
Method Article
Visualização 3D de populações de células imunes em tecidos infectados pelo HIV por meio de microscopia de clareamento, imunocoloração, confocal e fluorescência de folha de luz
Neste Artigo
Resumo
A limpeza do tecido, combinada com a microscopia de imunofluorescência, permite a visualização espacial e a quantificação de populações de células imunes e proteínas virais dentro de tecidos intactos. O corte óptico de tecidos limpos com microscopia de fluorescência confocal e de folha de luz pode gerar modelos 3D de ambientes complexos de tecidos e revelar a heterogeneidade espacial exibida durante a infecção pelo HIV.
Resumo
O Vírus da Imunodeficiência Humana (HIV), o agente causador da Síndrome da Imunodeficiência Adquirida (AIDS), é uma grande preocupação de saúde global, com quase 40 milhões de indivíduos infectados em todo o mundo e sem cura amplamente acessível. Apesar dos esforços intensivos, uma compreensão detalhada das interações entre vírus e células hospedeiras nos tecidos durante a infecção e em resposta à terapia permanece incompleta. Para resolver essas limitações, as técnicas de limpeza de tecidos à base de água CUBIC (Clear , Unobstructed Brain/Body Imaging Cocktails and Computational analysis) e CLARITY (Clear Lipid-exchanged Acrylamide-hybridized Rigid Imaging/Immunostaining/in situ-hybridization-compatible Tissue hYdrogel) são aplicadas para visualizar interações complexas entre células hospedeiras de vírus em tecidos infectados pelo HIV de modelos animais e humanos usando microscopia de fluorescência confocal e de folha de luz. O corte óptico de tecidos intactos e a análise de imagens permitem a reconstrução rápida de informações espaciais contidas em tecidos inteiros e a quantificação de populações de células imunes durante a infecção. Esses métodos são aplicáveis à maioria das fontes de tecidos e a diversas questões biológicas, incluindo doenças infecciosas e câncer.
Introdução
A crescente necessidade de imagens espaciais quantitativas de tecidos em pesquisas biológicas recentemente levou ao surgimento de técnicas de limpeza de tecidos para gerar imagens de maior volume (mm3 cm3) de tecidos intactos com resolução de célula única. Os tecidos incluem organizações complexas de biomoléculas com estruturas, composições e funções definidas de forma única. Infelizmente, muitas biomoléculas presentes nos tecidos (por exemplo, lipídios e cromóforos) se espalham, absorvem ou emitem luz quando visualizadas por microscopia de luz, dificultando a aquisição de imagens de grande volume. Além disso, os tecidos geralmente exibem um índice de refração incompatível com soluções de imagem padrão e lentes ópticas, resultando em distorções ópticas durante a imagem. Uma abordagem ideal para imagens de grandes volumes de tecido com um microscópio óptico deve envolver a correspondência do índice de refração dos tecidos, soluções de imagem e objetivas, permitindo a penetração profunda da luz no tecido sem interromper as características biológicas dos tecidos intactos durante o processamento. As tentativas iniciais de reduzir as diferenças no índice de refração entre tecidos e soluções de imagem por meio da limpeza de amostras de tecido opacas foram realizadas pelo anatomista alemão Werner Spalteholz no final de 18001. Essa técnica de limpeza de tecidos envolveu solventes químicos agressivos, que podem danificar amostras de tecido, mas, no entanto, representou a primeira imagem de maior volume relatada de tecidos intactos. Os métodos modernos de microscopia de luz, combinados com o poder de computação para captura e análise de imagens, recentemente trouxeram a limpeza de tecidos novamente à moda como um método para obter imagens de amostras de tecido grandes e intactas com resolução de célula única. Durante as últimas duas décadas, surgiram dezenas de técnicas avançadas de limpeza de tecidos, incluindo orgânicas e à base de água, cada uma com pontos fortes e fracos para aplicações específicas.
A imagem de tecido 3D pode sondar interações biológicas mais complexas que não podem ser reproduzidas em cultura de células. Por exemplo, padrões de sinalização celular2, distribuições espaciais de tipos de células distintos3 e conectividade cerebral4 foram previamente mapeados de maneira quantitativa usando métodos de imagem de tecido / órgão inteiro. Aqui está descrita uma aplicação de protocolos de limpeza de tecidos à base de água para limpar, imunocorar e visualizar populações distintas de células-alvo do HIV dentro de tecidos linfóides infectados pelo HIV intactos durante a infecção ativa. Dentro do corpo, o HIV infecta predominantemente células T CD4 + e integra uma cópia de seu genoma em genomas de células hospedeiras infectadas. O vírus subsequentemente sequestra a maquinaria da célula hospedeira infectada para se replicar, resultando em disseminação do vírus, morte da célula hospedeira, disfunção imunológica e progressão de longo prazo para a AIDS. É importante notar que os comportamentos das células T infectadas em cultura de tecidos e células são notavelmente discrepantes. Células T CD4 + cultivadas incubadas com HIV podem produzir sincícios maciços induzidos pelo HIV que podem incluir dezenas de núcleos5, enquanto experimentos semelhantes com células T CD4 + primárias cultivadas em hidrogéis de matriz extracelular (ECM) 3D ou amostras de tecido de camundongos humanizados infectados pelo HIV (camundongos hu) geralmente produzem sincício com 2-5 núcleos6. Compreender a transmissão local célula a célula e a disseminação sistêmica do vírus em indivíduos infectados pelo HIV é provavelmente ainda mais complicado, envolvendo o transporte do vírus por vários tipos de células infectadas de tecidos para vasos sanguíneos e novos tecidos, onde vírions livres e células produtoras de vírus podem acessar um grande número de linfócitos suscetíveis7. Atualmente, esses cenários não são possíveis de recapitular em sistemas de cultura de células, e tecidos de modelos animais e humanos continuam sendo um recurso importante para a compreensão da patogênese do vírus no contexto de um organismo complexo com um sistema imunológico funcional.
As terapias antirretrovirais atuais (ARTs) aumentam muito a expectativa e a qualidade de vida das pessoas com HIV (PWH), inibindo a replicação do HIV e interrompendo a progressão da doença em direção à AIDS. Infelizmente, a TARV não elimina as células imunes latentemente infectadas contendo uma inserção do genoma retroviral que estão quiescentes e não produzem vírus ativamente. Embora o vírus não seja detectável no sangue da maioria dos indivíduos em TARV, as cargas virais se recuperam rapidamente após a interrupção da TARV e a progressão da doença continua8. A natureza persistente da infecção pelo HIV causada pelo reservatório latente de células infectadas representa um enorme impedimento para o estabelecimento de uma cura para o HIV. Os reservatórios teciduais do HIV permanecem pouco compreendidos, e é crucial estabelecer uma compreensão mais profunda desses reservatórios nos tecidos linfóides antes, durante e após a TARV, para caracterizar completamente a patogênese do vírus e avaliar novos tratamentos que eliminem efetivamente as células infectadas latentes que não produzem vírus ativamente.
Aqui, CUBIC3 e CLARITY9, dois protocolos de limpeza de tecidos à base de água previamente adaptados, foram aplicados a populações de células imunes de imagem em vários tecidos linfóides intactos de camundongos infectados pelo HIV com sistemas imunológicos humanizados (camundongos hu), primatas não humanos infectados por SIV / SHIV (NHP) e humanos infectados pelo HIV. Esses protocolos são adaptáveis à microscopia de fluorescência confocal e de folha de luz, dependendo dos objetivos da imagem (maior resolução versus maior volume) e da instrumentação disponível. Embora a microscopia de luz não possa resolver vírions individuais, o uso de imunofluorescência pode identificar regiões de tecido contendo vírus e células produtoras de vírus que podem ser analisadas posteriormente com métodos de resolução mais alta. Os métodos apresentados aqui podem ser adaptados para visualizar quase qualquer tecido do corpo com resolução de célula única, a fim de quantificar as relações espaciais entre tipos específicos de células em diferentes condições durante a infecção e são facilmente traduzíveis para amostras de pacientes humanos altamente relevantes para o estudo de doenças infecciosas ou câncer.
Protocolo
Todos os experimentos com animais foram conduzidos de acordo com protocolos institucionais de cuidados com animais aprovados. Todos os tecidos humanos foram adquiridos de acordo com as diretrizes institucionais aprovadas de ética em pesquisa humana.
1. Colheita e fixação de tecidos (o mesmo para CUBIC e CLARITY)
- Identifique e disseque os tecidos linfóides conforme descrito anteriormente10.
- Extirpar os tecidos linfóides com tesouras e pinças de dissecação em minutos após a morte, quando possível com segurança.
- Coloque as amostras de tecido em um tampão fixador gelado recém-feito contendo 8% de paraformaldeído (PFA), 5% de sacarose em trihidrato de cacodilato de sódio 0,1 M para preservar adequadamente as amostras de tecido para microscopia de luz (LM), microscopia eletrônica (EM) ou imuno-EM. Alternativamente, fixe as amostras para LM com 4% de PFA em 0,1 M PBS. Corrija as amostras durante a noite antes de iniciar o processo de limpeza para garantir a desativação total do vírus.
CUIDADO: O paraformaldeído é tóxico por contato com a pele e inalação e também é um sólido inflamável; Manuseie com cuidado e guarde em um armário de armazenamento inflamável. O trihidrato de cacodilato de sódio é tóxico se ingerido ou inalado. - Faça uma imagem de referência do tecido antes de iniciar o processo de limpeza.
NOTA: As amostras LM podem ser armazenadas por pelo menos 1 ano nessas condições. Para trabalhar com amostras que expressam proteínas fluorescentes endógenas, mantenha sempre as amostras no escuro nas etapas subsequentes.
2. Limpeza de tecido cúbico
- Enxágue as amostras de tecido linfóide em PBS estéril 0,1 M três vezes com agitação em temperatura ambiente por 15 min para garantir a remoção do PFA durante cada troca de tampão.
NOTA: Descarte os líquidos que contêm PFA de acordo com as diretrizes institucionais. - Mergulhe a amostra de tecido linfóide no Reagente Cúbico 1 (consulte a Tabela de Materiais) a 37 ° C por 3 dias com agitação suave. Tire imagens de referência regulares para monitorar o processo de descoloração ao longo do tempo.
- Troque o Reagente-1 por mais 3-4 dias de imersão ou até que a descoloração do tecido esteja completa. O tempo necessário para a limpeza depende do volume e do tipo de tecido. Para acelerar o processo de descoloração do tecido, atualize o CUBIC Reagent-1 diariamente e use volumes maiores.
- Lave as amostras de tecido linfóide três vezes com PBS 0,1 M por 30 min em temperatura ambiente com agitação suave.
- Mergulhe as amostras de tecido linfóide no Reagente Cúbico-2 (ver Tabela de Materiais) a 37 ° C com agitação suave por 2-7 dias ou até que a transparência completa seja alcançada. Se as amostras não obtiverem transparência completa, repita as etapas 2.2-2.5 até que a limpeza não progrida mais. Tire imagens de referência regulares para monitorar o processo de limpeza ao longo do tempo.
- Lave as amostras de tecido linfóide três vezes com PBS 0,1 M por 30 min em temperatura ambiente com agitação suave.
- Armazene as amostras em CUBIC Reagent-2 com 0,01% de azida sódica a 0,01% de volume/volume (V/V) no escuro (consulte a Tabela de Materiais).
NOTA: As amostras podem ser armazenadas por pelo menos 6 meses usando este método.
CUIDADO: A azida de sódio é altamente tóxica e representa um sério risco de inalação. Recomenda-se a compra de soluções diluídas de azida sódica a 5% ou menos.
3. Bloqueio e imunocoloração de amostras cúbicas
- Lave as amostras de tecido linfóide três vezes com PBS 0,1 M por 30 min cada em temperatura ambiente com agitação suave.
- Para obter imagens usando um microscópio confocal, corte o tecido em fatias de ~ 0,5-1 mm de espessura usando uma matriz de corte de tecido. Para realizar a microscopia de fluorescência de folha de luz (LSFM), bloqueie toda a região do tecido.
- Bloquear as amostras com 5 ml de solução de bloqueio CUBIC durante a noite a 4 °C com agitação (ver Tabela de Materiais). Ao trabalhar com NHP ou amostras humanas, use FcR anti-humano. Ao trabalhar com amostras de mouse, use FcR anti-mouse na solução de bloqueio.
- Manchar as amostras com 5 mL de anticorpos primários (ver Tabela de Materiais) em solução de bloqueio (sem FcR específico da espécie) por 3 dias em temperatura ambiente com agitação (Opcional: centrifugue o estoque de anticorpos concentrados a 2.300 x g por 5 min antes de usar, para reduzir a adição de anticorpos agregados).
- Lave a amostra corada à temperatura ambiente com agitação por um período mínimo de 5 h no total com pelo menos cinco trocas de tampão de solução de lavagem (consulte a Tabela de Materiais).
- Manchar as amostras com anticorpos secundários (ver Tabela de Materiais) em solução de bloqueio (sem FcR específico da espécie) por 3 dias em temperatura ambiente com agitação (Opcional: centrifugue os anticorpos a 2.300 x g por 5 min antes de usar para minimizar a agregação de anticorpos).
- Lave a amostra corada cinco vezes com solução de lavagem à temperatura ambiente com agitação por pelo menos 5 h no total.
- Manchar as amostras com 5 mL de solução de coloração DAPI (ver Tabela de Materiais) em cada amostra de tecido e incubar por 10 min em temperatura ambiente. Deixar as amostras permanecerem na solução corante DAPI no escuro a 4 °C para obtenção de imagens posteriores.
- Lave as amostras de tecido linfóide com solução de lavagem três vezes à temperatura ambiente com agitação por 30 min cada.
- Mergulhe a amostra corada em CUBIC Reagent-2 durante a noite em temperatura ambiente no escuro antes da montagem da amostra.
4. Limpeza de tecido CLARITY
- Enxágue as amostras de tecido linfóide em PBS estéril 0,1 M três vezes agitando em temperatura ambiente por 15 min cada para remover o PFA.
- Coloque as amostras de tecido em 15 ml de solução de acrilamida recém-fabricada e incube a 4 °C durante a noite com agitação suave (ver Tabela de Materiais).
CUIDADO: A acrilamida não polimerizada é uma neurotoxina potente e prontamente absorvida pela pele. Evite qualquer contato com a pele e enxágue imediatamente se ocorrer contato. - Deixe as amostras de tecido aquecerem até a temperatura ambiente.
- OPCIONAL: Desgaseifique as amostras de tecido borbulhando nitrogênio na solução de acrilamida por 1 min. Tome cuidado para usar uma taxa de fluxo baixa que evite respingos de acrilamida não polimerizada tóxica (~ 1-2 bolhas / s).
- Coloque as amostras de tecido em banho-maria a 37 °C por 1-3 h para polimerizar, invertendo a cada 15 min. Remova as amostras assim que for detectada uma polimerização perceptível, conforme indicado por um líquido viscoso, o aparecimento de linhas de Schleren durante a mistura ou a formação de uma cápsula transparente ao redor do tecido.
NOTA: Se ocorrer a polimerização completa da solução de acrilamida, corte o excesso de hidrogel da amostra e continue o protocolo. - Lave as amostras de tecido com PBS estéril 0,1 M três vezes por 30 min cada em temperatura ambiente com agitação suave para remover a solução de acrilamida.
- Coloque as amostras de tecido em 15 mL de SDS a 8% em PBS 0,1 M a 37 °C com balanço suave por 2-5+ dias para permitir a limpeza. Atualize periodicamente a solução de SDS a 8% e use até 50 mL da solução para acelerar a limpeza, se necessário. Pare o processo de limpeza quando as amostras estiverem visualmente transparentes ou não estiverem mais progredindo. Tire imagens de referência regulares para monitorar o processo de limpeza ao longo do tempo.
- Lave as amostras de tecido com PBS estéril 0,1 M cinco vezes ao longo de 1 dia em temperatura ambiente com agitação suave.
- Mantenha as amostras temporariamente em PBS 0,1 M (mais 0,01% volume/volume (v/v) NaN3 para armazenamento de longo prazo) no escuro até que estejam prontas para obter imagens de fluorescência endógena.
- Coloque o tecido em 5 mL de meio de imagem RI-2 (consulte a Tabela de Materiais). Incubar durante a noite à temperatura ambiente no escuro para verificar a integridade do processo de limpeza antes da imunocoloração. Tire imagens de referência para monitorar a transparência do tecido.
5. Bloqueio e imunocoloração de amostras CLARITY
NOTA: Essas etapas são semelhantes ao bloqueio e imunocoloração de tecidos limpos CUBIC, mas usam formulações diferentes para bloquear, lavar e colorir soluções.
- Lave as amostras de tecido linfóide três vezes com 0,1 M PBS por 30 min de cada vez em temperatura ambiente com agitação suave.
- Para obter imagens usando um microscópio confocal, corte o tecido em fatias de ~ 0,5-1 mm de espessura usando um cortador de tecido de 0,5 mm e matriz. Para realizar LSFM, bloqueie toda a amostra de tecido.
- Bloquear as amostras com 5 ml de solução de bloqueio CLARITY (ver Tabela de Materiais) durante a noite a 4 °C com agitação.
- Manchar as amostras com 5 mL de anticorpos primários (ver Tabela de Materiais) em solução de bloqueio (sem FcR específico da espécie) por 3 dias em temperatura ambiente com agitação (Opcional: centrifugue os anticorpos a 2.300 x g por 5 min antes de usar para minimizar a agregação de anticorpos).
- Lave a amostra corada cinco vezes com a solução de lavagem à temperatura ambiente com agitação por pelo menos 5 h no total (consulte a Tabela de Materiais).
- Manchar as amostras com 5 mL de anticorpos secundários (ver Tabela de Materiais) em solução de bloqueio (sem FcR específico da espécie) por 3 dias em temperatura ambiente com agitação (Opcional: centrifugue os anticorpos a 2.300 x g por 5 min antes de usar para minimizar a agregação de anticorpos). Para encurtar a duração geral do protocolo, use anticorpos primários conjugados com fluoróforos para eliminar a necessidade de incubação com anticorpos secundários.
- Lave a amostra corada cinco vezes com a solução de lavagem à temperatura ambiente, agitando por pelo menos 5 h no total.
- Manchar as amostras com 5 mL de solução de coloração DAPI (ver Tabela de Materiais) em cada amostra de tecido e incubar por 10 min em temperatura ambiente. Deixe as amostras permanecerem a 4 ° C no escuro em solução de coloração DAPI para imagens posteriores.
- Lave as amostras de tecido linfóide com a solução de lavagem três vezes à temperatura ambiente, agitando por 30 minutos de cada vez.
- Coloque o tecido em 5 mL de meio de imagem RI-2 (RI = 1,46) e incube durante a noite em temperatura ambiente no escuro antes da montagem da amostra (consulte as etapas 6 e 7 do protocolo).
6. Montagem e imagem de amostras de tecido limpo para microscopia confocal
- Retire um lado da camada protetora de um isolador de silicone adesivo.
- Cole uma lamínula de microscópio (22 mm x 40 mm, 0,25 mm de espessura) no lado descascado do isolador de silicone para formar um espaço à prova de líquidos para a amostra.
- Retire o outro lado da camada protetora do isolador de silicone adesivo.
- Coloque a amostra para imagem no centro do isolador de silicone e, em seguida, adicione o CUBIC Reagent-2 ou o Imaging Media RI-2 conforme apropriado até que a superfície do líquido esteja tão alta quanto a borda do isolador.
- Para minimizar o aprisionamento de bolhas de ar dentro do isolador de silicone, alinhe e coloque suavemente a segunda lamínula de um lado usando uma pinça EM. Limpe o excesso de líquido. Pressione suavemente a lamínula ao redor do(s) poço(s) de amostra usando a parte de trás da pinça para selar o adesivo. Armazene as amostras montadas horizontalmente no escuro.
NOTA: As amostras podem ser visualizadas semanas a meses após serem montadas; no entanto, a qualidade da imagem geralmente diminui com o tempo. - Coloque a lâmina montada no microscópio stage e localize a amostra usando luz branca e uma objetiva de ampliação mais baixa (2-10x).
- Configure o perfil de aquisição de fluorescência com base nos fluoróforos individuais escolhidos.
NOTA: Recomenda-se adquirir individualmente canais de fluoróforo separados. Isso resulta em um tempo de aquisição mais longo, mas reduz a sobreposição espectral e a aquisição de sinal de fluorescência não específico. Um perfil de fluoróforo comum pode incluir DAPI (450 nm), Alexa488, Alexa594 e Alexa647 (ou combinações relacionadas) para minimizar a sobreposição espectral durante a aquisição da imagem. - Escolha uma objetiva de ampliação apropriada para criar imagens de regiões de interesse. Use objetivas de ampliação mais baixas (2-10x) para imagens de maior volume ou tecido total com resolução de célula única e use objetivas de ampliação mais altas (20-63x) para visualização de alta resolução de detalhes subcelulares em tecido limpo. Combine o índice de refração das objetivas, meios de imagem e tecidos o mais próximo possível para minimizar a introdução de distorções ópticas durante a aquisição da imagem.
- Escolha um tamanho de etapa para aquisição de pilha Z. Para objetivas de ampliação mais baixa (2-10x), selecione um tamanho de passo de ~3-5 μm para detectar fluorescência de uma célula individual em várias fatias Z contínuas para modelagem 3D, reduzindo o tempo total de aquisição e o tamanho geral do arquivo. Para objetivas de maior ampliação (20-63x), selecione um tamanho de passo de ~1 μm ou menos para minimizar a perda de informações subcelulares entre as fatias Z individuais.
- Amplie o campo de visão para visualizar toda a região do tecido a ser visualizada nas dimensões X e Y com o mínimo possível de área desocupada. Defina as coordenadas de aquisição do estágio Z superior e inferior que abrangem toda a região de interesse a ser fotografada.
- Adquira as imagens da pilha Z. Salve e exporte o arquivo para pós-processamento usando qualquer software de análise de imagem. Para determinados pacotes de software, converta os arquivos em tipos de arquivo específicos (por exemplo, .tiff, .ome-tiff, .jpeg, etc.). Realize a conversão usando qualquer software de aquisição de imagem de microscópio ou freeware de análise de imagem (por exemplo, ImageJ/Fiji).
7. Montagem e imagem das amostras na câmara LSFM ou cubeta
- Encha a câmara de imagem com CUBIC Reagent-2 ou RI-2, dependendo do protocolo específico usado. Evite a formação de bolhas durante a transferência do líquido. Remova o excesso de bolhas com uma pipeta.
- Mergulhe a amostra na câmara de imagem e restrinja o movimento da amostra.
NOTA: Dependendo do microscópio específico usado, isso pode incluir incorporar a amostra em agarose, suspender a amostra de um gancho ou adaptador de porco-espinho, imprimir em 3D um suporte de amostra ou anexar a amostra com adesivo a um prato de plástico. - Coloque a objetiva na solução de imagem, focada na amostra. Deixe a amostra montada na câmara de imagem por várias horas ou durante a noite para permitir o equilíbrio total das soluções e tecidos na cubeta.
- Adquira a pilha Z da região de interesse (consulte as etapas 6.7 a 6.11 para aquisição de imagem).
NOTA: Esta abordagem pode permitir a obtenção de imagens de volumes de tecido superiores a 1 cm3 com resolução de célula única.
8. Reconstrução de superfície e quantificação celular com o software de análise de imagem Imaris
NOTA: Essas etapas são específicas do software de análise de imagem Imaris, mas etapas semelhantes de processamento de imagem podem ser realizadas usando outros pacotes de software (por exemplo, ImageJ/Fiji, Aivia, Arivis, Amira, etc.).
- Use o Imaris File Converter para converter o arquivo de imagem Z-stack para o formato nativo Imaris .ims. Isso facilitará uma conversão de arquivos mais rápida, minimizando erros de conversão e possíveis problemas de software depois de abertos.
NOTA: Alguns LSFMs mais recentes permitem que o usuário salve arquivos diretamente no formato .ims. - Arraste o arquivo .ims a ser analisado para a área Arena do software Imaris. Ajuste o contraste ou a intensidade de cada canal de cor pelo painel Ajuste de exibição . Clique no ícone Adicionar novas superfícies no canto superior esquerdo.
- Clique em Avançar: Canal de origem (o ícone azul com uma seta apontando para a direita). Escolha o canal de origem da superfície a ser construída. Não altere os outros parâmetros.
- Clique em Avançar: Limite (o ícone azul com uma seta apontando para a direita).
- Para ajustar o limite (intensidade absoluta), arraste a linha de limite para a esquerda ou para a direita. Ative Dividir objetos em contato e insira o diâmetro médio da célula em mícrons como o padrão de divisão para que o sistema produza muitos pontos como origem para cada superfície individual.
- Não inclua sinais fluorescentes que sejam muito pequenos ou muito brilhantes, pois podem representar possíveis manchas ou artefatos de microscópio. Inclua apenas os pontos que têm tamanhos e intensidades de fluorescência aceitáveis, alterando o diâmetro médio da célula de acordo.
NOTA: O diâmetro médio da célula varia para tecidos ou tipos de células específicos, mas geralmente reside entre 5-15 μm.
- Não inclua sinais fluorescentes que sejam muito pequenos ou muito brilhantes, pois podem representar possíveis manchas ou artefatos de microscópio. Inclua apenas os pontos que têm tamanhos e intensidades de fluorescência aceitáveis, alterando o diâmetro médio da célula de acordo.
- Clique em Avançar: Classificar superfícies (o ícone azul com uma seta apontando para a direita). Ajuste as superfícies a serem incluídas arrastando a linha de limite para a esquerda ou para a direita. Certifique-se de que as superfícies se aproximem exatamente do sinal de fluorescência bruto, enquanto separa o sinal de fluorescência de células individuais.
- Clique em Concluir: Executar todas as etapas de criação e encerrar o assistente (o ícone verde com duas setas apontando para a direita). A superfície é oficialmente construída.
- Clique no sexto ícone rotulado Estatísticas no painel esquerdo para ver o número de células, nesta circunstância, o número de superfícies para o canal de cor específico analisado.
- Certifique-se de que as quatro variáveis Número de componentes desconectados por ponto no tempo, Número de superfícies por ponto no tempo, Número total de componentes desconectados e Número total de superfícies tenham o mesmo número, que é a contagem de células desse canal de cor.
Resultados
A limpeza do tecido envolve o tratamento de tecidos preservados com coquetéis químicos para extrair biomoléculas opacas do tecido, mantendo a arquitetura do tecido. Essas soluções de limpeza de tecido combinam o índice de refração do tecido com o meio de imagem circundante para minimizar distorções ópticas, melhorar a relação sinal-ruído profundamente nos tecidos e minimizar a autofluorescência de fundo. Dois protocolos à base de água para limpeza óptica de tecido, CUBIC3 e CLARITY9, foram usados para limpar amostras preservadas de tecido humano de camundongo / SIV infectado por HIV / SIV antes da coloração de imunofluorescência e imagem com microscopia de fluorescência confocal e de folha de luz.
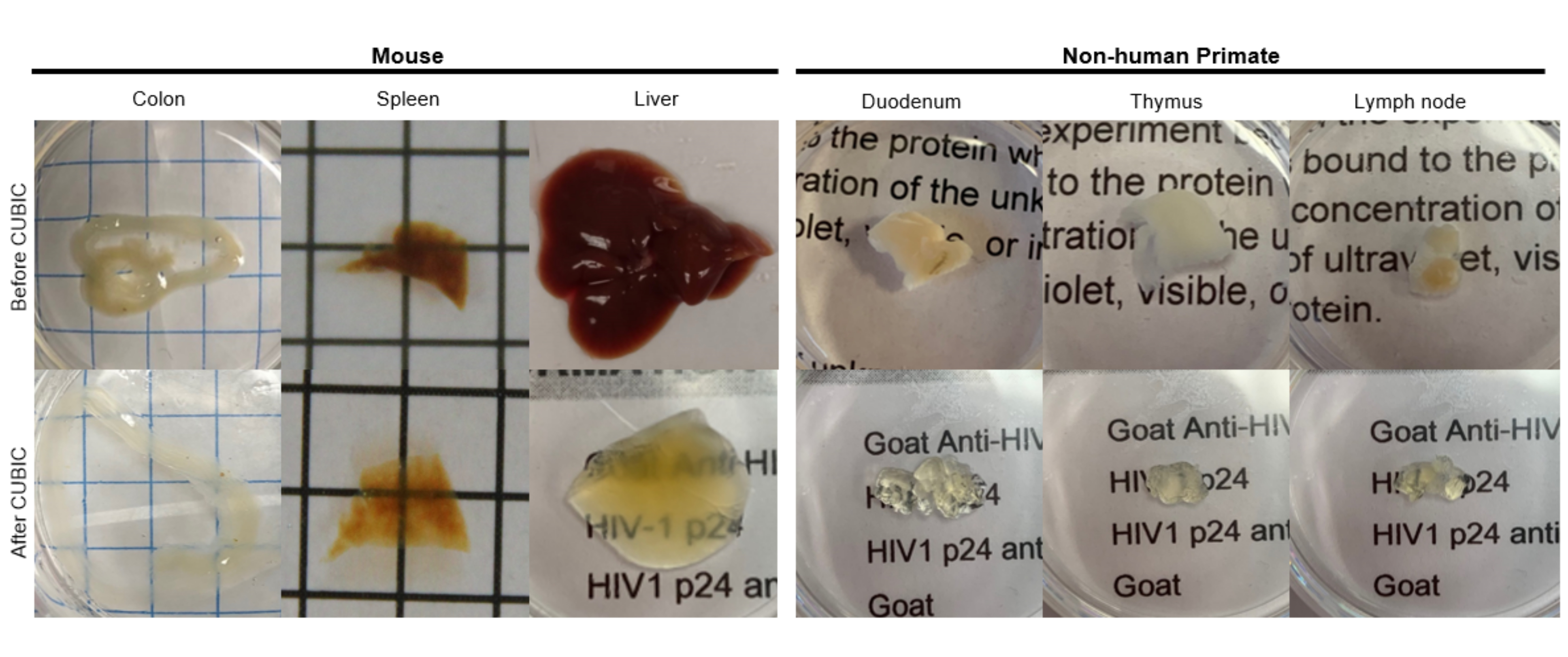
Para o protocolo CUBIC, os tecidos fixados foram lavados com PBS para remoção de fixadores e imersos em CUBIC Reagent-1, uma solução básica tamponada de aminoálcoois que elui cromóforos como o heme, resultando em descoloração e delipidação do tecido (Figura 1, topo). Volumes de tecido menores (~mm3) podem ser descoloridos após 3 dias de tratamento com CUBIC Reagent-1, mas volumes de tecido maiores (~cm3) ou tecidos com grande quantidade de heme (como fígado, baço ou coração) requerem tempos de incubação e volumes de solução mais longos (>1 mês e ~ 50 mL), bem como troca frequente da solução a cada 2-3 dias. Após a descoloração, os tecidos foram lavados e colocados no CUBIC Reagent-2, uma solução contendo sacarose com um índice de refração de aproximadamente 1,48-1,49, que corresponde ao índice de refração do tecido e aumenta a transmitância da luz. Os tecidos limpos foram imunomarcados e montados em uma solução de CUBIC Reagent-2 antes da imagem com um microscópio confocal ou de folha de luz. Os efeitos do procedimento de limpeza CUBIC foram visualizados para vários tecidos hu-mouse e NHP de vários tamanhos e concentrações de cromóforos (Figura 2). A limpeza óptica tornava os tecidos visivelmente transparentes a olho nu, permitindo que linhas de grade e texto em folhas de papel fossem vistos "através" do tecido. Tecidos ricos em cromóforos, como baço, fígado, medula óssea e coração, podem não descolorir completamente, mas permanecem adequados para imunocoloração e imagem (Figura 2 e Figura 5).
Para o protocolo CLARITY, os tecidos fixados foram lavados com PBS para remover os fixadores e, em seguida, incubados durante a noite a 4 ° C em uma solução de acrilamida a 40% com um iniciador térmico para formar ligações covalentes entre as proteínas da amostra e os monômeros de acrilamida (Figura 1, abaixo). No dia seguinte, após o tecido ter sido equilibrado à temperatura ambiente e aquecido em banho-maria a 37 °C, a polimerização da acrilamida foi iniciada e rapidamente envolveu a amostra em um hidrogel. A amostra foi tratada com uma solução de SDS a 8% ao longo de 2-5 dias para remover lipídios opacos. Imediatamente antes da coloração fluorescente, a amostra foi imersa em solução de correspondência de índice de refração (RIMS) para CLARITY (Imaging Media RI-2) contendo 90% de meio de gradiente de densidade não iônica. Para tecidos contendo grandes quantidades de heme, uma etapa de descoloração pode ser adicionada ao final da etapa de delipidação 9,11,12. A progressão da limpeza CUBIC e CLARITY foi comparada em diferentes seções da mesma amostra de baço humano (Figura 3). A limpeza CLARITY produz um gel de poliacrilamida visível que envolve a solução e normalmente exibe descoloração reduzida em comparação com a limpeza CUBIC, a menos que uma etapa adicional de descoloração seja adicionada 9,12.
Posteriormente, em ambos os protocolos, tecidos limpos e intactos foram imunomarcados para detectar populações específicas de células imunes. As amostras foram lavadas, bloqueadas com um reagente contendo α-FcR para reduzir a ligação de anticorpos inespecíficos e coradas por 3 dias quando se usou um anticorpo primário diretamente conjugado a um fluoróforo. Alternativamente, as amostras foram coradas por 3 dias com um anticorpo primário não conjugado, seguido por mais 3 dias com um anticorpo secundário conjugado a um fluoróforo. Os tecidos foram lavados novamente e, em seguida, incubados com coloração DAPI durante a noite a 4 °C para visualização nuclear. As amostras foram lavadas e incubadas em CUBIC Reagent-2 por 24-36 h ou Imaging Media RI-2 (CLARITY) durante a noite no escuro. Para a microscopia confocal, os tecidos foram montados em uma lâmina de microscópio no RIMS apropriado antes da imagem (Figura 4). Para microscopia de fluorescência de folha de luz (LSFM), as amostras foram completamente submersas com RIMS em uma cubeta de imagem durante a noite antes da imagem.
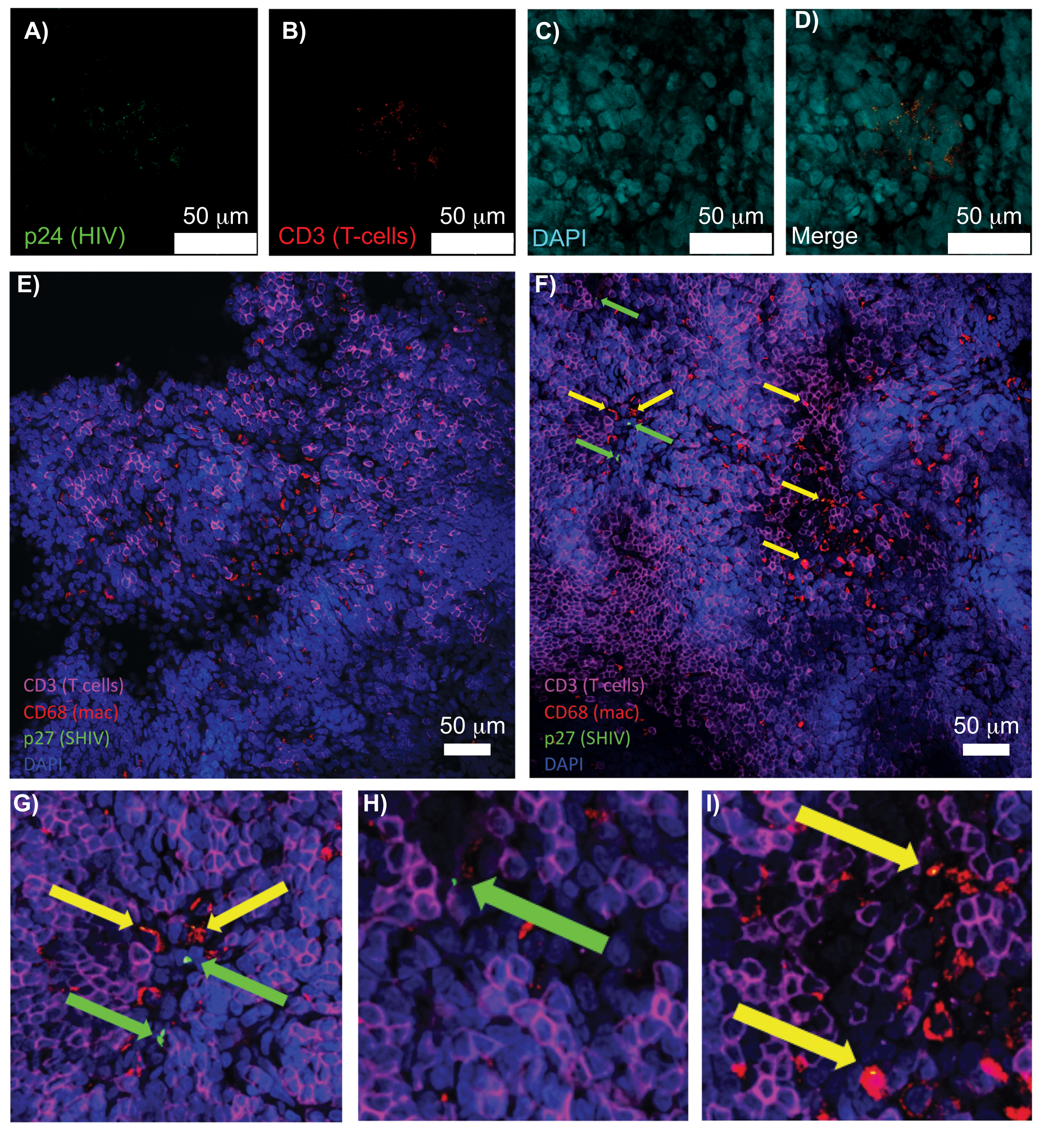
A microscopia confocal de tecidos linfóides intactos, limpos e imunomarcados permitiu a visualização simultânea de múltiplos sinais fluorescentes, incluindo núcleos, marcadores de células imunes e proteínas HIV/SIV CA (capsídeo) (Figura 5). As células produtoras de vírus foram determinadas por colocalização de fluorescência de marcadores de células imunes e proteínas do HIV. O baço humano infectado pelo HIV limpo e corado revelou múltiplas células T CD3 + co-localizadas com o HIV p24, indicando a presença de células produtoras de vírus dentro de uma região de tecido intacto (Figura 5A-D). Linfonodos NHP infectados por SHIV limpos e imunomarcados revelaram as distribuições de células T CD3 + e macrófagos CD68 + em regiões de tecido sem vírus detectado ( Figura 5E ), além de regiões com numerosas células produtoras de vírus ( Figura 5F ). Esses resultados mostraram que as células produtoras de vírus de diversas fontes de tecido eram distinguíveis de outras células dentro de um determinado campo de visão e permitiam a detecção de eventos biológicos raros em um ambiente de tecido complexo.
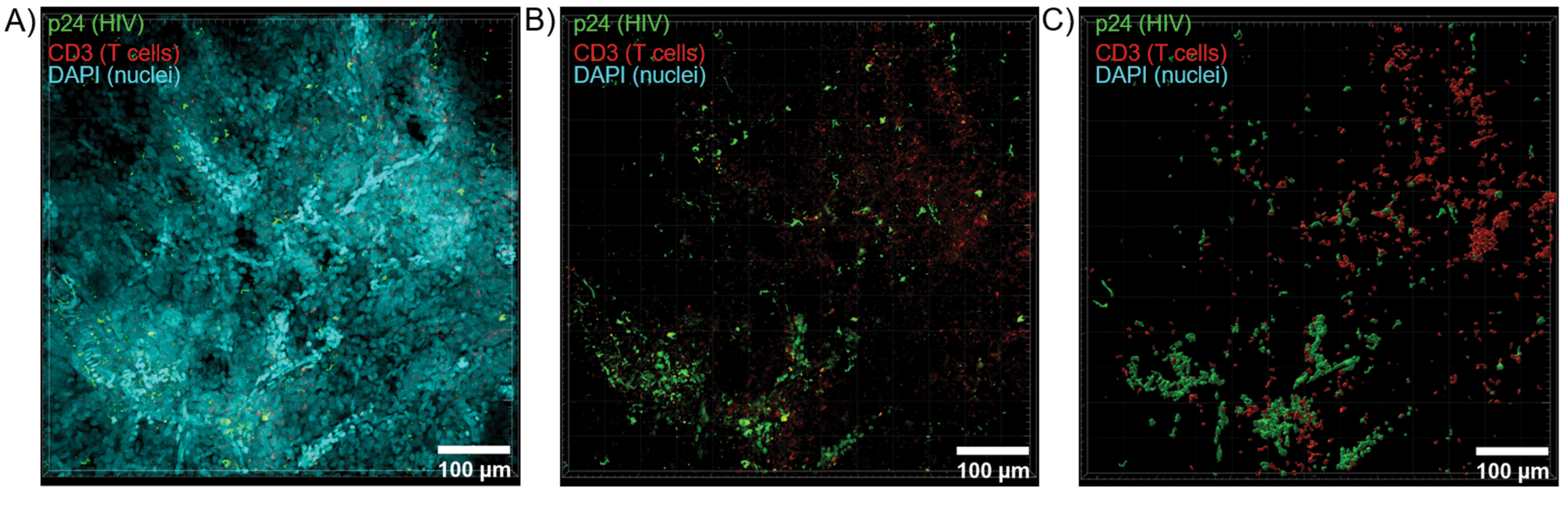
O corte óptico de tecidos limpos com um microscópio confocal foi aplicado para gerar pilhas Z e modelos de superfície 3D, que revelaram a heterogeneidade celular exibida durante a infecção pelo HIV (Figura 6). As pilhas Z foram recombinadas em uma imagem de projeção Z usando o pacote de software Imaris (Figura 6A) e o canal nuclear DAPI foi removido para visualização clara da fluorescência das células T CD3 + e da proteína do capsídeo do HIV (p24) em volumes inteiros de tecido (Figura 6B). A fluorescência de projeção Z foi segmentada automaticamente com o software Imaris para gerar um modelo de superfície 3D reconstruído para visualização espacial e quantificação do sinal de fluorescência em toda a pilha Z (Figura 6C). A análise do modelo de superfície 3D revelou 546 células T CD3+ e 218 células produtoras de HIV p24. Cumulativamente, a aquisição de imunofluorescência em pilha Z de tecidos linfóides limpos e infectados pelo HIV permitiu a geração de modelos 3D de composição celular dentro do tecido e quantificação automatizada de populações de células imunes dentro dos volumes de tecido.
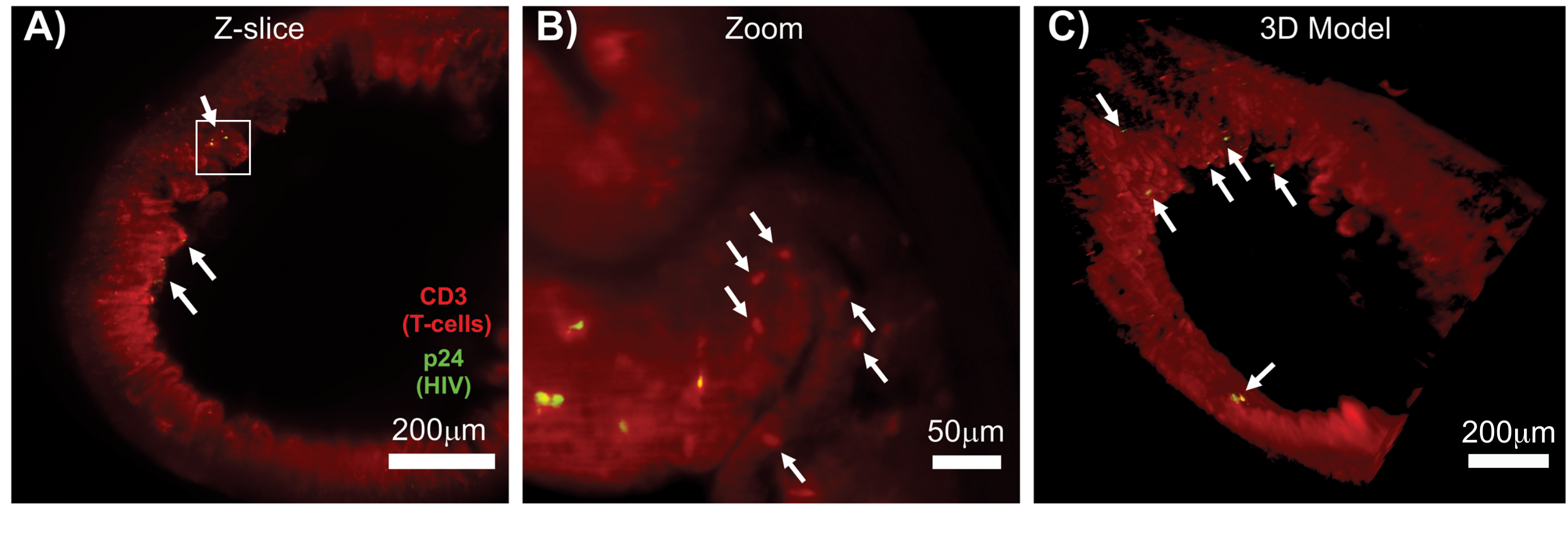
LSFM de tecidos linfóides intactos, limpos e imunomarcados permitiu imagens de imunofluorescência (IF) de maior volume de distribuição de células imunes e células produtoras de vírus nos tecidos linfóides (Figura 7). A imunomarcação do tecido do cólon de um camundongo hu infectado pelo HIV para células T hCD3 + e HIV p24 revelou focos de células produtoras de vírus dispersas entre grandes regiões de tecido sem evidência de infecção (Figura 7A). Uma visão ampliada de focos de células produtoras de vírus revelou várias células produtoras de vírus próximas a células-alvo em potencial (Figura 7B). A autofluorescência do tecido (névoa vermelha) foi usada para visualizar toda a arquitetura do tecido, distinguindo populações específicas de células imunológicas dentro do tecido que coraram mais intensamente do que a autofluorescência (ovais vermelhos). Um modelo 3D de todo o volume da pilha Z LSFM mostrou a distribuição espacial dos focos de células produtoras de vírus dentro de uma região de tecido intacto e permitiu o mapeamento dos locais de produção do vírus em relação à arquitetura geral do tecido (Figura 7C). Surpreendentemente, os focos de células produtoras de vírus eram frequentemente intercalados entre grandes regiões de tecido sem evidência de produção de vírus. Esses resultados podem permitir a quantificação de parâmetros de distribuição viral e densidade de células infectadas em diferentes tecidos e em diferentes momentos de infecção ou resposta a diferentes tratamentos.

Figura 1: Fluxo de trabalho de limpeza, imunocoloração e imagem de tecido CUBIC e CLARITY típicos. Os tempos de clareamento CUBIC (superior) e CLARITY (inferior) podem variar muito, dependendo do tamanho e tipo de tecido. Para a limpeza do CLARITY, é necessária uma etapa de incubação adicional com meios correspondentes ao índice de refração antes da imunocoloração para verificar se o tecido está limpo. A imunocoloração geralmente leva 3 dias quando os anticorpos primários são conjugados com fluoróforos e 6 dias se os anticorpos secundários fluorescentes são necessários. As amostras podem ser visualizadas com um confocal ou LSFM. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Limpeza CÚBICA de amostras de tecido hu-mouse e NHP. Dependendo das diferentes densidades de heme e lipídios das amostras de tecido, o tempo necessário para limpar cada tipo de tecido varia. Por exemplo, o cólon e o duodeno normalmente requerem períodos relativamente curtos (~ 7 dias), enquanto o baço e o fígado podem levar mais tempo para se tornarem transparentes (~ 30 dias). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Comparação longitudinal de métodos de limpeza de tecidos em amostras humanas. CUBIC (painéis superiores) e CLARITY (painéis inferiores) eliminaram o baço de um indivíduo infectado pelo HIV em terapia antirretroviral. Ambos os métodos limparam adequadamente o tecido no dia 32 para imunocoloração e imagem. A etapa de descoloração para o método CUBIC reduz visivelmente a autofluorescência causada pela presença de heme contido nas amostras do baço. Clique aqui para ver uma versão maior desta figura.
{kind=link}

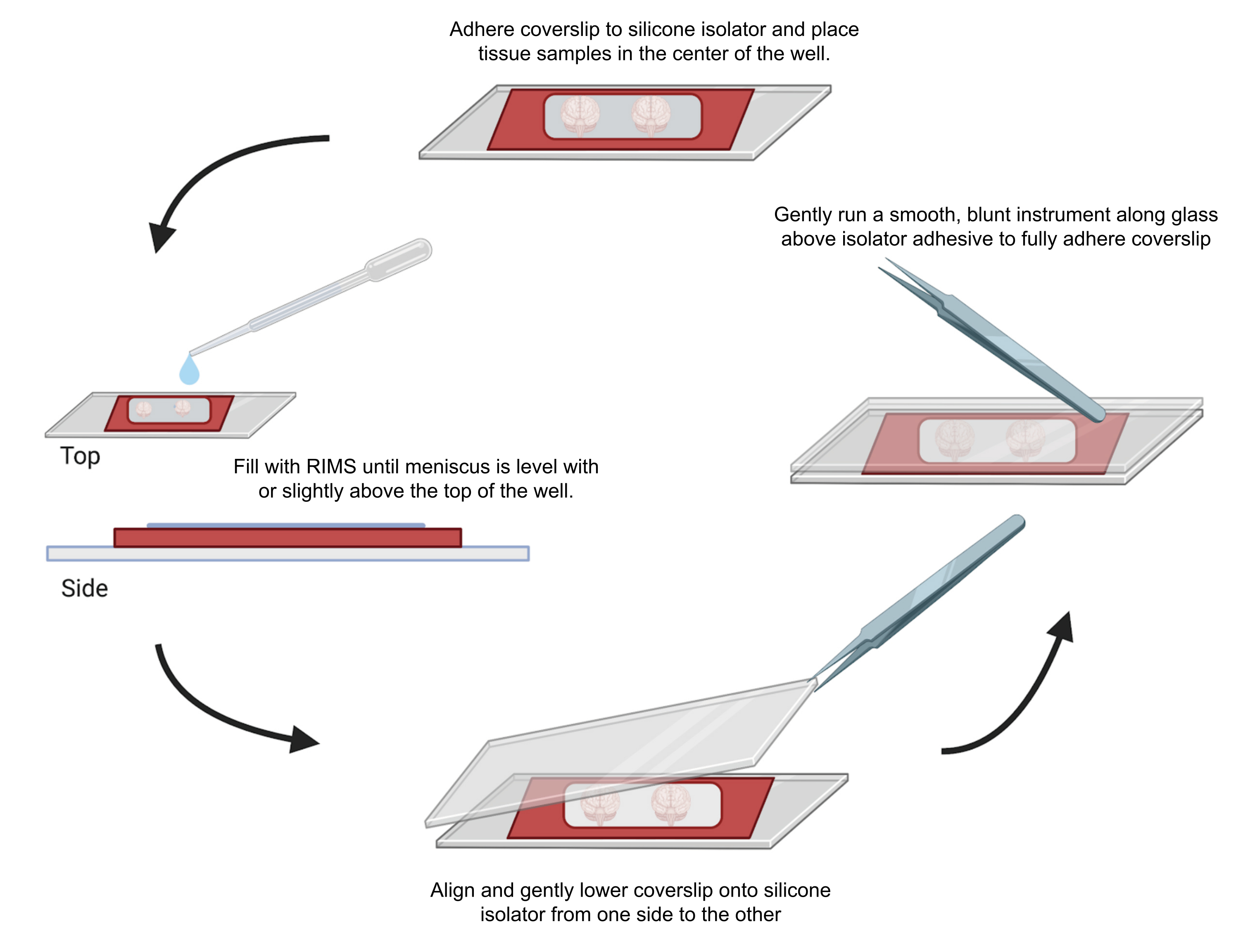
Figura 4: Montagem de amostras para microscopia confocal. As amostras foram montadas entre lamínulas separadas com isoladores de silicone adesivos de 0,5-1 mm. Isoladores de silicone foram aderidos à primeira lamínula e o tecido foi colocado no centro do poço (topo). O poço foi preenchido com RIMS até que o menisco estivesse nivelado ou ligeiramente acima do topo do poço (esquerda). A segunda lamínula foi cuidadosamente abaixada no lugar de um lado para o outro, evitando bolhas (parte inferior). As lamínulas foram totalmente aderidas ao isolador de silicone passando suavemente um instrumento rombudo ao redor do perímetro do poço (direita). As amostras foram fotografadas em um microscópio confocal padrão. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Microscopia confocal do baço humano limpo e intacto e dos linfonodos NHP. (A-D) O tecido humano infectado pelo HIV foi corado para HIV-1 p24 (verde), células T hCD3+ (vermelho) e núcleos (ciano). (E) Fatia Z confocal de linfonodo CUBIC limpo de um NHP infectado por SHIV 8 semanas após a infecção imunocorada para células T CD3+ (magenta), macrófagos CD68+ (mac/vermelho), SHIV p27 (verde) e núcleos (azul). O campo de visão contém células T, macrófagos e outros tipos de células, mas nenhuma evidência de células produtoras de SHIV (verde). F) Fatia Z confocal de uma região adjacente do mesmo linfonodo mostrando diferenças na densidade e número de células, juntamente com a presença de células T CD3+ produtoras de vírus (setas verdes) e macrófagos CD68+ (setas amarelas). (G-I) Vista ampliada das regiões selecionadas da coloração p27 de (F). As barras de escala são de 50 μm. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Volume da pilha Z e superfície reconstruída em 3D do baço humano infectado pelo HIV. (A) Imagem de projeção Z de 600 μm x 600 μm x 100 μm de pilha Z de tecido baço humano infectado pelo HIV corado para HIV-1 p24 (verde), células T hCD3 + (vermelho) e núcleos (ciano). (B) A mesma imagem de projeção Z sem coloração DAPI nuclear. (C) Modelo de superfície 3D reconstruído de fluorescência CD3 (vermelho) e p24 (verde) de todo o volume da pilha Z. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 7: LSFM e reconstrução 3D de volumes de superfície de tecidos infectados pelo HIV (A) Z-slice (1.000 μm x 1.000 μm) de cólon de um camundongo hu infectado pelo HIV imunomarcado para células T CD3 + (vermelho) e HIV p24 (verde). A névoa vermelha opaca representa a autofluorescência do tecido, enquanto os pontos vermelhos distintos indicam células T. As vilosidades são visíveis ao redor da periferia apontando para o lúmen central com vários focos de produção ativa de vírus (setas brancas) dispersos entre grandes áreas sem vírus. A caixa indica a região aproximada de interesse para o painel B. (B) Região ampliada do tecido mostrando células T hCD3+ produtoras de vírus individuais (amarelo) próximo a células T não infectadas (vermelho). A imagem foi girada e alterada para uma fatia Z próxima para mostrar um foco de células positivas p24 em um único plano Z. A autofluorescência vermelha de fundo mostra a arquitetura geral do tecido, além da coloração específica de células T hCD3+ (pontos vermelhos; setas brancas). (C) Modelo de superfície 3D do volume completo (1.000 μm x 1.000 μm x 200 μm) gerado com o software Imaris girado para mostrar focos de infecção pelo HIV (amarelo) em locais distintos do intestino. As setas brancas indicam focos individuais dentro do volume. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Os tecidos linfóides de interesse devem ser coletados rapidamente post-mortem e imediatamente colocados em tampões fixadores pré-resfriados para evitar necrose tecidual (tecido escuro ou preto) que pode afetar negativamente a coloração e a imagem. Após a colheita dos tecidos desejados, mergulhe imediatamente os tecidos em paraformaldeído (PFA) gelado a 4% a 8% durante a noite para fixação, o que também inativa possíveis patógenos associados às amostras. 4% de PFA é ideal para fixar amostras de LM, enquanto 8% de PFA pode preservar adequadamente os tecidos para LM e EM. Seguir esses procedimentos e armazenar amostras em fixador a 4 ° C no escuro pode preservar efetivamente os tecidos para imagens de LM por vários anos. Uma ressalva é que o armazenamento de longo prazo no fixador pode levar à introdução de artefatos de coloração, especialmente o mascaramento de antígenos, que é causado pela reticulação de proteínas adjacentes à proteína de interesse, o que pode obstruir a acessibilidade dos anticorpos de coloração ao epítopo13,14. Se os tecidos contiverem proteínas fluorescentes expressas endogenamente, tome medidas para evitar a exposição dos tecidos à luz sempre que possível durante todo o protocolo. Normalmente, as proteínas fluorescentes endógenas retêm a fluorescência por 6 a 12 meses após a fixação, mas as amostras de tecido individuais podem variar por períodos de tempo mais longos ou mais curtos. Se a fluorescência endógena for perdida devido à degradação da proteína, as proteínas fluorescentes podem ser detectadas usando um anticorpo primário específico para a proteína de interesse. A perfusão é outra opção para fixar rapidamente os tecidos antes da limpeza12; no entanto, devido a preocupações ao trabalhar com patógenos como o HIV, optou-se pela via de necropsia tecidual seguida de imersão em fixador gelado para preparar as amostras da forma mais segura possível.
Uma vantagem dos protocolos de limpeza à base de água descritos é que eles geralmente são mais suaves do que os protocolos de base orgânica, que às vezes podem danificar tecidos mais frágeis, como o fígado. Os protocolos de limpeza à base de água geralmente requerem mais tempo para atingir a limpeza completa da amostra (semanas vs. dias) em comparação com os protocolos de limpeza à base de orgânicos. Os protocolos CLARITY e CUBIC podem ser conduzidos mais rapidamente usando perfusão para limpar simultaneamente todos os órgãos dentro de um roedor11,12; no entanto, essa não era uma opção viável para PNH e autópsias humanas. As amostras processadas com CLARITY tendem a apresentar alguma expansão de volume, enquanto CUBIC revelou influência reduzida no volume de amostra9. Embora geralmente mais rápidos, muitos protocolos de limpeza de tecidos de base orgânica fazem com que os tecidos sofram encolhimento15, o que pode dificultar a detecção de detalhes de uma única célula ou subcelular em tecidos densos de células, como linfonodos e baço. A expansão induzida pela limpeza pode efetivamente aumentar a resolução da imagem, facilitando a observação de aspectos que seriam difíceis de observar no tamanho original do tecido. Alternativamente, o encolhimento do tecido pode efetivamente diminuir o tamanho geral da amostra, o que pode possibilitar a imagem de órgãos inteiros sem dissecção. Um benefício do protocolo CLARITY e CUBIC é que eles preservam a fluorescência de proteínas fluorescentes expressas endogenamente no tecido, mantendo-se passíveis de coloração por imunofluorescência11,12. A imunocoloração pode ser realizada usando métodos de limpeza de tecidos aquosos ou orgânicos; no entanto, a experiência pessoal mostrou uma proporção maior de compatibilidade de anticorpos usando protocolos à base de água em comparação com protocolos baseados em orgânicos. Os pesquisadores precisam considerar qual método de limpeza de tecido usar com base no(s) tecido(s) fotografado(s) e nas questões biológicas abordadas (por exemplo, imagens de órgãos inteiros versus imagens de regiões específicas de interesse). Não existe uma técnica universal de limpeza de tecidos que permita uma análise robusta e pronta para uso para todas as questões de imagem de grande volume e os métodos disponíveis exibem vantagens e desvantagens distintas, dependendo da aplicação biológica.
Ao realizar a coloração de anticorpos, vários aspectos precisam ser considerados. Como as amostras CLARITY são incorporadas em hidrogel de acrilamida, elas tendem a exigir tempos mais longos para incubação12. O tempo necessário para a incubação do anticorpo também depende do volume e da espessura de cada amostra. A maioria das amostras descritas aqui tinha ~ 2-3 milímetros de espessura e 3 dias foram suficientes para a coloração completa em todo o tecido. Se o objetivo for obter imagens de um cérebro de camundongo inteiro, o tempo de incubação do anticorpo pode levar 1 semana ou mais12. A escolha de um método de limpeza de tecido aquoso versus orgânico para imagens de imunofluorescência pode depender da compatibilidade de anticorpos. Em geral, para CUBIC ou CLARITY, a taxa de acerto para anticorpos que funcionam em células e tecidos cultivados é de ~ 70%. Seja utilizando um método de limpeza de tecido aquoso ou orgânico, é necessário avaliar a compatibilidade e eficácia de todos os anticorpos com o método específico usado. Conforme mostrado nesta seção de protocolo, a imunocoloração para amostras processadas CUBIC e CLARITY ocorre após o término da limpeza. Pelo contrário, esta etapa ocorre antes do procedimento de compensação para alguns protocolos de base orgânica, seguido de pós-fixação.
É extremamente importante que os tecidos estejam completamente imersos em um meio de imagem que corresponda ao seu índice de refração. Não fazer isso introduzirá aberrações esféricas durante a imagem e distorcerá a luz capturada durante a aquisição da imagem. Deve-se tomar cuidado para remover todas as bolhas de ar do meio de imagem ao montar amostras para confocal e LSFM, pois as bolhas podem interromper o caminho da luz para dentro ou para fora da amostra. As bolhas podem ser removidas manualmente com uma pipeta antes da montagem final da amostra. Para imagens de amostras mais espessas com um microscópio confocal, vários espaçadores de silicone podem ser colocados em camadas uns sobre os outros para acomodar tecidos com mais de 0,5 mm de espessura. Uma recomendação é equilibrar todos os tecidos no RIMS por várias horas a durante a noite enquanto montado no microscópio sem movimento adicional da amostra. O equilíbrio total do tecido e do meio de imagem evitará a mistura de soluções com índices de refração incompatíveis que podem gerar aberrações durante a imagem. É importante lembrar que, ao montar amostras de tecido limpas, não há um único método de montagem pronto para uso para obter imagens de todas as amostras em todos os microscópios. Este protocolo discute as opções de montagem de amostras que funcionaram de maneira ideal em um contexto, mas existem inúmeras abordagens para montagem de amostras, dependendo do microscópio individual usado e da questão biológica abordada. Essas abordagens podem incluir, mas não estão limitadas a, incorporar a amostra em agarose, suspender a amostra de um gancho ou linha de plástico correspondente ao índice de refração, usar adaptador de porco-espinho, imprimir em 3D um suporte de amostra ou anexar a amostra com adesivo a um prato de plástico.
Os microscópios confocais podem funcionar bem para obter imagens de volumes de tecido ~ 1 mm3-1 cm3. Para microscópios confocais, use uma objetiva de 2 a 10x para localizar inicialmente as regiões de interesse e adquirir pilhas Z de maior volume ou tecido inteiro com resolução de célula única. Mude para objetivas de 20-63x para adquirir imagens de alta resolução de regiões específicas de interesse com informações subcelulares. A objetiva ideal para imagens de tecidos CLÚBICOS e CLARITY é uma objetiva específica CLARITY/Scale que corresponde com precisão ao índice de refração do tecido e da solução de imagem. Se esse tipo de objetiva não estiver disponível, é ideal obter imagens de amostras com uma objetiva de imersão em glicerol ou óleo (por exemplo, objetiva de imersão em óleo LD LCI Plan-Apochromat 25 x 0,8 NA Imm Corr DIC M27: distância de trabalho = 0,57 mm) em vez de uma objetiva de ar. Isso minimizará a introdução de distorções ópticas devido a índices de refração incompatíveis durante a captura de imagem. As objetivas de 20 a 25x podem equilibrar a aquisição de imagens de maior volume com a obtenção de detalhes de coloração de células individuais em um ambiente de tecido complexo. É importante ressaltar que a maioria dos microscópios confocais contém módulos que permitem o ladrilho 3D de volumes de imagem. Esse tipo de aquisição de imagem pode idealmente gerar pilhas Z de maior volume que contêm informações subcelulares.
A imagem LSFM pode permitir a visualização 3D de populações de células específicas no contexto de grandes volumes de tecido (>1 cm3) e até órgãos inteiros. Durante os últimos 10 anos, a limpeza de tecidos combinada com LSFM se concentrou amplamente na compreensão da conectividade cerebral em roedores; no entanto, aplicações mais recentes incluem a visualização de paisagens metastáticas tumorais16, distribuição celular dentro de compartimentos anatômicos 9,17 e dispersão de patógenos18. Em comparação com as células cultivadas, a maioria dos eventos biológicos nos tecidos não é uniforme e o LSFM pode ser particularmente hábil para visualizar e quantificar a heterogeneidade espacial do tecido desses eventos (por exemplo, replicação do vírus, sinalização imunológica, distribuição celular, etc.).
Os conjuntos de dados 3D adquiridos via confocal ou LSFM podem ser pós-processados com várias plataformas de análise de imagem. O pacote de software Imaris pode ser usado para construção de superfícies, geração de animação 3D e quantificação de células; no entanto, existem vários sistemas de análise de imagem disponíveis que permitem pós-processamento e análise de imagem eficientes. O freeware ImageJ/Fiji19 é uma plataforma alternativa de processamento de imagem atraente acessível à maioria dos laboratórios, mas não existe um software de análise único que se destaque em todas as formas de análise e visualização de imagens. Muitos pacotes de software de análise de imagem podem ser proibitivamente caros se não estiverem disponíveis por meio de recursos de uso compartilhado. Finalmente, um aspecto crítico do LSFM ou grandes conjuntos de dados 3D confocais lado a lado é o gerenciamento de dados. Essas plataformas de imagem podem gerar arquivos massivos (>1 Tb) que exigem estações de trabalho de computador de última geração para visualização e quantificação de dados. Em última análise, esse fluxo de trabalho de imagem pode agilizar a aquisição e quantificação de populações de células espacialmente distintas em tecidos inteiros e é amplamente aplicável à maioria das fontes de tecidos e sistemas biológicos.
Divulgações
Os autores não têm conflitos de interesse a divulgar.
Agradecimentos
Obrigado às instalações centrais do Instituto de Biologia Genômica da Universidade de Illinois em Urbana-Champaign pelo uso de microscópios de fluorescência confocais e de folha de luz. Obrigado aos incríveis indivíduos da coorte "The Last Gift" para amostras de tecido humano, que foi financiada pelas seguintes doações: I147821, DA051915, AI131385 e P30 AI036214. Obrigado a Nancy Haigwood e Ann Hessell por amostras de tecido NHP infectadas com SHIV.
Materiais
| Name | Company | Catalog Number | Comments |
| Acrylamide Solution (in 0.1 M PBS, 40 mL in total) | |||
| 40% Acrylamide: 4 mL | Bio-Rad | 1610144 | |
| VA-044 Thermal Initiator: 0.1g | Fujifilm | 011-19365 | |
| CLARITY Blocking solution (in 0.1 M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Tween-20: 5 µL | Fisher Scientific | BP337-500 | |
| CLARITY wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Tween-20: 50 µL | Fisher Scientific | BP337-500 | |
| CUBIC Blocking solution (in 0.1M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Triton X-100: 5 µL | VWR | M143-1L | |
| CUBIC Reagent-1 (in 0.1M PBS, 50 mL in total) | |||
| N, N, N’, N’-tetrakis (2-hydroxypropyl) ethylenediamine: 12.5 g | Aldrich | 122262 | |
| Triton X-100: 7.5 g | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC Reagent-2 (in 0.1M PBS, 50 mL in total) | |||
| Sucrose: 25 g | Sigma | S1888-500G | |
| Sodium azide (in powder form): 10 g | Sigma | 71289-50G | |
| Sodium azide stock solution (in DI H2O, 50 mL in total) | Sigma | 71289-50G | |
| Triethanolamine: 5 g | Sigma | 90270-500mL | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| DAPI staining solution (0.5 µg/mL) | |||
| DAPI stock solution: 1 µL | |||
| Wash solution: 10 mL | |||
| DAPI stock solution (5 mg/mL) | |||
| DAPI powder: 5 mg | Sigma-Aldrich | D9542-1MG | |
| DMSO (100%): 1 mL | ThermoFisher | D12345 | |
| Imaging Media RI-2 (in dH2O) | |||
| 90% Histodenz | Sigma | D2158-100G | |
| 0.01% Sodium azide | Sigma | 71289-50G | |
| 0.02 Sodium Phosphate Buffer, pH 7.5 | Sigma-Aldrich | S9638-250G | |
| 0.1% Tween-20 | Fisher Scientific | BP337-500 | |
| Primary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Goat anti-HIV p24: 10 µL (1:200) | Creative Diagnostics | DPATB-H81692 | |
| Mouse anti-human CD68: 10 µL(1:200) | Dako | M0876 | |
| Rabbit anti-human CD3: 10 µL (1:200) | Dako | A0452 | |
| 8% SDS Solution (in 0.1 M PBS, 50 mL in total) | |||
| SDS powder: 4 g | Sigma-Aldrich | L3771-500G | |
| Secondary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Donkey anti-goat conjugated with AlexaFluor647: 2 µL | Invitrogen | A21447 | |
| Donkey anti-mouse conjugated with AlexaFluor594: 2 µL | Invitrogen | A21203 | |
| Donkey anti-rabbit conjugated with AlexaFluor488: 2 µL | Invitrogen | A21206 |
Referências
- Spalteholz, W., Barker, L. F., Mall, F. P. . Hand-Atlas of Human Anatomy. , (1907).
- Jacob, T., Gray, J. W., Troxell, M., Vu, T. Q. Multiplexed imaging reveals heterogeneity of PI3K/MAPK network signaling in breast lesions of known PIK3CA genotype. Breast Cancer Research and Treatment. 159 (3), 575-583 (2016).
- Kieffer, C., Ladinsky, M. S., Ninh, A., Galimidi, R. P., Bjorkman, P. J. Longitudinal imaging of HIV-1 spread in humanized mice with parallel 3d immunofluorescence and electron tomography. eLife. 6, 23282 (2017).
- Chung, K., Deisseroth, K. CLARITY for mapping the nervous system. Nature Methods. 10 (6), 508-513 (2013).
- Compton, A. A., Schwartz, O. They might be giants: Does syncytium formation sink or spread HIV infection. PLoS Pathogens. 13 (2), 2-8 (2017).
- Symeonides, M., et al. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses. 7 (12), 6590-6603 (2015).
- Sharova, N., Swingler, C., Sharkey, M., Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. The EMBO Journal. 24 (13), 2481-2489 (2005).
- Colby, D. J., et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nature Medicine. 24 (7), 923-926 (2018).
- Ladinsky, M. S., et al. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. eLife. 8, 46916 (2019).
- Buettner, M., Bode, U. Lymph node dissection--understanding the immunological function of lymph nodes. Clinical and Experimental Immunology. 169 (3), 205-212 (2012).
- Treweek, J. B., et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nature Protocols. 10, 1860-1896 (2015).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159, 911-924 (2014).
- Sompuram, S. R., Vani, K., Bogen, S. A. A molecular model of antigen retrieval using a peptide array. American Journal of Clinical Pathology. 125 (1), 91-98 (2006).
- Scalia, C. R., et al. Antigen masking during fixation and embedding, dissected. The journal of histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 65 (1), 5-20 (2017).
- Jing, D., et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Research. 28 (8), 803-818 (2018).
- Guldner, I. H., et al. An Integrative platform for three-dimensional quantitative analysis of spatially heterogeneous metastasis landscapes. Scientific Reports. 6, 24201 (2016).
- Muntifering, M., et al. Clearing for deep tissue imaging. Current Protocols in Cytometry. 86 (1), 38 (2018).
- DePas, W. H., et al. Exposing the three-dimensional biogeography and metabolic states of pathogens in cystic fibrosis sputum via hydrogel embedding, clearing, and rRNA labeling. mBio. 7 (5), 00796 (2018).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image. Nature Methods. 9 (7), 676-682 (2012).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados