la fixation, ce qui inactive également les agents pathogènes potentiels associés aux échantillons. 4 % de PFA est optimal pour la fixation des échantillons de LM, tandis que 8 % de PFA peut préserver de manière adéquate les tissus pour la LM et l’EM. Le respect de ces procédures et le stockage des échantillons dans un fixateur à 4 °C dans l’obscurité peuvent préserver efficacement les tissus pour l’imagerie LM pendant plusieurs années. Une mise en garde est que le stockage à long terme dans un fixateur peut conduire à l’introduction d’artefacts de coloration, en particulier le masquage de l’antigène qui est causé par la réticulation des protéines adjacentes à la protéine d’intérêt, ce qui peut masquer l’accessibilité des anticorps de coloration à l’épitope13,14. Si les tissus contiennent des protéines fluorescentes exprimées de manière endogène, prenez des mesures pour éviter d’exposer les tissus à la lumière dans la mesure du possible pendant toute la durée du protocole. En règle générale, les protéines fluorescentes endogènes conservent leur fluorescence pendant 6 à 12 mois après la fixation, mais les échantillons de tissus individuels peuvent varier pendant des périodes plus ou moins longues. Si la fluorescence endogène est perdue en raison de la dégradation des protéines, les protéines fluorescentes peuvent souvent être détectées à l’aide d’un anticorps primaire spécifique de la protéine d’intérêt. La perfusion est une autre option pour fixer rapidement les tissus avant de disparaître12 ; cependant, en raison de préoccupations liées au travail avec des agents pathogènes tels que le VIH, la voie de la nécropsie tissulaire suivie d’une immersion dans un fixateur glacé a été choisie pour préparer les échantillons de la manière la plus sûre possible.
L’un des avantages des protocoles de nettoyage à base d’eau décrits est qu’ils sont généralement plus doux que les protocoles à base organique, ce qui peut parfois endommager des tissus plus fragiles, comme le foie. Les protocoles de nettoyage à base d’eau nécessitent généralement plus de temps pour obtenir un nettoyage complet des échantillons (semaines contre jours) par rapport aux protocoles de nettoyage à base organique. Les protocoles CLARITY et CUBIC peuvent être menés plus rapidement en utilisant la perfusion pour éliminer simultanément tous les organes d’un rongeur11,12 ; cependant, cette option n’était pas réalisable pour les PSN et les autopsies humaines. Les échantillons traités avec CLARITY ont tendance à montrer une certaine expansion de volume, tandis que CUBIC a révélé une influence réduite sur le volumed’échantillon 9. Bien que généralement plus rapides, de nombreux protocoles d’élimination des tissus organiques provoquent un rétrécissement des tissus15, ce qui peut rendre la détection de détails unicellulaires ou subcellulaires plus difficile à observer dans les tissus denses en cellules tels que les ganglions lymphatiques et la rate. L’expansion induite par la clairance peut augmenter efficacement la résolution de l’imagerie, ce qui facilite l’observation d’aspects qui seraient difficiles à observer dans la taille d’origine du tissu. Alternativement, le rétrécissement tissulaire peut réduire efficacement la taille globale de l’échantillon, ce qui peut rendre possible l’imagerie d’un organe entier sans dissection. L’un des avantages des protocoles CLARITY et CUBIC est qu’ils préservent la fluorescence des protéines fluorescentes exprimées de manière endogène dans les tissus tout en restant sensibles à la coloration par immunofluorescence11,12. L’immunocoloration peut être réalisée à l’aide de méthodes de clarification des tissus aqueux ou organiques ; Cependant, l’expérience personnelle a montré une proportion plus élevée de compatibilité d’anticorps en utilisant des protocoles à base d’eau par rapport aux protocoles à base organique. Les chercheurs doivent déterminer la méthode d’élimination des tissus à utiliser en fonction des tissus imagés et des questions biologiques abordées (p. ex., imagerie d’un organe entier par rapport à l’imagerie d’une région d’intérêt spécifique). Il n’existe pas de technique universelle de clarification tissulaire permettant une analyse clé en main robuste pour toutes les questions d’imagerie de grand volume, et les méthodes disponibles présentent des avantages et des inconvénients distincts en fonction de l’application biologique.
Lors de la coloration des anticorps, de nombreux aspects doivent être pris en compte. Étant donné que les échantillons CLARITY sont intégrés dans de l’hydrogel d’acrylamide, ils ont tendance à nécessiter des temps d’incubation12 plus longs. Le temps nécessaire à l’incubation des anticorps dépend également du volume et de l’épaisseur de chaque échantillon. La plupart des échantillons décrits ici avaient une épaisseur de ~2 à 3 millimètres, et 3 jours ont suffi pour une coloration complète dans tout le tissu. Si l’objectif est d’imager le cerveau d’une souris entière, le temps d’incubation des anticorps peut prendre 1 semaine ou plus12. Le choix d’une méthode de nettoyage des tissus aqueux ou organiques pour l’imagerie par immunofluorescence peut dépendre de la compatibilité des anticorps. En général, pour CUBIC ou CLARITY, le taux de réussite des anticorps qui agissent dans les cellules et les tissus cultivés est de ~70 %. Qu’il s’agisse d’une méthode d’élimination aqueuse ou organique des tissus, il est nécessaire d’évaluer la compatibilité et l’efficacité de tous les anticorps avec la méthode spécifique utilisée. Comme le montre la section de ce protocole, l’immunocoloration des échantillons traités par CUBIC et CLARITY a lieu une fois le nettoyage terminé. Au contraire, cette étape a lieu avant la procédure de déblaiement pour certains protocoles organiques, suivie d’un post-fixage.
Il est extrêmement important que les tissus soient complètement immergés dans un support d’imagerie qui correspond à leur indice de réfraction. Le non-respect de cette consigne introduira des aberrations sphériques lors de l’imagerie et déformera la lumière capturée lors de l’acquisition de l’image. Il faut prendre soin d’éliminer toutes les bulles d’air du support d’imagerie lors du montage d’échantillons pour confocal et LSFM, car les bulles peuvent perturber le chemin de la lumière vers ou loin de l’échantillon. Les bulles peuvent être retirées manuellement à l’aide d’une pipette avant le montage final de l’échantillon. Pour l’imagerie d’échantillons plus épais avec un microscope confocal, plusieurs espaceurs en silicone peuvent être superposés les uns sur les autres pour s’adapter aux tissus de plus de 0,5 mm d’épaisseur. Une recommandation est d’équilibrer tous les tissus dans le RIMS pendant plusieurs heures à toute la nuit lorsqu’il est monté sur le microscope sans mouvement supplémentaire de l’échantillon. L’équilibrage complet des tissus et des milieux d’imagerie empêchera le mélange de solutions dont les indices de réfraction ne correspondent pas et qui peuvent générer des aberrations pendant l’imagerie. Il est important de se rappeler que lors du montage d’échantillons de tissus clairs, il n’existe pas une seule méthode de montage clé en main pour imager tous les échantillons dans tous les microscopes. Ce protocole traite des options de montage d’échantillons qui ont fonctionné de manière optimale dans un contexte, mais il existe de nombreuses approches pour le montage d’échantillons en fonction du microscope utilisé et de la question biologique abordée. Ces approches peuvent inclure, sans s’y limiter, l’intégration de l’échantillon dans de l’agarose, la suspension de l’échantillon à un crochet ou à une ligne en plastique à indice de réfraction, l’utilisation d’un adaptateur de porc-épic, l’impression 3D d’un porte-échantillon ou la fixation de l’échantillon avec un adhésif à une parabole en plastique.
Les microscopes confocaux peuvent bien fonctionner pour l’imagerie de volumes tissulaires ~1 mm3-1 cm3. Pour les microscopes confocaux, utilisez un objectif 2-10x pour localiser initialement les régions d’intérêt et acquérir des empilements Z de plus grand volume ou de tissus entiers avec une résolution de cellule unique. Passez à des objectifs 20-63x pour acquérir des images à haute résolution de régions d’intérêt spécifiques avec des informations subcellulaires. L’objectif idéal pour l’imagerie des tissus clairsemés CUBIC et CLARITY est un objectif spécifique à CLARITY/Scale qui est adapté avec précision à l’indice de réfraction des tissus et de la solution d’imagerie. Si ce type d’objectif n’est pas disponible, il est optimal d’imager des échantillons avec un objectif à immersion dans le glycérol ou l’huile (par exemple, LD LCI Plan-Apochromat 25 x 0,8 NA Imm Corr DIC M27 objectif multi-immersion : distance de travail = 0,57 mm) plutôt qu’avec un objectif à air. Cela minimisera l’introduction de distorsions optiques dues à des indices de réfraction inadaptés lors de la capture d’image. Les objectifs 20-25x peuvent équilibrer l’acquisition d’images de plus grand volume avec l’obtention de détails de coloration à partir de cellules individuelles dans un environnement tissulaire complexe. Il est important de noter que la plupart des microscopes confocaux contiennent des modules qui permettent la mosaïque 3D de volumes d’imagerie. Ce type d’acquisition d’images peut idéalement générer des piles Z de plus grand volume contenant des informations subcellulaires.
L’imagerie LSFM peut permettre la visualisation 3D de populations cellulaires spécifiques dans le contexte d’un grand volume de tissu (>1cm3) et même d’organes entiers. Au cours des 10 dernières années, le nettoyage des tissus combiné à la LSFM s’est largement concentré sur la compréhension de la connectivité cérébrale chez les rongeurs ; Cependant, des applications plus récentes incluent la visualisation des paysages métastatiques tumoraux16, la distribution cellulaire dans les compartiments anatomiques 9,17 et la dispersion des agents pathogènes18. Par rapport aux cellules cultivées, la plupart des événements biologiques dans les tissus ne sont pas uniformes et la LSFM peut être particulièrement apte à visualiser et à quantifier l’hétérogénéité tissulaire spatiale de ces événements (par exemple, la réplication du virus, la signalisation immunitaire, la distribution cellulaire, etc.).
Les ensembles de données 3D acquis via confocal ou LSFM peuvent être post-traités avec de nombreuses plateformes d’analyse d’images. La suite logicielle Imaris peut être utilisée pour la construction de surfaces, la génération d’animations 3D et la quantification de cellules ; Cependant, il existe de nombreux systèmes d’analyse d’images qui permettent un post-traitement et une analyse efficaces des images. Le logiciel gratuit ImageJ/Fiji19 est une plate-forme alternative attrayante de traitement d’images accessible à la plupart des laboratoires, mais il n’existe pas de logiciel d’analyse unique qui excelle dans toutes les formes d’analyse et de visualisation d’images. De nombreuses suites logicielles d’analyse d’images peuvent être d’un coût prohibitif si elles ne sont pas disponibles dans des installations d’utilisation partagée. Enfin, la gestion des données est un aspect essentiel de la LSFM ou des grands ensembles de données 3D confocales en mosaïque. Ces plateformes d’imagerie peuvent générer des fichiers volumineux (>1 To) qui nécessitent des postes de travail informatiques haut de gamme pour la visualisation et la quantification des données. En fin de compte, ce flux de travail d’imagerie peut rationaliser l’acquisition et la quantification de populations cellulaires spatialement distinctes au sein de tissus entiers et est largement applicable à la plupart des sources tissulaires et des systèmes biologiques.
Déclarations de divulgation
Les auteurs n’ont pas de conflits d’intérêts à divulguer.
Remerciements
Merci à l’Institut de biologie génomique de l’Université de l’Illinois à Urbana-Champaign pour l’utilisation des microscopes confocaux et à fluorescence à feuillet de lumière. Merci aux personnes extraordinaires de la cohorte « The Last Gift » pour les échantillons de tissus humains, qui a été financée par les subventions suivantes : I147821, DA051915, AI131385 et P30 AI036214. Merci à Nancy Haigwood et Ann Hessell pour les échantillons de tissus de PSN infectés par le SHIV.
matériels
| Name | Company | Catalog Number | Comments |
| Acrylamide Solution (in 0.1 M PBS, 40 mL in total) | |||
| 40% Acrylamide: 4 mL | Bio-Rad | 1610144 | |
| VA-044 Thermal Initiator: 0.1g | Fujifilm | 011-19365 | |
| CLARITY Blocking solution (in 0.1 M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Tween-20: 5 µL | Fisher Scientific | BP337-500 | |
| CLARITY wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Tween-20: 50 µL | Fisher Scientific | BP337-500 | |
| CUBIC Blocking solution (in 0.1M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Triton X-100: 5 µL | VWR | M143-1L | |
| CUBIC Reagent-1 (in 0.1M PBS, 50 mL in total) | |||
| N, N, N’, N’-tetrakis (2-hydroxypropyl) ethylenediamine: 12.5 g | Aldrich | 122262 | |
| Triton X-100: 7.5 g | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC Reagent-2 (in 0.1M PBS, 50 mL in total) | |||
| Sucrose: 25 g | Sigma | S1888-500G | |
| Sodium azide (in powder form): 10 g | Sigma | 71289-50G | |
| Sodium azide stock solution (in DI H2O, 50 mL in total) | Sigma | 71289-50G | |
| Triethanolamine: 5 g | Sigma | 90270-500mL | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| DAPI staining solution (0.5 µg/mL) | |||
| DAPI stock solution: 1 µL | |||
| Wash solution: 10 mL | |||
| DAPI stock solution (5 mg/mL) | |||
| DAPI powder: 5 mg | Sigma-Aldrich | D9542-1MG | |
| DMSO (100%): 1 mL | ThermoFisher | D12345 | |
| Imaging Media RI-2 (in dH2O) | |||
| 90% Histodenz | Sigma | D2158-100G | |
| 0.01% Sodium azide | Sigma | 71289-50G | |
| 0.02 Sodium Phosphate Buffer, pH 7.5 | Sigma-Aldrich | S9638-250G | |
| 0.1% Tween-20 | Fisher Scientific | BP337-500 | |
| Primary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Goat anti-HIV p24: 10 µL (1:200) | Creative Diagnostics | DPATB-H81692 | |
| Mouse anti-human CD68: 10 µL(1:200) | Dako | M0876 | |
| Rabbit anti-human CD3: 10 µL (1:200) | Dako | A0452 | |
| 8% SDS Solution (in 0.1 M PBS, 50 mL in total) | |||
| SDS powder: 4 g | Sigma-Aldrich | L3771-500G | |
| Secondary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Donkey anti-goat conjugated with AlexaFluor647: 2 µL | Invitrogen | A21447 | |
| Donkey anti-mouse conjugated with AlexaFluor594: 2 µL | Invitrogen | A21203 | |
| Donkey anti-rabbit conjugated with AlexaFluor488: 2 µL | Invitrogen | A21206 |
Références
- Spalteholz, W., Barker, L. F., Mall, F. P. . Hand-Atlas of Human Anatomy. , (1907).
- Jacob, T., Gray, J. W., Troxell, M., Vu, T. Q. Multiplexed imaging reveals heterogeneity of PI3K/MAPK network signaling in breast lesions of known PIK3CA genotype. Breast Cancer Research and Treatment. 159 (3), 575-583 (2016).
- Kieffer, C., Ladinsky, M. S., Ninh, A., Galimidi, R. P., Bjorkman, P. J. Longitudinal imaging of HIV-1 spread in humanized mice with parallel 3d immunofluorescence and electron tomography. eLife. 6, 23282 (2017).
- Chung, K., Deisseroth, K. CLARITY for mapping the nervous system. Nature Methods. 10 (6), 508-513 (2013).
- Compton, A. A., Schwartz, O. They might be giants: Does syncytium formation sink or spread HIV infection. PLoS Pathogens. 13 (2), 2-8 (2017).
- Symeonides, M., et al. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses. 7 (12), 6590-6603 (2015).
- Sharova, N., Swingler, C., Sharkey, M., Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. The EMBO Journal. 24 (13), 2481-2489 (2005).
- Colby, D. J., et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nature Medicine. 24 (7), 923-926 (2018).
- Ladinsky, M. S., et al. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. eLife. 8, 46916 (2019).
- Buettner, M., Bode, U. Lymph node dissection--understanding the immunological function of lymph nodes. Clinical and Experimental Immunology. 169 (3), 205-212 (2012).
- Treweek, J. B., et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nature Protocols. 10, 1860-1896 (2015).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159, 911-924 (2014).
- Sompuram, S. R., Vani, K., Bogen, S. A. A molecular model of antigen retrieval using a peptide array. American Journal of Clinical Pathology. 125 (1), 91-98 (2006).
- Scalia, C. R., et al. Antigen masking during fixation and embedding, dissected. The journal of histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 65 (1), 5-20 (2017).
- Jing, D., et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Research. 28 (8), 803-818 (2018).
- Guldner, I. H., et al. An Integrative platform for three-dimensional quantitative analysis of spatially heterogeneous metastasis landscapes. Scientific Reports. 6, 24201 (2016).
- Muntifering, M., et al. Clearing for deep tissue imaging. Current Protocols in Cytometry. 86 (1), 38 (2018).
- DePas, W. H., et al. Exposing the three-dimensional biogeography and metabolic states of pathogens in cystic fibrosis sputum via hydrogel embedding, clearing, and rRNA labeling. mBio. 7 (5), 00796 (2018).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image. Nature Methods. 9 (7), 676-682 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.