
Method Article
3D-Visualisierung von Immunzellpopulationen in HIV-infizierten Geweben mittels Clearing-, Immunfärbung-, Konfokal- und Lichtblatt-Fluoreszenzmikroskopie
In diesem Artikel
Zusammenfassung
Die Gewebereinigung, kombiniert mit Immunfluoreszenzmikroskopie, ermöglicht die räumliche Visualisierung und Quantifizierung von Immunzellpopulationen und Virusproteinen in intaktem Gewebe. Die optische Schnittung von geklärtem Gewebe mit konfokaler und Lichtblatt-Fluoreszenzmikroskopie kann 3D-Modelle komplexer Gewebeumgebungen erzeugen und die räumliche Heterogenität während einer HIV-Infektion aufdecken.
Zusammenfassung
Das humane Immundefizienz-Virus (HIV), der Erreger des erworbenen Immunschwächesyndroms (AIDS), ist ein großes globales Gesundheitsproblem, da weltweit fast 40 Millionen Menschen infiziert sind und es kein allgemein zugängliches Heilmittel gibt. Trotz intensiver Bemühungen ist ein detailliertes Verständnis der Wechselwirkungen zwischen Viren und Wirtszellen in Geweben während der Infektion und als Reaktion auf die Therapie unvollständig. Um diese Einschränkungen zu beheben, werden die wasserbasierten Gewebereinigungstechniken CUBIC (Clear, Unobstructed Brain/Body Imaging Cocktails and Computational analysis) und CLARITY (Clear Lipid-exchange Acrylamide-hybridized Rigid Imaging/Immunostaining/in situ-hybridization-compatible Tissue hYdrogel) eingesetzt, um komplexe Virus-Wirt-Zell-Interaktionen in HIV-infizierten Geweben aus Tiermodellen und Menschen mittels konfokaler und Lichtblatt-Fluoreszenzmikroskopie sichtbar zu machen. Die optische Schnittung von intaktem Gewebe und die Bildanalyse ermöglichen eine schnelle Rekonstruktion der räumlichen Informationen, die in ganzen Geweben enthalten sind, und die Quantifizierung von Immunzellpopulationen während der Infektion. Diese Methoden sind auf die meisten Gewebequellen und verschiedene biologische Fragestellungen anwendbar, einschließlich Infektionskrankheiten und Krebs.
Einleitung
Der wachsende Bedarf an quantitativer räumlicher Gewebebildgebung in der biologischen Forschung führte in jüngster Zeit zur Entstehung von Gewebereinigungstechniken zur Erzeugung von großvolumigeren (mm3-cm 3) Bildern von intaktem Gewebe mit Einzelzellauflösung. Gewebe umfassen komplexe Organisationen von Biomolekülen mit einzigartig definierten Strukturen, Zusammensetzungen und Funktionen. Leider streuen, absorbieren oder emittieren viele Biomoleküle, die in Geweben vorhanden sind (z. B. Lipide und Chromophore), Licht, wenn sie mit Hilfe der Lichtmikroskopie abgebildet werden, was die Bildgebung großer Mengen erschwert. Darüber hinaus weisen Gewebe oft einen Brechungsindex auf, der nicht mit Standard-Bildgebungslösungen und optischen Linsen übereinstimmt, was zu optischen Verzerrungen während der Bildgebung führt. Ein optimaler Ansatz für die Bildgebung großer Gewebemengen mit einem Lichtmikroskop sollte darin bestehen, den Brechungsindex von Geweben, Bildgebungslösungen und Objektiven abzugleichen und gleichzeitig ein tiefes Eindringen von Licht in das Gewebe zu ermöglichen, ohne die biologischen Eigenschaften des intakten Gewebes während der Verarbeitung zu stören. Erste Versuche, die Unterschiede im Brechungsindex zwischen Geweben und bildgebenden Lösungen durch die Klärung undurchsichtiger Gewebeproben zu verringern, wurden von dem deutschen Anatomen Werner Spalteholz in den späten 1800er Jahren unternommen1. Bei dieser Gewebereinigungstechnik wurden aggressive chemische Lösungsmittel verwendet, die Gewebeproben beschädigen können, stellten aber dennoch die erste berichtete Bildgebung von intaktem Gewebe mit größerem Volumen dar. Moderne lichtmikroskopische Methoden, kombiniert mit Rechenleistung für die Bilderfassung und -analyse, haben in jüngster Zeit das Tissue Clearing als Methode zur Abbildung großer, intakter Gewebeproben mit Einzelzellauflösung wieder in Mode gebracht. In den letzten zwei Jahrzehnten sind Dutzende von fortschrittlichen Gewebereinigungstechniken entstanden, darunter sowohl organische als auch wasserbasierte, jede mit Stärken und Schwächen für spezifische Anwendungen.
Mit der 3D-Gewebebildgebung können komplexere biologische Wechselwirkungen untersucht werden, die in Zellkulturen nicht reproduzierbar sind. So wurden beispielsweise Zellsignalmuster2, räumliche Verteilungen verschiedener Zelltypen3 und Gehirnkonnektivität4 zuvor mit bildgebenden Verfahren des gesamten Gewebes/Organs quantitativ kartiert. Hier wird eine Anwendung von wasserbasierten Gewebereinigungsprotokollen beschrieben, um unterschiedliche HIV-Zielzellpopulationen in intakten HIV-infizierten lymphatischen Geweben während der aktiven Infektion zu reinigen, immunzu färben und zu visualisieren. Im Körper infiziert HIV überwiegend CD4+ T-Zellen und integriert eine Kopie seines Genoms in das Genom infizierter Wirtszellen. Anschließend kapert das Virus die infizierte Wirtszellmaschinerie, um sich selbst zu replizieren, was zur Virusverbreitung, zur Abtötung von Wirtszellen, zu Immunstörungen und zum langfristigen Fortschreiten von AIDS führt. Es ist wichtig zu beachten, dass das Verhalten infizierter T-Zellen in Gewebe- und Zellkulturen bemerkenswert unterschiedlich ist. Kultivierte CD4+ T-Zellen, die mit HIV inkubiert wurden, können eine massive HIV-induzierte Synzytie erzeugen, die Dutzende von Zellkernen umfassen kann5, während ähnliche Experimente mit primären CD4+ T-Zellen, die in 3D-Hydrogelen der extrazellulären Matrix (ECM) kultiviert wurden, oder Gewebeproben von HIV-infizierten humanisierten Mäusen (Hu-Mäusen) im Allgemeinen Synzytien mit 2-5 Zellkernenergeben 6. Das Verständnis der lokalen Zell-zu-Zell-Übertragung und der systemischen Verbreitung des Virus innerhalb von HIV-infizierten Personen ist wahrscheinlich noch komplizierter, da es um den Transport des Virus durch mehrere infizierte Zelltypen von Geweben über Blutgefäße zu neuen Geweben geht, wo freie Virionen und virusproduzierende Zellen Zugang zu einer großen Anzahl anfälliger Lymphozyten haben7. Diese Szenarien lassen sich derzeit nicht in Zellkultursystemen rekapitulieren, und Gewebe aus Tiermodellen und Menschen sind nach wie vor eine wichtige Ressource für das Verständnis der Viruspathogenese im Kontext eines komplexen Organismus mit einem funktionierenden Immunsystem.
Die derzeitigen antiretroviralen Therapien (ARTs) erhöhen die Lebenserwartung und -qualität von Menschen mit HIV (PWH) erheblich, indem sie die HIV-Replikation hemmen und das Fortschreiten der Krankheit zu AIDS stoppen. Leider eliminiert die ART latent infizierte Immunzellen, die eine Insertion des retroviralen Genoms enthalten und ruhig sind und keine aktiv Viren produzieren, nicht. Obwohl das Virus bei den meisten Personen mit ART im Blut nicht nachweisbar ist, steigt die Viruslast nach der Unterbrechung der ART schnell wieder an und das Fortschreiten der Krankheit setzt sich fort8. Die Persistenz der HIV-Infektion, die durch das latente Reservoir infizierter Zellen verursacht wird, stellt ein massives Hindernis für die Etablierung einer HIV-Heilung dar. Die Gewebereservoire von HIV sind nach wie vor wenig verstanden, und es ist von entscheidender Bedeutung, ein tieferes Verständnis dieser Reservoire in lymphatischen Geweben vor, während und nach der ART zu erlangen, um die Viruspathogenese vollständig zu charakterisieren und neuartige Behandlungen zu bewerten, die latent infizierte Zellen, die nicht aktiv Viren produzieren, effektiv eliminieren.
Hier wurden CUBIC3 und CLARITY9, zwei zuvor angepasste wasserbasierte Gewebereinigungsprotokolle, angewendet, um Immunzellpopulationen in zahlreichen intakten lymphatischen Geweben von HIV-infizierten Mäusen mit humanisiertem Immunsystem (hu-Mäuse), SIV/SHIV-infizierten nicht-menschlichen Primaten (NHP) und HIV-infizierten Menschen abzubilden. Diese Protokolle können sowohl an die konfokale als auch an die Lichtblatt-Fluoreszenzmikroskopie angepasst werden, abhängig von den Zielen der Bildgebung (höhere Auflösung im Vergleich zu größerem Volumen) und den verfügbaren Instrumenten. Obwohl die Lichtmikroskopie einzelne Virionen nicht auflösen kann, kann der Einsatz von Immunfluoreszenz Regionen im Gewebe identifizieren, die Viren und virusproduzierende Zellen enthalten, die mit Methoden mit höherer Auflösung weiter analysiert werden können. Die hier vorgestellten Methoden können so angepasst werden, dass sie nahezu jedes Gewebe im Körper mit Einzelzellauflösung sichtbar machen, um die räumlichen Beziehungen zwischen bestimmten Zelltypen unter verschiedenen Bedingungen während der Infektion zu quantifizieren, und sind leicht auf hochrelevante menschliche Patientenproben für die Untersuchung von Infektionskrankheiten oder Krebs übertragbar.
Protokoll
Alle Tierversuche wurden nach genehmigten institutionellen Tierhaltungsprotokollen durchgeführt. Alle menschlichen Gewebe wurden gemäß den anerkannten Ethikrichtlinien der institutionellen Humanforschung erworben.
1. Gewebeentnahme und -fixierung (gleiches gilt für CUBIC und CLARITY)
- Identifizieren und präparieren Sie das lymphatische Gewebe, wie zuvor beschrieben10.
- Entfernen Sie das lymphatische Gewebe mit einer Präparierschere und einer Pinzette innerhalb von Minuten nach dem Tod, wenn dies sicher möglich ist.
- Geben Sie die Gewebeproben in einen frisch hergestellten, eiskalten Fixierpuffer, der 8 % Paraformaldehyd (PFA) und 5 % Saccharose in 0,1 M Natriumcacodylat-Trihydrat enthält, um die Gewebeproben für die Lichtmikroskopie (LM), Elektronenmikroskopie (EM) oder Immun-EM angemessen zu konservieren. Alternativ können Sie die Proben für LM mit 4 % PFA in 0,1 M PBS fixieren. Fixieren Sie die Proben über Nacht, bevor Sie mit dem Räumvorgang beginnen, um eine vollständige Deaktivierung des Virus zu gewährleisten.
ACHTUNG: Paraformaldehyd ist giftig bei Hautkontakt und Einatmen und ist auch ein brennbarer Feststoff; Vorsichtig handhaben und in einem brennbaren Vorratsschrank aufbewahren. Natriumcacodylattrihydrat ist giftig, wenn es verschluckt oder eingeatmet wird. - Machen Sie ein Referenzbild des Gewebes, bevor Sie mit dem Reinigungsprozess beginnen.
HINWEIS: LM-Proben können unter diesen Bedingungen mindestens 1 Jahr gelagert werden. Um mit Proben zu arbeiten, die endogene fluoreszierende Proteine exprimieren, halten Sie die Proben in den folgenden Schritten immer im Dunkeln.
2. CUBIC Gewebereinigung
- Spülen Sie die lymphatischen Gewebeproben dreimal in sterilem 0,1 M PBS und schütteln Sie sie 15 Minuten lang bei Raumtemperatur, um die Entfernung von PFA bei jedem Pufferwechsel sicherzustellen.
HINWEIS: Entsorgen Sie die PFA-haltigen Flüssigkeiten gemäß den institutionellen Richtlinien. - Tauchen Sie die lymphatische Gewebeprobe 3 Tage lang unter leichtem Schütteln in CUBIC Reagenz-1 (siehe Materialtabelle) bei 37 °C. Nehmen Sie regelmäßig Referenzbilder auf, um den Entfärbungsprozess im Laufe der Zeit zu überwachen.
- Tauschen Sie das Reagenz-1 für weitere 3-4 Tage Eintauchen aus oder bis die Entfärbung des Gewebes abgeschlossen ist. Die Zeit, die für die Reinigung benötigt wird, hängt sowohl vom Volumen als auch von der Art des Gewebes ab. Um den Entfärbungsprozess des Gewebes zu beschleunigen, frischen Sie das CUBIC Reagenz-1 täglich auf und verwenden Sie größere Volumina.
- Waschen Sie die lymphatischen Gewebeproben dreimal mit 0,1 M PBS für 30 min bei Raumtemperatur unter leichtem Schütteln.
- Tauchen Sie die lymphatischen Gewebeproben 2-7 Tage lang in CUBIC Reagenz-2 (siehe Materialtabelle) bei 37 °C unter leichtem Schütteln oder bis eine vollständige Transparenz erreicht ist. Wenn die Proben keine vollständige Transparenz erreichen, wiederholen Sie die Schritte 2.2-2.5, bis der Löschvorgang nicht mehr voranschreitet. Nehmen Sie regelmäßig Referenzbilder auf, um den Räumvorgang im Laufe der Zeit zu überwachen.
- Waschen Sie die lymphatischen Gewebeproben dreimal mit 0,1 M PBS für 30 min bei Raumtemperatur unter leichtem Schütteln.
- Lagern Sie die Proben in CUBIC Reagenz-2 mit 0,01 % Volumen/Volumen (V/V) Natriumazid im Dunkeln (siehe Materialtabelle).
HINWEIS: Die Proben können mit dieser Methode mindestens 6 Monate gelagert werden.
ACHTUNG: Natriumazid ist hochgiftig und stellt eine ernsthafte Gefahr für das Einatmen dar. Der Kauf von verdünnten Lösungen mit 5% Natriumazid oder weniger wird empfohlen.
3. Blockierung und Immunfärbung von kubischen Proben
- Waschen Sie die lymphatischen Gewebeproben dreimal mit 0,1 M PBS für jeweils 30 min bei Raumtemperatur unter leichtem Schütteln.
- Um mit einem konfokalen Mikroskop abzubilden, schneiden Sie das Gewebe mit einer Gewebeschneider-Matrize in ~0,5-1 mm dicke Scheiben. Um eine Lichtblatt-Fluoreszenzmikroskopie (LSFM) durchzuführen, blockieren Sie den gesamten Gewebebereich.
- Die Proben werden über Nacht bei 4 °C unter Schütteln mit 5 mL CUBIC-Blockierungslösung verstopft (siehe Materialtabelle). Wenn Sie mit NHP oder menschlichen Proben arbeiten, verwenden Sie Anti-Human-FcR. Wenn Sie mit Mausproben arbeiten, verwenden Sie Anti-Maus-FcR in der Blockierungslösung.
- Färben Sie die Proben mit 5 mL Primärantikörpern (siehe Materialtabelle) in Blockierungslösung (ohne speziesspezifisches FcR) für 3 Tage bei Raumtemperatur unter Schütteln (Optional: Zentrifugieren Sie den konzentrierten Antikörperstamm bei 2.300 x g für 5 min vor der Verwendung, um die Zugabe von aggregierten Antikörpern zu reduzieren).
- Waschen Sie die gefärbte Probe bei Raumtemperatur unter Schütteln für einen Zeitraum von insgesamt mindestens 5 Stunden mit mindestens fünfmaligem Austausch des Waschlösungspuffers (siehe Materialtabelle).
- Färben Sie die Proben mit Sekundärantikörpern (siehe Materialtabelle) in Blockierlösung (ohne speziesspezifisches FcR) für 3 Tage bei Raumtemperatur unter Schütteln (Optional: Zentrifugieren Sie die Antikörper bei 2.300 x g für 5 min vor der Verwendung, um die Antikörperaggregation zu minimieren).
- Waschen Sie die verschmutzte Probe fünfmal mit Waschlösung bei Raumtemperatur und schütteln Sie sie insgesamt mindestens 5 Stunden lang.
- Färben Sie die Proben mit 5 mL DAPI-Färbelösung (siehe Materialtabelle) auf jede Gewebeprobe und inkubieren Sie sie 10 Minuten lang bei Raumtemperatur. Lassen Sie die Proben in der DAPI-Färbelösung im Dunkeln bei 4 °C verbleiben, um sie später zu bebildern.
- Waschen Sie die lymphatischen Gewebeproben dreimal bei Raumtemperatur mit Waschlösung und schütteln Sie sie jeweils 30 Minuten lang.
- Tauchen Sie die gefärbte Probe über Nacht bei Raumtemperatur im Dunkeln in CUBIC Reagenz-2, bevor Sie die Probe einbetten.
4. KLARHEIT Gewebereinigung
- Spülen Sie die lymphatischen Gewebeproben dreimal in sterilem 0,1 M PBS und schütteln Sie es jeweils 15 Minuten lang bei Raumtemperatur, um PFA zu entfernen.
- Die Gewebeproben werden in 15 mL frisch hergestellte Acrylamidlösung gegeben und über Nacht bei 4 °C unter leichtem Rühren inkubiert (siehe Materialtabelle).
ACHTUNG: Unpolymerisiertes Acrylamid ist ein starkes Neurotoxin und wird leicht über die Haut aufgenommen. Vermeiden Sie jeglichen Kontakt mit der Haut und spülen Sie sie sofort aus, wenn es zu einem Kontakt kommt. - Lassen Sie die Gewebeproben auf Raumtemperatur erwärmen.
- OPTIONAL: Entgasen Sie die Gewebeproben, indem Sie Stickstoff 1 min lang in die Acrylamidlösung blasen. Achten Sie auf eine niedrige Durchflussrate, die das Spritzen von giftigem unpolymerisiertem Acrylamid (~1-2 Blasen/s) vermeidet.
- Legen Sie die Gewebeproben für 1-3 h in ein 37 °C heißes Wasserbad, um zu polymerisieren, und kehren Sie sie alle 15 Minuten um. Entnehmen Sie die Proben, sobald eine auffällige Polymerisation festgestellt wird, die durch eine viskose Flüssigkeit, das Auftreten von Schleren-Linien beim Mischen oder die Bildung einer klaren Kapsel um das Gewebe angezeigt wird.
HINWEIS: Wenn eine vollständige Polymerisation der Acrylamidlösung auftritt, schneiden Sie das überschüssige Hydrogel aus der Probe ab und setzen Sie das Protokoll fort. - Waschen Sie die Gewebeproben dreimal für jeweils 30 Minuten bei Raumtemperatur mit sterilem 0,1 M PBS unter leichtem Schütteln, um die Acrylamidlösung zu entfernen.
- Legen Sie die Gewebeproben in 15 mL mit 8 % SDS in 0,1 M PBS bei 37 °C und schütteln Sie sie 2-5+ Tage lang, um eine Reinigung zu ermöglichen. Frischen Sie die 8%ige SDS-Lösung regelmäßig auf und verwenden Sie bei Bedarf bis zu 50 ml der Lösung, um die Beseitigung zu beschleunigen. Stoppen Sie den Räumvorgang, wenn die Proben visuell transparent sind oder nicht mehr fortschreiten. Nehmen Sie regelmäßig Referenzbilder auf, um den Räumvorgang im Laufe der Zeit zu überwachen.
- Waschen Sie die Gewebeproben mit sterilem 0,1 M PBS fünfmal über 1 Tag bei Raumtemperatur unter leichtem Schütteln.
- Bewahren Sie die Proben vorübergehend in 0,1 M PBS (plus 0,01 % Volumen/Volumen (v/v)NaN 3 für eine längerfristige Lagerung) im Dunkeln auf, bis sie für die Abbildung der endogenen Fluoreszenz bereit sind.
- Legen Sie das Gewebe in 5 ml Bildgebungsmedium RI-2 (siehe Materialtabelle). Über Nacht bei Raumtemperatur im Dunkeln inkubieren, um die Vollständigkeit des Reinigungsprozesses vor der Immunfärbung zu überprüfen. Nehmen Sie Referenzbilder auf, um die Gewebetransparenz zu überwachen.
5. Blockierung und Immunfärbung von CLARITY-Proben
HINWEIS: Diese Schritte ähneln dem Blockieren und Immunfärben von CUBIC-geklärtem Gewebe, verwenden jedoch unterschiedliche Formulierungen zum Blockieren, Waschen und Färben von Lösungen.
- Waschen Sie die lymphatischen Gewebeproben dreimal mit 0,1 M PBS für jeweils 30 min bei Raumtemperatur unter leichtem Schütteln.
- Um Bilder mit einem konfokalen Mikroskop zu machen, schneiden Sie das Gewebe mit einem 0,5-mm-Gewebeschneider und einer Matrix in ~0,5-1 mm dicke Scheiben. Um LSFM durchzuführen, blockieren Sie die gesamte Gewebeprobe.
- Die Proben werden über Nacht mit 5 mL CLARITY Blockierungslösung (siehe Materialtabelle) bei 4 °C unter Schütteln verstopft.
- Färben Sie die Proben mit 5 mL Primärantikörpern (siehe Materialtabelle) in Blockierungslösung (ohne speziesspezifisches FcR) für 3 Tage bei Raumtemperatur unter Schütteln (Optional: Zentrifugieren Sie die Antikörper bei 2.300 x g für 5 min vor der Verwendung, um die Antikörperaggregation zu minimieren).
- Waschen Sie die verschmutzte Probe fünfmal mit der Waschlösung bei Raumtemperatur und schütteln Sie sie insgesamt mindestens 5 Stunden lang (siehe Materialtabelle).
- Färben Sie die Proben mit 5 mL Sekundärantikörpern (siehe Materialtabelle) in Blockierlösung (ohne speziesspezifisches FcR) für 3 Tage bei Raumtemperatur unter Schütteln (Optional: zentrifugieren Sie die Antikörper vor der Verwendung 5 min lang bei 2.300 x g , um die Antikörperaggregation zu minimieren). Um die Gesamtlänge des Protokolls zu verkürzen, verwenden Sie primäre Antikörper, die mit Fluorophoren konjugiert sind, um eine Inkubation mit Sekundärantikörpern zu vermeiden.
- Waschen Sie die verschmutzte Probe fünfmal mit der Waschlösung bei Raumtemperatur und schütteln Sie sie insgesamt mindestens 5 Stunden lang.
- Färben Sie die Proben mit 5 mL DAPI-Färbelösung (siehe Materialtabelle) auf jede Gewebeprobe und inkubieren Sie sie 10 Minuten lang bei Raumtemperatur. Lassen Sie die Proben im Dunkeln in der DAPI-Färbelösung bei 4 °C bleiben, um sie später zu bebildern.
- Waschen Sie die lymphatischen Gewebeproben dreimal bei Raumtemperatur mit der Waschlösung und schütteln Sie sie jeweils 30 Minuten lang.
- Legen Sie das Gewebe in 5 ml Bildgebungsmedium RI-2 (R.I. = 1,46) und inkubieren Sie es über Nacht bei Raumtemperatur im Dunkeln, bevor Sie die Probe einbetten (siehe Protokollschritt 6 und 7).
6. Befestigung und Bildgebung von freigegebenen Gewebeproben für die konfokale Mikroskopie
- Ziehen Sie eine Seite der Schutzschicht eines selbstklebenden Silikonisolators ab.
- Kleben Sie ein Mikroskop-Deckglas (22 mm x 40 mm, 0,25 mm dick) auf die geschälte Seite des Silikonisolators, um einen flüssigkeitsdichten Raum für die Probe zu schaffen.
- Ziehen Sie die andere Seite der Schutzschicht des selbstklebenden Silikonisolators ab.
- Legen Sie die Probe für die Bildgebung in die Mitte des Silikonisolators und fügen Sie dann je nach Bedarf CUBIC Reagenz-2 oder das Bildgebungsmedium RI-2 hinzu, bis die Flüssigkeitsoberfläche so hoch wie der Rand des Isolators ist.
- Um das Einfangen von Luftblasen im Silikonisolator zu minimieren, richten Sie das zweite Deckglas aus und schichten Sie es vorsichtig von einer Seite mit einer EM-Pinzette nach unten. Wischen Sie überschüssige Flüssigkeit ab. Drücken Sie das Deckglas vorsichtig mit der Rückseite der Pinzette um die Probenvertiefung(en), um den Klebstoff zu versiegeln. Lagern Sie die eingefassten Proben horizontal im Dunkeln.
HINWEIS: Proben können Wochen bis Monate nach dem Einhängen abgebildet werden. Die Bildqualität nimmt jedoch im Allgemeinen mit der Zeit ab. - Legen Sie den montierten Objektträger auf den Mikroskoptisch und lokalisieren Sie die Probe mit weißem Licht und einem Objektiv mit geringerer Vergrößerung (2-10x).
- Richten Sie das Fluoreszenzerfassungsprofil basierend auf den einzelnen ausgewählten Fluorophoren ein.
HINWEIS: Es wird empfohlen, separate Fluorophorkanäle einzeln zu erwerben. Dies führt zu einer längeren Erfassungszeit, reduziert aber die spektrale Überlappung und die Erfassung von unspezifischem Fluoreszenzsignal. Ein gängiges Fluorophorprofil kann DAPI (450 nm), Alexa488, Alexa594 und Alexa647 (oder verwandte Kombinationen) umfassen, um die spektrale Überlappung während der Bildaufnahme zu minimieren. - Wählen Sie ein geeignetes Vergrößerungsobjektiv, um interessante Bereiche abzubilden. Verwenden Sie Objektive mit geringerer Vergrößerung (2-10x) für die Bildgebung größerer Volumina oder Ganzgewebe mit Einzelzellauflösung und Objektive mit höherer Vergrößerung (20-63x) für eine höher aufgelöste Visualisierung subzellulärer Details in geklärtem Gewebe. Passen Sie den Brechungsindex von Objektiven, Bildgebungsmedien und Gewebe so genau wie möglich an, um die Einführung optischer Verzerrungen während der Bildaufnahme zu minimieren.
- Wählen Sie eine Schrittweite für die Z-Stapel-Erfassung. Für Objektive mit geringerer Vergrößerung (2-10x) wählen Sie eine Schrittweite von ~3-5 μm, um die Fluoreszenz einer einzelnen Zelle in mehreren kontinuierlichen Z-Schichten für die 3D-Modellierung zu detektieren und gleichzeitig die Gesamterfassungszeit und die Gesamtdateigröße zu reduzieren. Für Objektive mit höherer Vergrößerung (20-63x) wählen Sie eine Schrittweite von ~1 μm oder weniger, um den Verlust subzellulärer Informationen zwischen den einzelnen Z-Schichten zu minimieren.
- Zoomen Sie das Sichtfeld, um den gesamten abzubildenden Gewebebereich in den Dimensionen X und Y mit so wenig unbesetztem Bereich wie möglich zu visualisieren. Legen Sie die oberen und unteren Erfassungskoordinaten der Z-Stufe fest, die den gesamten zu bebildenden Interessenbereich umfassen.
- Erfassen Sie die Z-Stack-Images. Speichern und exportieren Sie die Datei für die Nachbearbeitung mit einer beliebigen Bildanalysesoftware. Konvertieren Sie bei bestimmten Software-Suites die Dateien in bestimmte Dateitypen (z. B. .tiff, .ome-tiff, .jpeg usw.). Führen Sie die Konvertierung mit einer beliebigen Mikroskop-Bilderfassungssoftware oder Bildanalyse-Freeware (z. B. ImageJ/Fiji) durch.
7. Einbetten und Abbilden der Proben in einer LSFM-Kammer oder Küvette
- Füllen Sie die Bildgebungskammer mit CUBIC Reagent-2 oder RI-2, je nach verwendetem Protokoll. Vermeiden Sie die Bildung von Blasen beim Umfüllen der Flüssigkeit. Entfernen Sie die überschüssigen Blasen mit einer Pipette.
- Tauchen Sie die Probe in die Bildgebungskammer ein und schränken Sie die Bewegung der Probe ein.
HINWEIS: Je nach verwendetem Mikroskop kann dies das Einbetten der Probe in Agarose, das Aufhängen der Probe an einem Haken oder Stachelschweinadapter, den 3D-Druck eines Probenhalters oder das Befestigen der Probe mit Klebstoff an einer Kunststoffschale umfassen. - Platzieren Sie das Objektiv in der Bildgebungslösung und konzentrieren Sie sich auf die Probe. Lassen Sie die eingefasste Probe mehrere Stunden oder über Nacht in der Bildgebungskammer, um eine vollständige Äquilibrierung der Lösungen und des Gewebes in der Küvette zu ermöglichen.
- Erfassen Sie den Z-Stapel des interessierenden Bereichs (siehe Schritte 6.7-6.11 für die Bildaufnahme).
HINWEIS: Dieser Ansatz ermöglicht die Bildgebung von Gewebevolumina von mehr als 1 cm3 mit Einzelzellauflösung.
8. Oberflächenrekonstruktion und Zellquantifizierung mit der Bildanalysesoftware Imaris
HINWEIS: Diese Schritte sind spezifisch für die Bildanalysesoftware Imaris, aber ähnliche Bildverarbeitungsschritte können mit anderen Software-Suiten (z. B. ImageJ/Fiji, Aivia, Arivis, Amira usw.) durchgeführt werden.
- Verwenden Sie den Imaris File Converter, um die Z-Stack-Image-Datei in das native Imrais-Format .ims zu konvertieren. Dies ermöglicht eine schnellere Dateikonvertierung und minimiert gleichzeitig Konvertierungsfehler und potenzielle Softwareprobleme nach dem Öffnen.
HINWEIS: Einige neuere LSFMs ermöglichen es dem Benutzer, Dateien direkt im .ims-Format zu speichern. - Ziehen Sie die zu analysierende .ims-Datei in den Arena-Bereich der Imaris-Software. Passen Sie den Kontrast oder die Intensität der einzelnen Farbkanäle über das Bedienfeld "Anzeigeanpassung" an. Klicken Sie oben links auf das Symbol Neue Flächen hinzufügen .
- Klicken Sie auf Weiter: Quellkanal (das blaue Symbol mit einem Pfeil nach rechts). Wählen Sie den Quellkanal der zu konstruierenden Fläche aus. Ändern Sie die anderen Parameter nicht.
- Klicken Sie auf Weiter: Schwellenwert (das blaue Symbol mit einem Pfeil nach rechts).
- Um den Schwellenwert (absolute Intensität) anzupassen, ziehen Sie die Linie des Schwellenwerts nach links oder rechts. Aktivieren Sie " Berührende Objekte teilen " und geben Sie den durchschnittlichen Zelldurchmesser in Mikrometern als Teilungsstandard ein, damit das System viele Punkte als Ursprung für jede einzelne Oberfläche ergibt.
- Fügen Sie keine Fluoreszenzsignale hinzu, die zu klein oder zu hell sind, da sie potenzielle Färbungen oder Mikroskopartefakte darstellen können. Fügen Sie nur die Punkte hinzu, die akzeptable Größen und Fluoreszenzintensitäten haben, indem Sie den durchschnittlichen Zelldurchmesser entsprechend ändern.
HINWEIS: Der durchschnittliche Zelldurchmesser variiert je nach Gewebe oder Zelltyp, liegt aber im Allgemeinen zwischen 5 und 15 μm.
- Fügen Sie keine Fluoreszenzsignale hinzu, die zu klein oder zu hell sind, da sie potenzielle Färbungen oder Mikroskopartefakte darstellen können. Fügen Sie nur die Punkte hinzu, die akzeptable Größen und Fluoreszenzintensitäten haben, indem Sie den durchschnittlichen Zelldurchmesser entsprechend ändern.
- Klicken Sie auf Weiter: Oberflächen klassifizieren (das blaue Symbol mit einem nach rechts zeigenden Pfeil ist beschriftet). Passen Sie die einzuschließenden Flächen an, indem Sie die Linie des Schwellenwerts nach links oder rechts ziehen. Stellen Sie sicher, dass die Oberflächen dem rohen Fluoreszenzsignal genau entsprechen, während das Fluoreszenzsignal von den einzelnen Zellen getrennt wird.
- Klicken Sie auf Fertigstellen: Alle Erstellungsschritte ausführen und Beenden Sie den Assistenten (das grüne Symbol mit zwei Pfeilen, die nach rechts zeigen, beschriftet). Die Oberfläche ist offiziell gebaut.
- Klicken Sie auf das sechste Symbol mit der Bezeichnung Statistik im linken Bereich, um die Anzahl der Zellen anzuzeigen, in diesem Fall die Anzahl der Oberflächen für den spezifischen analysierten Farbkanal.
- Stellen Sie sicher, dass die vier Variablen Anzahl der getrennten Komponenten pro Zeitpunkt, Anzahl der Oberflächen pro Zeitpunkt, Gesamtzahl der getrennten Komponenten und Gesamtzahl der Oberflächen dieselbe Zahl haben, d. h. die Zellenanzahl dieses Farbkanals.
Ergebnisse
Bei der Gewebereinigung wird konserviertes Gewebe mit chemischen Cocktails behandelt, um undurchsichtige Biomoleküle aus dem Gewebe zu extrahieren und gleichzeitig die Gewebearchitektur zu erhalten. Diese Gewebereinigungslösungen gleichen den Brechungsindex des Gewebes mit dem umgebenden Bildgebungsmedium ab, um optische Verzerrungen zu minimieren, das Signal-Rausch-Verhältnis tief im Gewebe zu verbessern und die Autofluoreszenz im Hintergrund zu minimieren. Zwei wasserbasierte Protokolle für die optische Gewebereinigung, CUBIC3 und CLARITY9, wurden verwendet, um konservierte HIV/SIV-infizierte Hu-Maus-, nicht-menschliche Primaten- und menschliche Gewebeproben vor der Immunfluoreszenzfärbung und Bildgebung mit konfokaler und Lichtblatt-Fluoreszenzmikroskopie zu reinigen.
Für das CUBIC-Protokoll wurde das fixierte Gewebe mit PBS gewaschen, um Fixiermittel zu entfernen, und in CUBIC Reagenz-1 getaucht, eine basische gepufferte Lösung von Aminoalkoholen, die Chromophore wie Häm eluiert, was zu einer Entfärbung und Entfettung des Gewebes führt (Abbildung 1, oben). Kleinere Gewebevolumina (~mm 3) können nach 3 Tagen Behandlung mit CUBIC Reagenz-1 entfärbt werden, aber größere Gewebevolumina (~cm 3) oder Gewebe mit einer großen Häm-Menge (wie Leber, Milz oder Herz) erfordern längere Inkubationszeiten und Lösungsvolumina (>1 Monat und ~50 ml) sowie einen häufigen Austausch der Lösung alle 2-3 Tage. Nach der Entfärbung wurden die Gewebe gewaschen und in CUBIC Reagenz-2 gelegt, eine Saccharose-haltige Lösung mit einem Brechungsindex von etwa 1,48-1,49, die dem Brechungsindex des Gewebes entspricht und die Lichtdurchlässigkeit erhöht. Das gereinigte Gewebe wurde immungefärbt und in einer Lösung aus CUBIC Reagenz-2 montiert, bevor es mit einem konfokalen oder Lichtblattmikroskop fotografiert wurde. Die Auswirkungen des CUBIC-Clearing-Verfahrens wurden für mehrere Hu-Maus- und NHP-Gewebe unterschiedlicher Größe und Konzentration von Chromophoren abgebildet (Abbildung 2). Durch die optische Klärung wurden die Gewebe für das bloße Auge sichtbar gemacht, so dass Rasterlinien und Text auf Papierbögen "durch" das Gewebe gesehen werden konnten. Chromophorreiche Gewebe wie Milz, Leber, Knochenmark und Herz verfärben sich möglicherweise nicht vollständig, bleiben aber für die Immunfärbung und Bildgebung geeignet (Abbildung 2 und Abbildung 5).
Für das CLARITY-Protokoll wurden fixierte Gewebe mit PBS gewaschen, um Fixiermittel zu entfernen, und dann über Nacht bei 4 °C in einer 40%igen Acrylamidlösung mit einem thermischen Initiator inkubiert, um kovalente Bindungen zwischen Proteinen in der Probe und Monomeren von Acrylamid zu bilden (Abbildung 1, unten). Am folgenden Tag, nachdem das Gewebe auf Raumtemperatur äquilibriert und anschließend in einem 37 °C warmen Wasserbad erwärmt worden war, wurde die Acrylamidpolymerisation eingeleitet und die Probe schnell in ein Hydrogel eingehüllt. Die Probe wurde über einen Zeitraum von 2-5 Tagen mit einer Lösung von 8% SDS behandelt, um undurchsichtige Lipide zu entfernen. Unmittelbar vor der Fluoreszenzfärbung wurde die Probe in eine Brechungsindex-Matching-Lösung (RIMS) für CLARITY (Imaging Media RI-2) getaucht, die 90 % nichtionisches Dichtegradientenmedium enthielt. Bei Geweben, die große Mengen an Häm enthalten, kann am Ende des Entfettungsschritts 9,11,12 ein Entfärbungsschritt hinzugefügt werden. Der Verlauf des CUBIC und CLARITY Clearings wurde an verschiedenen Schnitten derselben menschlichen Milzprobe verglichen (Abbildung 3). CLARITY Clearing erzeugt ein sichtbares Polyacrylamid-Gel, das die Lösung umhüllt und typischerweise eine reduzierte Entfärbung im Vergleich zu CUBIC Clearing aufweist, es sei denn, es wird ein zusätzlicher Entfärbungsschritt hinzugefügt 9,12.
Anschließend wurde in beiden Protokollen geklärtes, intaktes Gewebe immungefärbt, um spezifische Immunzellpopulationen nachzuweisen. Die Proben wurden gewaschen, mit einem α-FcR-haltigen Reagenz blockiert, um die unspezifische Antikörperbindung zu reduzieren, und 3 Tage lang gefärbt, wenn ein primärer Antikörper verwendet wurde, der direkt an ein Fluorophor konjugiert war. Alternativ wurden die Proben 3 Tage lang mit einem unkonjugierten Primärantikörper gefärbt, gefolgt von weiteren 3 Tagen mit einem Sekundärantikörper, der an ein Fluorophor konjugiert war. Das Gewebe wurde erneut gewaschen und dann über Nacht bei 4 °C mit DAPI-Färbung zur nuklearen Visualisierung inkubiert. Die Proben wurden gewaschen und entweder in CUBIC Reagenz-2 für 24-36 h oder Imaging Media RI-2 (CLARITY) über Nacht im Dunkeln inkubiert. Für die konfokale Mikroskopie wurden Gewebe vor der Bildgebung in den entsprechenden RIMS auf einen Objektträger montiert (Abbildung 4). Für die Lichtblatt-Fluoreszenzmikroskopie (LSFM) wurden die Proben vor der Bildgebung über Nacht vollständig mit RIMS in eine Bildgebungsküvette getaucht.
Die konfokale Mikroskopie von intaktem, geklärtem und immungefärbtem lymphatischem Gewebe ermöglichte die gleichzeitige Visualisierung mehrerer fluoreszierender Signale, einschließlich Zellkernen, Immunzellmarkern und HIV/SIV CA (Kapsid)-Proteinen (Abbildung 5). Virusproduzierende Zellen wurden durch Fluoreszenzkolokalisation von Immunzellmarkern und HIV-Proteinen bestimmt. Gereinigte und gefärbte HIV-infizierte menschliche Milz zeigte mehrere CD3+ T-Zellen, die mit HIV p24 kolokalisiert waren, was auf das Vorhandensein von virusproduzierenden Zellen in einer Region intakten Gewebes hinweist (Abbildung 5A-D). Geklärte und immungefärbte SHIV-infizierte NHP-Lymphknoten zeigten die Verteilungen von CD3+ T-Zellen und CD68+ Makrophagen in Geweberegionen, in denen kein Virus nachgewiesen wurde (Abbildung 5E), zusätzlich zu Regionen mit zahlreichen virusproduzierenden Zellen (Abbildung 5F). Diese Ergebnisse zeigten, dass virusproduzierende Zellen aus verschiedenen Gewebequellen von anderen Zellen innerhalb eines bestimmten Sichtfeldes unterscheidbar waren und die Detektion seltener biologischer Ereignisse in einer komplexen Gewebeumgebung ermöglichten.
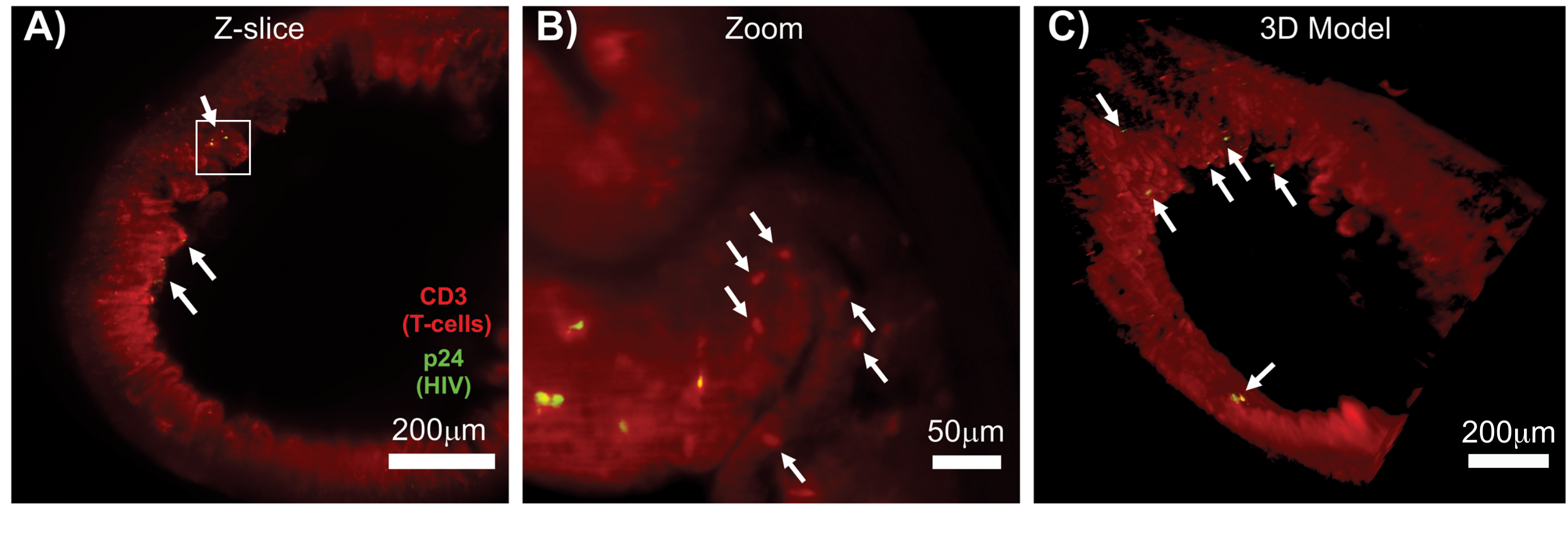
Optische Schnitte von geklärtem Gewebe mit einem konfokalen Mikroskop wurden angewendet, um Z-Stacks und 3D-Oberflächenmodelle zu generieren, die die zelluläre Heterogenität während einer HIV-Infektion aufzeigten (Abbildung 6). Die Z-Stacks wurden mit der Imaris Software Suite (Abbildung 6A) zu einem Z-Projektionsbild rekombiniert und der DAPI-Kernkanal wurde entfernt, um eine klare Visualisierung der CD3+ T-Zellen und der Fluoreszenz des HIV-Kapsidproteins (p24) über das gesamte Gewebevolumen zu ermöglichen (Abbildung 6B). Die Z-Projektionsfluoreszenz wurde mit der Imaris-Software automatisiert segmentiert, um ein rekonstruiertes 3D-Oberflächenmodell für die räumliche Visualisierung und Quantifizierung des Fluoreszenzsignals über den gesamten Z-Stack zu generieren (Abbildung 6C). Die Analyse des 3D-Oberflächenmodells ergab 546 CD3+ T-Zellen und 218 Zellen, die HIV p24 produzieren. Kumulativ ermöglichte die Z-Stack-Erfassung von Immunfluoreszenz aus geklärten, HIV-infizierten lymphatischen Geweben die Erstellung von 3D-Modellen der zellulären Zusammensetzung im Gewebe und die automatisierte Quantifizierung von Immunzellpopulationen innerhalb des Gewebevolumens.
LSFM von intaktem, geklärtem und immungefärbtem lymphatischem Gewebe ermöglichte eine großvolumige Immunfluoreszenz (IF)-Bildgebung der Immunzell- und virusproduzierenden Zellverteilung in den lymphatischen Geweben (Abbildung 7). Die Immunfärbung von Dickdarmgewebe einer HIV-infizierten Hu-Maus auf hCD3+ T-Zellen und HIV p24 ergab Herde von virusproduzierenden Zellen, die über große Geweberegionen verteilt waren, ohne dass eine Infektion nachgewiesen wurde (Abbildung 7A). Eine vergrößerte Ansicht eines Herdes von virusproduzierenden Zellen zeigte mehrere virusproduzierende Zellen in unmittelbarer Nähe zu potenziellen Zielzellen (Abbildung 7B). Die Autofluoreszenz des Gewebes (roter Schleier) wurde verwendet, um die gesamte Gewebearchitektur zu visualisieren und gleichzeitig spezifische Immunzellpopulationen innerhalb des Gewebes zu unterscheiden, die heller färbten als die Autofluoreszenz (rote Ovale). Ein 3D-Modell des gesamten LSFM-Z-Stack-Volumens zeigte die räumliche Verteilung der Herde virusproduzierender Zellen innerhalb einer Region mit intaktem Gewebe und ermöglichte die Kartierung von Orten der Virusproduktion relativ zur gesamten Gewebearchitektur (Abbildung 7C). Überraschenderweise waren Herde von virusproduzierenden Zellen oft in großen Geweberegionen verstreut, ohne dass es Hinweise auf eine Virusproduktion gab. Diese Ergebnisse können die Quantifizierung von Parametern der Virusverteilung und der infizierten Zelldichte in verschiedenen Geweben und zu unterschiedlichen Zeitpunkten der Infektion oder des Ansprechens auf verschiedene Behandlungen ermöglichen.

Abbildung 1: Arbeitsablauf der typischen CUBIC und CLARITY Gewebereinigung, Immunfärbung und Bildgebung. Die Clearingzeiten für CUBIC (oben) und CLARITY (unten) können je nach Größe und Art des Gewebes stark variieren. Für das CLARITY-Clearing ist vor der Immunfärbung ein zusätzlicher Inkubationsschritt mit Brechungsindex-angepassten Medien erforderlich, um zu überprüfen, ob das Gewebe klar ist. Die Immunfärbung dauert in der Regel 3 Tage, wenn Primärantikörper mit Fluorophoren konjugiert werden, und 6 Tage, wenn fluoreszierende Sekundärantikörper erforderlich sind. Die Proben können entweder mit einem konfokalen oder einem LSFM abgebildet werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: CUBIC Clearing von Hu-Maus- und NHP-Gewebeproben. Abhängig von den unterschiedlichen Dichten von Häm und Lipiden der Gewebeproben variiert die Zeit, die für die Reinigung der einzelnen Gewebetypen benötigt wird. Zum Beispiel benötigen der Dickdarm und der Zwölffingerdarm in der Regel relativ kurze Zeiträume (~7 Tage), während Milz und Leber länger brauchen können, um transparent zu werden (~30 Tage). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
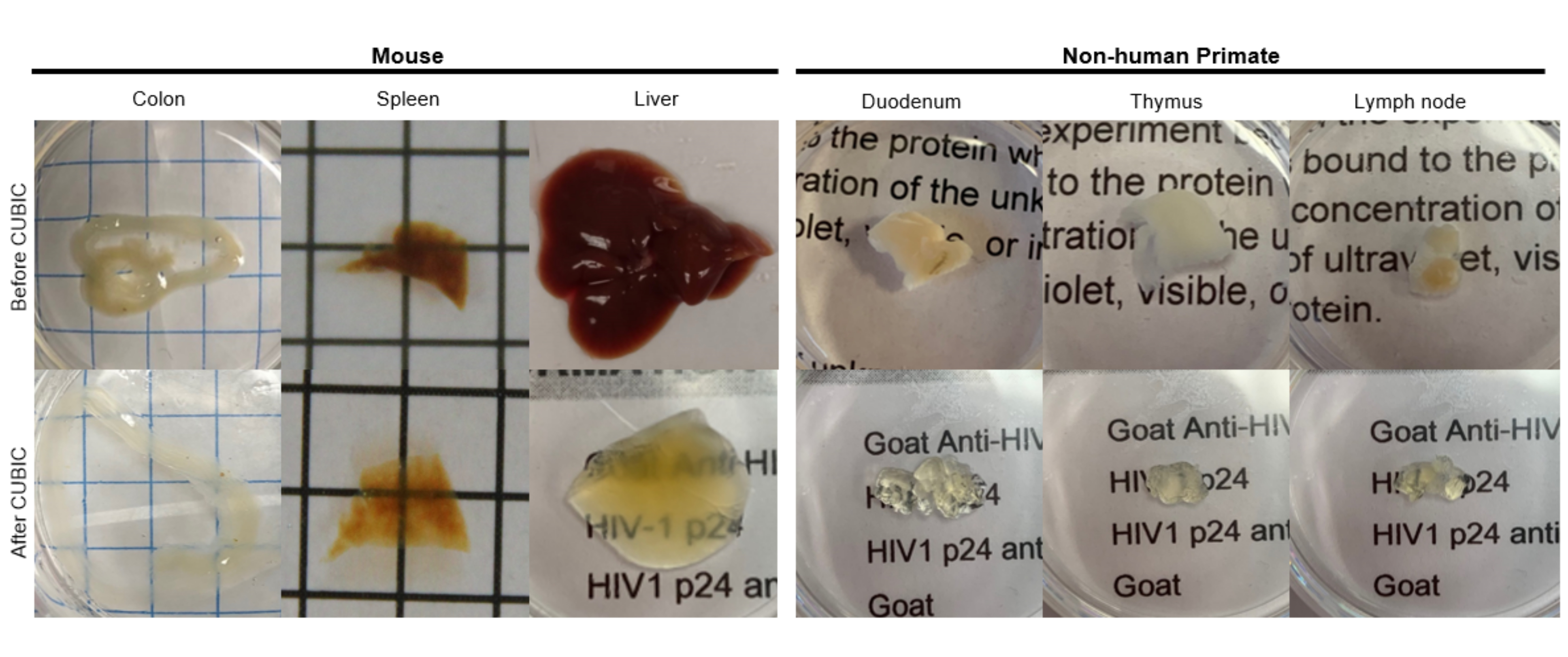{kind=link}

Abbildung 3: Längsschnittvergleich von Gewebereinigungsmethoden an menschlichen Proben. CUBIC (obere Felder) und CLARITY (untere Felder) befreiten die Milz einer HIV-infizierten Person unter antiretroviraler Therapie. Bei beiden Methoden wurde das Gewebe bis zum 32. Tag für die Immunfärbung und Bildgebung ausreichend gereinigt. Der Entfärbungsschritt für die CUBIC-Methode reduziert sichtbar die Autofluoreszenz, die durch das Vorhandensein von Häm in Milzproben verursacht wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Probenaufnahme für die konfokale Mikroskopie. Die Proben wurden zwischen Deckgläsern montiert, die mit selbstklebenden 0,5-1 mm Silikonisolatoren getrennt waren. Silikonisolatoren wurden auf das erste Deckglas geklebt und Gewebe in die Mitte der Vertiefung (oben) gelegt. Die Vertiefung wurde mit RIMS gefüllt, bis der Meniskus auf gleicher Höhe oder leicht über der Oberseite der Vertiefung lag (links). Das zweite Deckglas wurde vorsichtig von einer Seite zur anderen abgesenkt, um Blasenbildung zu vermeiden (unten). Deckgläser wurden vollständig auf den Silikonisolator geklebt, indem ein stumpfes Instrument vorsichtig um den Umfang der Vertiefung geführt wurde (rechts). Die Proben wurden in einem konfokalen Standardmikroskop abgebildet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
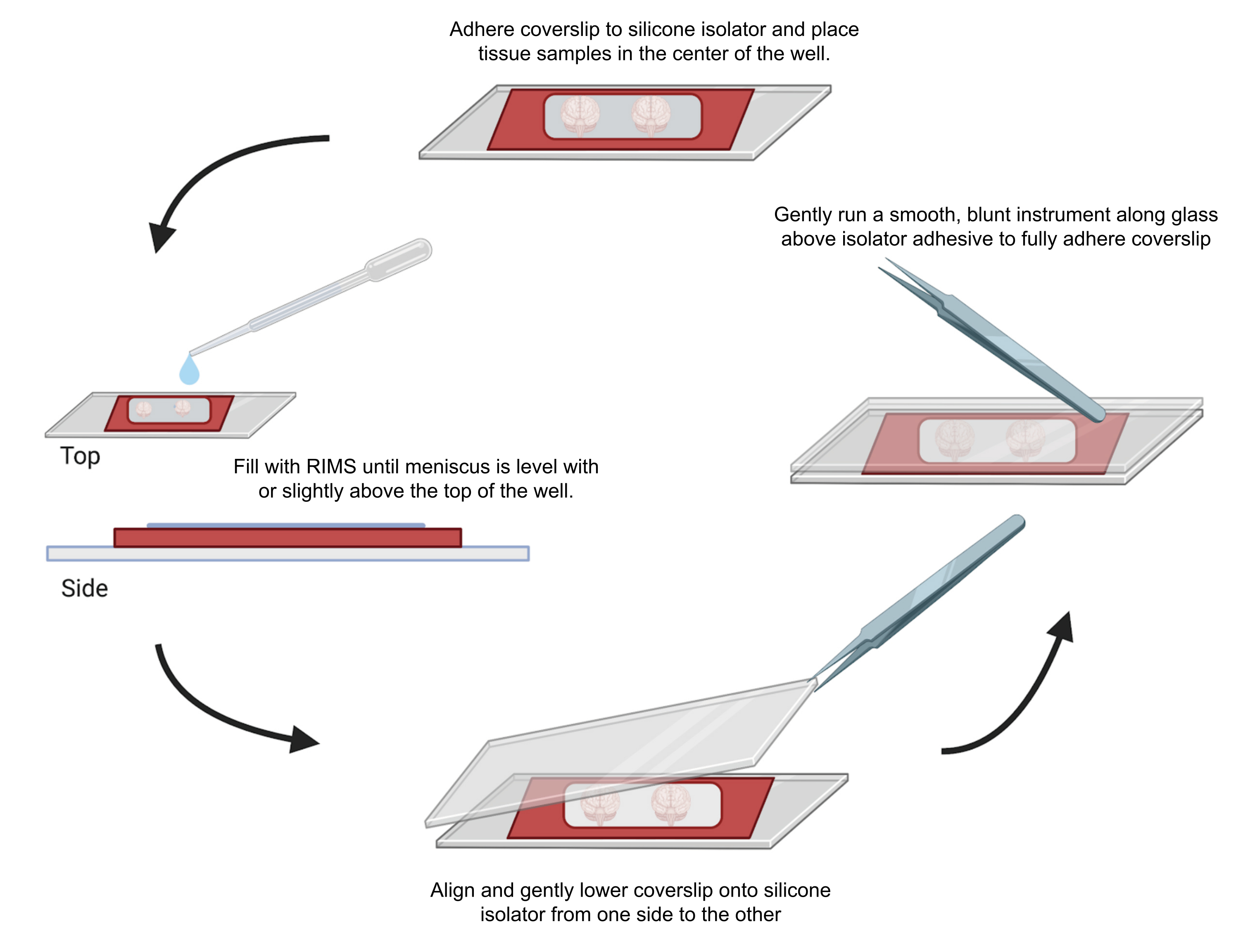{kind=link}

Abbildung 5: Konfokale Mikroskopie von gereinigten, intakten menschlichen Milz- und NHP-Lymphknoten. (A-D) Geklärtes HIV-infiziertes menschliches Gewebe wurde auf HIV-1 p24 (grün), hCD3+ T-Zellen (rot) und Zellkerne (cyan) gefärbt. (E) Konfokale Z-Scheibe eines CUBIC entfernten Lymphknotens aus einem SHIV-infizierten NHP 8 Wochen nach der Infektion, immungefärbt für CD3+ T-Zellen (magenta), CD68+ Makrophagen (mac/rot), SHIV p27 (grün) und Zellkerne (blau). Das Sichtfeld enthält T-Zellen, Makrophagen und andere Zelltypen, aber keine Hinweise auf SHIV-produzierende Zellen (grün). F) Konfokale Z-Scheibe einer angrenzenden Region desselben Lymphknotens mit Unterschieden in der Zelldichte und -anzahl sowie dem Vorhandensein von virusproduzierenden CD3+ T-Zellen (grüne Pfeile) und CD68+ Makrophagen (gelbe Pfeile). (G-I) Vergrößerte Ansicht ausgewählter Bereiche der p27-Färbung von (F). Die Maßstabsstäbe sind 50 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
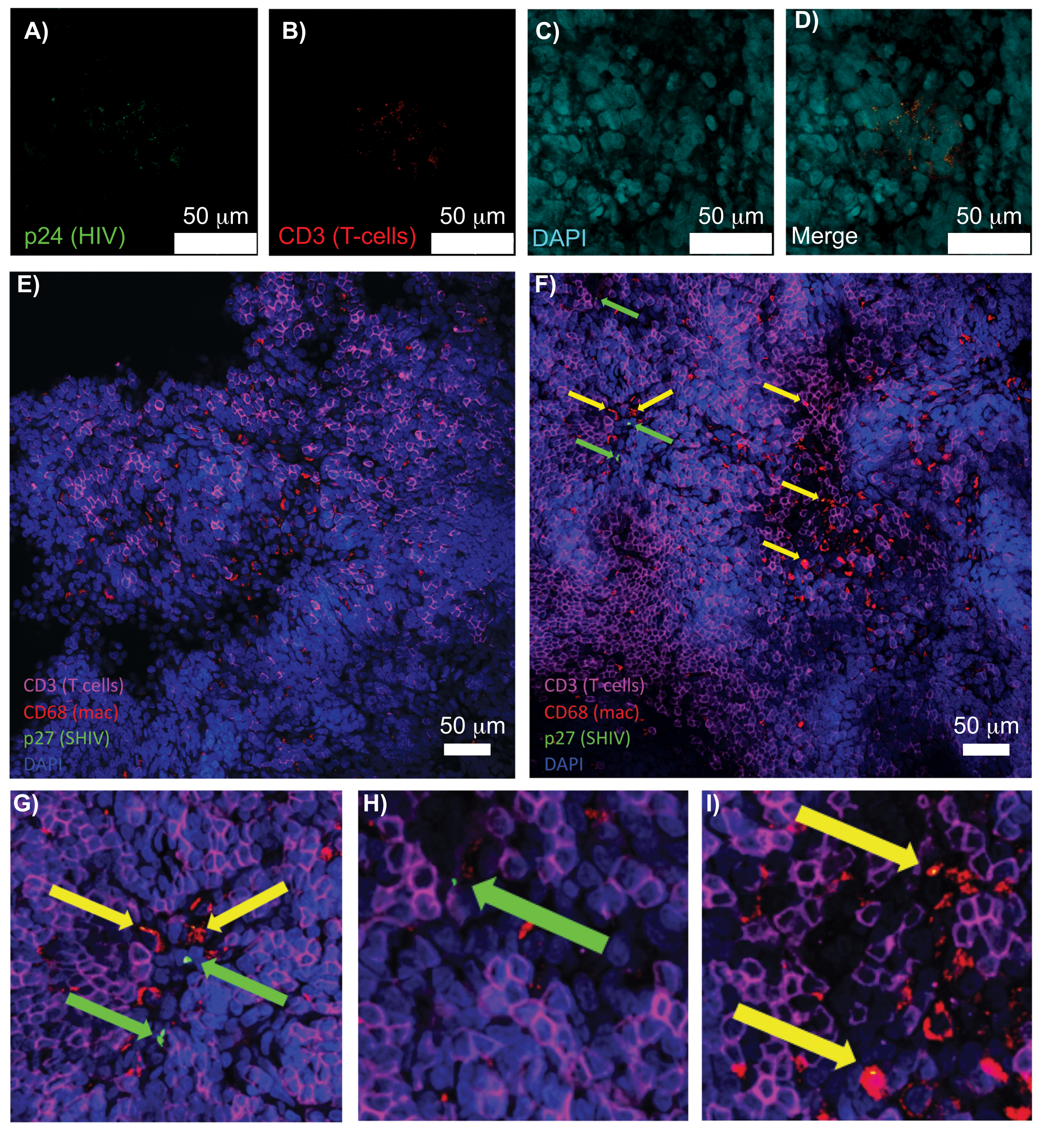{kind=link}

Abbildung 6: Z-Stack-Volumen und 3D-rekonstruierte Oberfläche von HIV-infizierter menschlicher Milz. (A) Z-Projektionsbild von einem 600 μm x 600 μm x 100 μm großen Z-Stapel von HIV-infiziertem menschlichem Milzgewebe, das auf HIV-1 p24 (grün), hCD3+ T-Zellen (rot) und Zellkerne (cyan) gefärbt wurde. (B) Das gleiche Z-Projektionsbild ohne nukleare DAPI-Färbung. (C) Rekonstruiertes 3D-Oberflächenmodell der CD3 (rot) und p24 (grün) Fluoreszenz aus dem gesamten Z-Stack-Volumen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
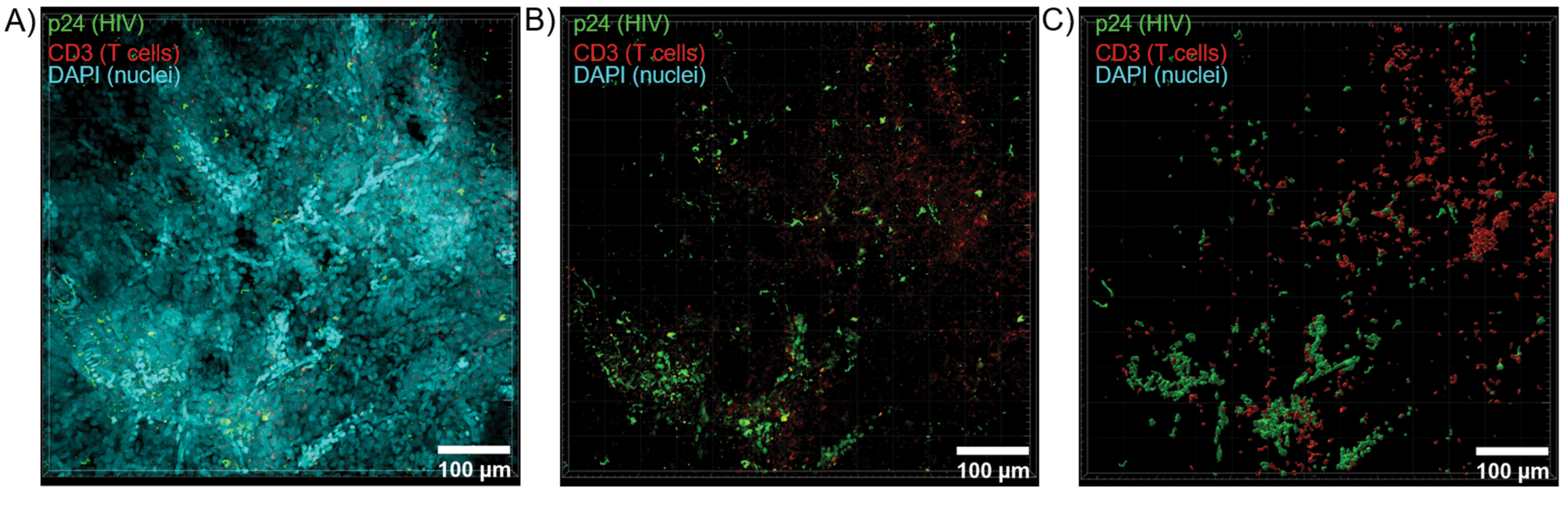{kind=link}

Abbildung 7: LSFM- und 3D-Rekonstruktion des Oberflächenvolumens von HIV-infizierten Geweben (A) Z-Scheibe (1.000 μm x 1.000 μm) des Dickdarms einer HIV-infizierten Hu-Maus, immungefärbt auf CD3+ T-Zellen (rot) und HIV p24 (grün). Der mattrote Schleier steht für die Autofluoreszenz des Gewebes, während die deutlichen roten Punkte auf T-Zellen hinweisen. An der Peripherie sind Zotten sichtbar, die auf das zentrale Lumen zeigen, mit mehreren Herden aktiver Virusproduktion (weiße Pfeile), die über große Bereiche verteilt sind, die kein Virus enthalten. Der Kasten zeigt den ungefähren Bereich von Interesse für Panel B. (B) Vergrößerter Bereich des Gewebes, der einzelne Virus-produzierende hCD3+ T-Zellen (gelb) in der Nähe von nicht infizierten T-Zellen (rot) zeigt. Das Bild wurde gedreht und in eine nahe gelegene Z-Schicht geändert, um einen Fokus von p24 positiven Zellen in einer einzigen Z-Ebene zu zeigen. Die rote Autofluoreszenz im Hintergrund zeigt die allgemeine Gewebearchitektur zusätzlich zur spezifischen hCD3+ T-Zell-Färbung (rote Punktzahl; weiße Pfeile). (C) 3D-Oberflächenmodell des gesamten Volumens (1.000 μm x 1.000 μm x 200 μm), das mit der Imaris-Software erstellt und gedreht wurde, um HIV-Infektionsherde (gelb) an verschiedenen Stellen des Darms anzuzeigen. Weiße Pfeile zeigen einzelne Brennpunkte innerhalb des Volumens an. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Lymphatisches Gewebe von Interesse sollte schnell post mortem entnommen und sofort in vorgekühlte Fixierpuffer gegeben werden, um Gewebenekrosen (dunkles oder schwarzes Gewebe) zu vermeiden, die sich negativ auf die Färbung und Bildgebung auswirken können. Tauchen Sie das Gewebe nach der Entnahme des gewünschten Gewebes sofort über Nacht in eiskaltes 4%-8%iges Paraformaldehyd (PFA) zur Fixierung, wodurch auch potenzielle Krankheitserreger, die mit den Proben in Verbindung gebracht werden, inaktiviert werden. 4 % PFA ist optimal für die Fixierung von LM-Proben, während 8 % PFA Gewebe sowohl für LM als auch für EM angemessen konservieren können. Nach diesen Verfahren und der Lagerung der Proben in einem Fixiermittel bei 4 °C im Dunkeln kann das Gewebe für die LM-Bildgebung mehrere Jahre lang effektiv konserviert werden. Ein Vorbehalt besteht darin, dass eine längerfristige Lagerung in Fixiermitteln zur Einführung von Färbeartefakten führen kann, insbesondere zur Antigenmaskierung, die durch Vernetzung benachbarter Proteine mit dem interessierenden Protein verursacht wird, was den Zugang von färbenden Antikörpern zum Epitop versperren kann13,14. Wenn das Gewebe endogen exprimierte fluoreszierende Proteine enthält, ergreifen Sie Maßnahmen, um zu vermeiden, dass das Gewebe während des gesamten Protokolls nach Möglichkeit Licht ausgesetzt wird. Typischerweise behalten endogene fluoreszierende Proteine die Fluoreszenz für 6-12 Monate nach der Fixierung bei, aber einzelne Gewebeproben können über längere oder kürzere Zeiträume variieren. Wenn die endogene Fluoreszenz aufgrund des Proteinabbaus verloren geht, können fluoreszierende Proteine oft mit einem primären Antikörper nachgewiesen werden, der für das betreffende Protein spezifisch ist. Perfusion ist eine weitere Option zur schnellen Fixierung von Gewebe vor der Klärung12; Aufgrund von Bedenken bei der Arbeit mit Krankheitserregern wie HIV wurde jedoch der Weg der Gewebenekpsie mit anschließendem Eintauchen in eiskaltes Fixiermittel gewählt, um die Proben so sicher wie möglich vorzubereiten.
Ein Vorteil der beschriebenen wasserbasierten Clearing-Protokolle besteht darin, dass sie im Allgemeinen milder sind als organische Protokolle, die manchmal empfindlicheres Gewebe wie die Leber schädigen können. Wasserbasierte Clearing-Protokolle benötigen im Vergleich zu biobasierten Clearing-Protokollen in der Regel eine längere Zeit, um eine vollständige Probenreinigung zu erreichen (Wochen vs. Tage). Die Protokolle CLARITY und CUBIC können schneller durchgeführt werden, indem die Perfusion verwendet wird, um alle Organe innerhalb eines Nagetiers gleichzeitig zu reinigen11,12; Dies war jedoch keine praktikable Option für NHP und menschliche Autopsien. Proben, die mit CLARITY verarbeitet wurden, neigen dazu, eine gewisse Volumenausdehnung zu zeigen, während CUBIC einen reduzierten Einfluss auf das Probenvolumen9 zeigte. Obwohl sie im Allgemeinen schneller sind, führen viele organische Gewebereinigungsprotokolle zu einer Schrumpfung des Gewebes15, was die Erkennung von einzelzelligen oder subzellulären Details in zelldichten Geweben wie Lymphknoten und Milz erschweren kann. Die durch das Clearing induzierte Expansion kann die Auflösung der Bildgebung effektiv erhöhen, wodurch es einfacher wird, Aspekte zu beobachten, die in der ursprünglichen Größe des Gewebes schwer zu beobachten wären. Alternativ kann die Gewebeschrumpfung die Gesamtgröße der Probe effektiv verringern, was die Bildgebung ganzer Organe ohne Dissektion ermöglichen kann. Ein Vorteil sowohl des CLARITY- als auch des CUBIC-Protokolls besteht darin, dass sie die Fluoreszenz von endogen exprimierten fluoreszierenden Proteinen im Gewebe erhalten, während sie für die Immunfluoreszenzfärbung zugänglich bleiben11,12. Die Immunfärbung kann mit wässrigen oder organischen Gewebereinigungsmethoden durchgeführt werden. Persönliche Erfahrungen zeigten jedoch einen höheren Anteil an Antikörperkompatibilität bei der Verwendung von wasserbasierten Protokollen im Vergleich zu organischen Protokollen. Die Forscher müssen überlegen, welche Gewebereinigungsmethode auf der Grundlage der abgebildeten Gewebe und der angesprochenen biologischen Fragestellungen (z. B. Bildgebung des gesamten Organs im Vergleich zur Bildgebung der spezifischen Region of Interest) verwendet werden soll. Es gibt keine universelle Gewebereinigungstechnik, die eine robuste, schlüsselfertige Analyse für alle großvolumigen Bildgebungsfragen ermöglicht, und die verfügbaren Methoden weisen je nach biologischer Anwendung deutliche Vor- und Nachteile auf.
Bei der Durchführung von Antikörperfärbungen sind zahlreiche Aspekte zu beachten. Da CLARITY-Proben in Acrylamid-Hydrogel eingebettet sind, benötigen sie in der Regel längere Inkubationszeiten12. Die Zeit, die für die Antikörperinkubation benötigt wird, hängt auch vom Volumen und der Dicke jeder Probe ab. Die meisten hier beschriebenen Proben waren ~2-3 Millimeter dick, und 3 Tage reichten für eine vollständige Färbung im gesamten Gewebe aus. Wenn das Ziel darin besteht, ein ganzes Mäusegehirn abzubilden, kann die Inkubationszeit des Antikörpers 1 Woche oder länger dauern12. Die Wahl einer wässrigen oder organischen Gewebereinigungsmethode für die Immunfluoreszenzbildgebung kann von der Antikörperkompatibilität abhängen. Im Allgemeinen beträgt die Trefferquote für Antikörper, die in kultivierten Zellen und Geweben wirken, für CUBIC oder CLARITY ~70%. Unabhängig davon, ob eine wässrige oder organische Gewebereinigungsmethode verwendet wird, ist es notwendig, die Verträglichkeit und Wirksamkeit aller Antikörper mit der verwendeten Methode zu bewerten. Wie in diesem Protokollabschnitt gezeigt, erfolgt die Immunfärbung für CUBIC- und CLARITY-prozessierte Proben nach Abschluss des Clearings. Im Gegenteil, dieser Schritt findet vor dem Clearing-Verfahren für einige Protokolle auf organischer Basis statt, gefolgt von einem Post-Fixing.
Es ist von entscheidender Bedeutung, dass das Gewebe vollständig in ein Bildgebungsmedium eingetaucht ist, das seinem Brechungsindex entspricht. Andernfalls führt dies zu sphärischen Aberrationen während der Bildgebung und verzerrt das während der Bildaufnahme aufgenommene Licht. Es muss darauf geachtet werden, dass alle Luftblasen von den Bildgebungsmedien entfernt werden, wenn Proben sowohl für konfokale als auch für LSFM aufgenommen werden, da Blasen den Weg des Lichts zur oder von der Probe weg stören können. Blasen können vor der endgültigen Probenmontage manuell mit einer Pipette entfernt werden. Für die Bildgebung dickerer Proben mit einem konfokalen Mikroskop können mehrere Silikonspacer übereinander geschichtet werden, um Gewebe mit einer Dicke von mehr als 0,5 mm aufzunehmen. Eine Empfehlung ist, alle Gewebe in RIMS für mehrere Stunden bis über Nacht zu äquilibrieren, während sie am Mikroskop montiert sind, ohne dass sich die Probe zusätzlich bewegt. Die vollständige Äquilibrierung des Gewebes und der Bildgebungsmedien verhindert das Vermischen von Lösungen mit nicht übereinstimmenden Brechungsindizes, die während der Bildgebung zu Aberrationen führen können. Es ist wichtig, sich daran zu erinnern, dass es bei der Aufnahme von freigegebenen Gewebeproben keine einzige schlüsselfertige Einbettungsmethode gibt, um alle Proben in allen Mikroskopen abzubilden. In diesem Protokoll werden Optionen für die Probeneinfassung diskutiert, die in einem Kontext optimal funktioniert haben, aber es gibt zahlreiche Ansätze für die Probeneinbettung, abhängig vom verwendeten Mikroskop und der angesprochenen biologischen Fragestellung. Diese Ansätze können unter anderem das Einbetten der Probe in Agarose, das Aufhängen der Probe an einem Haken oder einer auf den Brechungsindex abgestimmten Kunststofflinie, die Verwendung eines Stachelschweinadapters, den 3D-Druck eines Probenhalters oder das Befestigen der Probe mit Klebstoff an einer Kunststoffschale umfassen.
Konfokale Mikroskope eignen sich gut für die Abbildung von Gewebevolumina von ~1 mm3-1 cm3. Verwenden Sie bei konfokalen Mikroskopen ein 2- bis 10-fach-Objektiv, um zunächst interessante Regionen zu lokalisieren und Z-Stapel mit größerem Volumen oder ganzem Gewebe mit Einzelzellauflösung zu erfassen. Wechseln Sie zu 20- bis 63-fachen Objektiven, um Bilder mit höherer Auflösung bestimmter Regionen von Interesse mit subzellulären Informationen aufzunehmen. Das ideale Objektiv für die Bildgebung von CUBIC und CLARITY geklärtem Gewebe ist ein CLARITY/Scale-spezifisches Objektiv, das genau auf den Brechungsindex des Gewebes und der Bildgebungslösung abgestimmt ist. Wenn dieser Objektivtyp nicht verfügbar ist, ist es optimal, Proben mit einem Glycerin- oder Öl-Immersionsobjektiv (z. B. LD LCI Plan-Apochromat 25 x 0,8 NA Imm Corr DIC M27 Multi-Immersionsobjektiv: Arbeitsabstand = 0,57 mm) anstelle eines Luftobjektivs abzubilden. Dadurch wird die Einführung optischer Verzerrungen aufgrund von nicht übereinstimmenden Brechungsindizes während der Bildaufnahme minimiert. Die 20-25x-Objektive können die Aufnahme größerer Volumina mit der Gewinnung von Färbedetails aus einzelnen Zellen in einer komplexen Gewebeumgebung in Einklang bringen. Wichtig ist, dass die meisten konfokalen Mikroskope Module enthalten, die eine 3D-Kachelung von Bildgebungsvolumina ermöglichen. Diese Art der Bildaufnahme kann im Idealfall Z-Stapel mit größerem Volumen erzeugen, die subzelluläre Informationen enthalten.
Die LSFM-Bildgebung kann die 3D-Visualisierung spezifischer Zellpopulationen im Kontext eines großen Gewebevolumens (>1 cm3) und sogar ganzer Organe ermöglichen. In den letzten 10 Jahren konzentrierte sich die Gewebereinigung in Kombination mit LSFM weitgehend auf das Verständnis der Gehirnkonnektivität bei Nagetieren; Zu den neueren Anwendungen gehören jedoch die Visualisierung von Tumormetastasen16, die Zellverteilung innerhalb anatomischer Kompartimente 9,17 und die Ausbreitung von Krankheitserregern18. Im Vergleich zu kultivierten Zellen sind die meisten biologischen Ereignisse in Geweben uneinheitlich und LSFM kann besonders gut geeignet sein, die räumliche Gewebeheterogenität dieser Ereignisse zu visualisieren und zu quantifizieren (z. B. Virusreplikation, Immunsignalisierung, Zellverteilung usw.).
3D-Datensätze, die über konfokale oder LSFM-Daten erfasst wurden, können mit zahlreichen Bildanalyseplattformen nachbearbeitet werden. Die Imaris Software-Suite kann für die Konstruktion von Oberflächen, die Generierung von 3D-Animationen und die Zellquantifizierung verwendet werden. Es stehen jedoch zahlreiche Bildanalysesysteme zur Verfügung, die eine effiziente Bildnachbearbeitung und -analyse ermöglichen. ImageJ/Fiji Freeware19 ist eine ansprechende alternative Bildverarbeitungsplattform, die für die meisten Labore zugänglich ist, aber es gibt keine einheitliche Analysesoftware, die sich durch alle Formen der Bildanalyse und -visualisierung auszeichnet. Viele Bildanalyse-Software-Suiten können unerschwinglich teuer sein, wenn sie nicht über gemeinsam genutzte Einrichtungen verfügbar sind. Ein kritischer Aspekt von LSFM oder großen gekachelten konfokalen 3D-Datensätzen ist das Datenmanagement. Diese Imaging-Plattformen können riesige Dateien (>1 TB) erzeugen, die höherwertige Computer-Workstations für die Datenvisualisierung und -quantifizierung erfordern. Letztendlich kann dieser Bildgebungs-Workflow die Erfassung und Quantifizierung räumlich unterschiedlicher Zellpopulationen innerhalb ganzer Gewebe rationalisieren und ist auf die meisten Gewebequellen und biologischen Systeme anwendbar.
Offenlegungen
Die Autoren haben keine Interessenkonflikte offenzulegen.
Danksagungen
Vielen Dank an die Core Facilities des University of Illinois am Urbana-Champaign Institute for Genomic Biology für den Einsatz von konfokalen und Lichtblatt-Fluoreszenzmikroskopen. Vielen Dank an die großartigen Individuen aus der "The Last Gift"-Kohorte für menschliche Gewebeproben, die durch die folgenden Zuschüsse finanziert wurde: I147821, DA051915, AI131385 und P30 AI036214. Vielen Dank an Nancy Haigwood und Ann Hessell für SHIV-infizierte NHP-Gewebeproben.
Materialien
| Name | Company | Catalog Number | Comments |
| Acrylamide Solution (in 0.1 M PBS, 40 mL in total) | |||
| 40% Acrylamide: 4 mL | Bio-Rad | 1610144 | |
| VA-044 Thermal Initiator: 0.1g | Fujifilm | 011-19365 | |
| CLARITY Blocking solution (in 0.1 M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Tween-20: 5 µL | Fisher Scientific | BP337-500 | |
| CLARITY wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Tween-20: 50 µL | Fisher Scientific | BP337-500 | |
| CUBIC Blocking solution (in 0.1M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Triton X-100: 5 µL | VWR | M143-1L | |
| CUBIC Reagent-1 (in 0.1M PBS, 50 mL in total) | |||
| N, N, N’, N’-tetrakis (2-hydroxypropyl) ethylenediamine: 12.5 g | Aldrich | 122262 | |
| Triton X-100: 7.5 g | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC Reagent-2 (in 0.1M PBS, 50 mL in total) | |||
| Sucrose: 25 g | Sigma | S1888-500G | |
| Sodium azide (in powder form): 10 g | Sigma | 71289-50G | |
| Sodium azide stock solution (in DI H2O, 50 mL in total) | Sigma | 71289-50G | |
| Triethanolamine: 5 g | Sigma | 90270-500mL | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| DAPI staining solution (0.5 µg/mL) | |||
| DAPI stock solution: 1 µL | |||
| Wash solution: 10 mL | |||
| DAPI stock solution (5 mg/mL) | |||
| DAPI powder: 5 mg | Sigma-Aldrich | D9542-1MG | |
| DMSO (100%): 1 mL | ThermoFisher | D12345 | |
| Imaging Media RI-2 (in dH2O) | |||
| 90% Histodenz | Sigma | D2158-100G | |
| 0.01% Sodium azide | Sigma | 71289-50G | |
| 0.02 Sodium Phosphate Buffer, pH 7.5 | Sigma-Aldrich | S9638-250G | |
| 0.1% Tween-20 | Fisher Scientific | BP337-500 | |
| Primary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Goat anti-HIV p24: 10 µL (1:200) | Creative Diagnostics | DPATB-H81692 | |
| Mouse anti-human CD68: 10 µL(1:200) | Dako | M0876 | |
| Rabbit anti-human CD3: 10 µL (1:200) | Dako | A0452 | |
| 8% SDS Solution (in 0.1 M PBS, 50 mL in total) | |||
| SDS powder: 4 g | Sigma-Aldrich | L3771-500G | |
| Secondary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Donkey anti-goat conjugated with AlexaFluor647: 2 µL | Invitrogen | A21447 | |
| Donkey anti-mouse conjugated with AlexaFluor594: 2 µL | Invitrogen | A21203 | |
| Donkey anti-rabbit conjugated with AlexaFluor488: 2 µL | Invitrogen | A21206 |
Referenzen
- Spalteholz, W., Barker, L. F., Mall, F. P. . Hand-Atlas of Human Anatomy. , (1907).
- Jacob, T., Gray, J. W., Troxell, M., Vu, T. Q. Multiplexed imaging reveals heterogeneity of PI3K/MAPK network signaling in breast lesions of known PIK3CA genotype. Breast Cancer Research and Treatment. 159 (3), 575-583 (2016).
- Kieffer, C., Ladinsky, M. S., Ninh, A., Galimidi, R. P., Bjorkman, P. J. Longitudinal imaging of HIV-1 spread in humanized mice with parallel 3d immunofluorescence and electron tomography. eLife. 6, 23282 (2017).
- Chung, K., Deisseroth, K. CLARITY for mapping the nervous system. Nature Methods. 10 (6), 508-513 (2013).
- Compton, A. A., Schwartz, O. They might be giants: Does syncytium formation sink or spread HIV infection. PLoS Pathogens. 13 (2), 2-8 (2017).
- Symeonides, M., et al. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses. 7 (12), 6590-6603 (2015).
- Sharova, N., Swingler, C., Sharkey, M., Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. The EMBO Journal. 24 (13), 2481-2489 (2005).
- Colby, D. J., et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nature Medicine. 24 (7), 923-926 (2018).
- Ladinsky, M. S., et al. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. eLife. 8, 46916 (2019).
- Buettner, M., Bode, U. Lymph node dissection--understanding the immunological function of lymph nodes. Clinical and Experimental Immunology. 169 (3), 205-212 (2012).
- Treweek, J. B., et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nature Protocols. 10, 1860-1896 (2015).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159, 911-924 (2014).
- Sompuram, S. R., Vani, K., Bogen, S. A. A molecular model of antigen retrieval using a peptide array. American Journal of Clinical Pathology. 125 (1), 91-98 (2006).
- Scalia, C. R., et al. Antigen masking during fixation and embedding, dissected. The journal of histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 65 (1), 5-20 (2017).
- Jing, D., et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Research. 28 (8), 803-818 (2018).
- Guldner, I. H., et al. An Integrative platform for three-dimensional quantitative analysis of spatially heterogeneous metastasis landscapes. Scientific Reports. 6, 24201 (2016).
- Muntifering, M., et al. Clearing for deep tissue imaging. Current Protocols in Cytometry. 86 (1), 38 (2018).
- DePas, W. H., et al. Exposing the three-dimensional biogeography and metabolic states of pathogens in cystic fibrosis sputum via hydrogel embedding, clearing, and rRNA labeling. mBio. 7 (5), 00796 (2018).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image. Nature Methods. 9 (7), 676-682 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten