
Method Article
クリアリング、免疫染色、共焦点、およびライトシート蛍光顕微鏡によるHIV感染組織における免疫細胞集団の3D可視化
要約
組織クリアリングと免疫蛍光顕微鏡を組み合わせることで、無傷の組織内の免疫細胞集団とウイルスタンパク質の空間的な可視化と定量が可能になります。共焦点およびライトシート蛍光顕微鏡による透明組織の光学切片化は、複雑な組織環境の3Dモデルを生成し、HIV感染中に示される空間的不均一性を明らかにすることができます。
要約
後天性免疫不全症候群(AIDS)の原因物質であるヒト免疫不全ウイルス(HIV)は、世界中で約4,000万人が感染しており、広く利用可能な治療法がないという世界的な主要な健康問題です。集中的な努力にもかかわらず、感染中および治療に応答した組織内のウイルスと宿主細胞の相互作用の詳細な理解は不完全なままです。これらの制限に対処するために、水性組織透明化技術CUBIC(Clear, Unobstructed Brain/Body Imaging Cocktails and Computational analysis)およびCLARITY(Clear Lipid-exchanged Acrylamide-hybridized Rigid Imaging/Immunostaining/in situ-hybridization-compatible Tissue hYdrogel)を適用して、共焦点およびライトシート蛍光顕微鏡を使用して、動物モデルとヒトのHIV感染組織における複雑なウイルス宿主間相互作用を視覚化します。無傷組織の光学切片化と画像解析により、組織全体に含まれる空間情報の迅速な再構築と、感染中の免疫細胞集団の定量化が可能になります。これらの方法は、ほとんどの組織源や、感染症やがんなどの多様な生物学的問題に適用できます。
概要
生物学研究における定量的な空間組織イメージングの必要性の高まりは、最近、単一細胞の解像度で無傷の組織のより大きなボリューム(mm3-cm 3)画像を生成するための組織透明化技術の出現につながりました。組織には、独自に定義された構造、組成、および機能を持つ生体分子の複雑な組織が含まれます。残念ながら、組織に存在する多くの生体分子(脂質や発色団など)は、光学顕微鏡でイメージングすると光を散乱、吸収、または放出するため、大量のイメージングが困難になります。さらに、組織は、標準的なイメージングソリューションや光学レンズと一致しない屈折率を示すことが多く、イメージング中に光学的な歪みが生じます。光学顕微鏡で大量の組織をイメージングするための最適なアプローチには、組織の屈折率、イメージングソリューション、および対物レンズを一致させると同時に、処理中に無傷の組織の生物学的特徴を損なうことなく組織の深部に光を浸透させることが必要です。不透明な組織サンプルの透明化を通じて、組織とイメージング溶液との間の屈折率の差を減少させる最初の試みは、1800年代後半にドイツの解剖学者ヴェルナー・シュパルテホルツによって行われました1。この組織透明化技術には、組織サンプルを損傷する可能性のある刺激の強い化学溶剤が含まれていましたが、それでも、無傷の組織の大容量イメージングが最初に報告されました。最新の光学顕微鏡法は、画像キャプチャと分析のための計算能力と組み合わされ、最近、単一細胞の解像度で大きな無傷の組織サンプルをイメージングする方法として、組織透明化が再び流行しました。過去20年間で、有機ベースと水性の両方を含む数十の高度な組織透明化技術が登場し、それぞれに特定のアプリケーションに対する長所と短所があります。
3D組織イメージングは、細胞培養では再現できない、より複雑な生物学的相互作用を探ることができます。例えば、細胞シグナル伝達パターン2、異なる細胞型の空間分布3、および脳の接続性4は、以前に全組織/臓器イメージング法を用いて定量的にマッピングされていた。ここで説明するのは、活動性感染中に無傷のHIV感染リンパ組織内の異なるHIV標的細胞集団を透明化、免疫染色、および視覚化するための水性組織透明化プロトコルのアプリケーションです。体内では、HIVは主にCD4+ T細胞に感染し、そのゲノムのコピーを感染した宿主細胞のゲノムに統合します。その後、ウイルスは感染した宿主細胞機構を乗っ取って自己複製し、ウイルスの拡散、宿主細胞の死滅、免疫機能障害、およびエイズへの長期的な進行をもたらします。組織培養と細胞培養における感染T細胞の挙動は著しく異なることに注意することが重要です。HIVとインキュベートした培養CD4+ T細胞は、数十個の核を含む可能性のある大量のHIV誘発性合胞体を産生することができます5が、3D細胞外マトリックス(ECM)ハイドロゲルまたはHIV感染ヒト化マウス(huマウス)の組織サンプルで培養した初代CD4+ T細胞を用いた同様の実験では、一般に2〜5個の核6を有する合胞体が得られます。HIV感染者内でのウイルスの局所的な細胞間伝播と全身拡散を理解することは、おそらくさらに複雑であり、複数の感染細胞タイプによるウイルスの組織から血管への輸送、そして遊離ビリオンとウイルス産生細胞が多数の感受性リンパ球にアクセスできる場所7.これらのシナリオは、現在のところ細胞培養系で再現することは不可能であり、動物モデルやヒトの組織は、免疫系が機能している複雑な生物の文脈でウイルスの病因を理解するための重要なリソースであり続けています。
現在の抗レトロウイルス療法(ART)は、HIVの複製を阻害し、エイズへの疾患の進行を止めることにより、HIV感染者(PWH)の平均余命と質を大幅に向上させます。残念ながら、ARTは、レトロウイルスゲノムの挿入物を含む潜伏感染した免疫細胞のうち、休止状態でウイルスを活発に産生していないものを排除するものではありません。ARTを服用しているほとんどの人の血液中にウイルスは検出されませんが、ARTが中断されるとウイルス量は急速に回復し、疾患の進行が続きます8。HIV感染は、感染した細胞の潜伏層によって引き起こされる持続的な性質が、HIV治療法の確立に大きな障害となっています。HIVの組織リザーバーは依然として十分に理解されておらず、ART前、ART中、ART後にリンパ組織におけるこれらのリザーバーをより深く理解して、ウイルスの病因を完全に特徴付け、ウイルスを活発に産生しない潜伏感染細胞を効果的に排除する新しい治療法を評価することが重要です。
ここでは、CUBIC3 およびCLARITY9、以前に適応した2つの水性組織透明化プロトコルを、ヒト化免疫系を有するHIV感染マウス(huマウス)、SIV/SHIV感染非ヒト霊長類(NHP)、およびHIV感染ヒトの多数の無傷リンパ組織内の免疫細胞集団の画像化に適用しました。これらのプロトコルは、イメージングの目的(高解像度と大容量)および利用可能な機器に応じて、共焦点およびライトシート蛍光顕微鏡の両方に適応できます。光学顕微鏡では個々のビリオンを分離することはできませんが、免疫蛍光法を使用すると、ウイルスを含む組織やウイルス産生細胞の領域を特定し、より高い分解能の方法でさらに分析することができます。ここで紹介する方法は、感染中のさまざまな条件での特定の細胞タイプ間の空間的関係を定量化するために、体内のほぼすべての組織をシングルセル分解能で視覚化するように適応させることができ、感染症やがんの研究に関連性の高いヒト患者サンプルに容易に変換できます。
プロトコル
すべての動物実験は、承認された施設の動物管理プロトコルに従って実施されました。すべてのヒト組織は、承認された機関のヒト研究倫理ガイドラインに従って取得されました。
1. ティッシュの採取と固定(CUBICとCLARITYで同じ)
- 前に説明したようにリンパ組織を特定して解剖します10.
- 安全に可能な場合は、死後数分以内に解剖はさみとピンセットでリンパ組織を切除します。
- 0.1 Mカコジル酸ナトリウム三水和物に8%パラホルムアルデヒド(PFA)、5%スクロースを含む、新たに調製した氷冷固定緩衝液に組織サンプルを入れ、光学顕微鏡(LM)、電子顕微鏡(EM)、または免疫電子顕微鏡のために組織サンプルを適切に保存します。あるいは、0.1 M PBS中の4% PFAでLMのサンプルを固定します。ウイルスが完全に不活性化されるように、クリアプロセスを開始する前にサンプルを一晩固定します。
注意:パラホルムアルデヒドは、皮膚接触や吸入によって有毒であり、可燃性の固体でもあります。取り扱いには注意し、可燃性の収納キャビネットに保管してください。カコジル酸ナトリウム三水和物は、飲み込んだり吸い込んだりすると有毒です。 - 透明化プロセスを開始する前に、組織の参照画像を撮影してください。
注:LMサンプルは、これらの条件下で少なくとも1年間保存できます。内因性蛍光タンパク質を発現するサンプルを扱うには、後続のステップで常にサンプルを暗くしてください。
2. キュービック組織クリアリング
- リンパ組織サンプルを滅菌0.1 M PBSで3回すすぎ、室温で15分間振とうし、各バッファー交換中にPFAが除去されるようにします。
注意: PFAを含む液体は、機関のガイドラインに従って廃棄してください。 - リンパ組織サンプルをCUBIC Reagent-1( 材料表参照)に37°Cで3日間、穏やかに振とうしながら浸漬します。定期的な参照画像を撮影して、脱色プロセスを経時的に監視します。
- Reagent-1をさらに3〜4日間浸漬するか、組織の脱色が完了するまで交換します。クリアリングに必要な時間は、組織の量と種類の両方によって異なります。組織の脱色プロセスをスピードアップするには、CUBIC Reagent-1を毎日更新し、より多くの量を使用してください。
- リンパ組織サンプルを0.1 M PBSで室温で30分間、穏やかに振とうしながら3回洗浄します。
- リンパ組織サンプルをCUBIC Reagent-2( 材料表を参照)に37°Cで2〜7日間穏やかに振とうしながら、または完全な透明化が得られるまで浸漬します。サンプルが完全な透明化を達成しない場合は、透明化が進行しなくなるまで手順2.2〜2.5を繰り返します。定期的な参照画像を撮影して、クリアプロセスを経時的に監視します。
- リンパ組織サンプルを0.1 M PBSで室温で30分間、穏やかに振とうしながら3回洗浄します。
- サンプルは、0.01%の容量/容量(V / V)アジ化ナトリウムを含むCUBIC試薬-2に暗所で保存します( 材料の表を参照)。
注:サンプルは、この方法を使用して少なくとも6ヶ月間保存できます。
注意: アジ化ナトリウムは非常に毒性が高く、深刻な吸入の危険をもたらします。5%アジ化ナトリウム以下の希釈溶液の購入をお勧めします。
3. キュービックサンプルのブロッキングと免疫染色
- リンパ組織サンプルを0.1 M PBSで3回、室温で30分間、穏やかに振とうしながら洗浄します。
- 共焦点顕微鏡を使用してイメージングするには、組織スライサーマトリックスを使用して組織を厚さ0.5〜1mmのスライスに切断します。ライトシート蛍光顕微鏡(LSFM)を行うには、組織領域全体をブロックします。
- 5 mLのCUBICブロッキング溶液でサンプルを4°Cで一晩、振とうしながらブロッキングします( 材料表を参照)。NHPまたはヒトサンプルを扱う場合は、抗ヒトFcRを使用してください。マウスサンプルを扱う場合は、ブロッキング溶液に抗マウスFcRを使用してください。
- サンプルをブロッキング溶液(種特異的FcRを含まない)で5 mLの一次抗体( 材料表を参照)で室温で3日間、振とうしながら染色します(オプション:濃縮抗体ストックを2,300 x g で5分間遠心分離してから使用してください。これにより、凝集抗体の添加が減ります)。
- 染色したサンプルを室温で最低5時間振とうしながら、少なくとも5回の洗浄液バッファーの交換を行いながら洗浄します( 材料の表を参照)。
- サンプルを二次抗体( 材料表を参照)でブロッキング溶液(種特異的FcRを含まない)で室温で3日間、振とうしながら染色します(オプション:抗体の凝集を最小限に抑えるために、使用前に抗体を2,300 x g で5分間遠心分離します)。
- 染色したサンプルを室温の洗浄液で5回洗浄し、合計で少なくとも5時間振とうします。
- 各組織サンプルに5 mLのDAPI染色溶液( 材料表を参照)でサンプルを染色し、室温で10分間インキュベートします。サンプルを暗所4°CのDAPI染色液に放置し、後でイメージングします。
- リンパ組織サンプルを洗浄液で室温で3回、それぞれ30分間振とうしながら洗浄します。
- 染色したサンプルをCUBIC Reagent-2に室温の暗所で一晩浸してから、サンプルをマウントします。
4. CLARITY組織クリアリング
- リンパ組織サンプルを滅菌済みの0.1 M PBSで3回すすぎ、室温でそれぞれ15分間振とうしてPFAを除去します。
- 組織サンプルを15 mLの新しくできたアクリルアミド溶液に入れ、穏やかに攪拌しながら4°Cで一晩インキュベートします( 材料の表を参照)。
注意:未重合アクリルアミドは強力な神経毒であり、皮膚から容易に吸収されます。皮膚との接触を避け、接触した場合はすぐに洗い流してください。 - 組織サンプルを室温まで温めます。
- オプション:アクリルアミド溶液に窒素を1分間バブリングして、組織サンプルを脱気します。有毒な未重合アクリルアミド(~1〜2バブル/秒)が飛散しないように、低流量を使用するように注意してください。
- 組織サンプルを37°Cのウォーターバスに1〜3時間入れて重合し、15分ごとに反転させます。粘性のある液体、混合中のシュレレン線の出現、または組織の周囲に透明なカプセルが形成されるなど、顕著な重合が検出されたらすぐにサンプルを取り出します。
注:アクリルアミド溶液の完全な重合が起こる場合は、サンプルから余分なヒドロゲルをトリミングし、プロトコールを続行します。 - 組織サンプルを滅菌済みの0.1 M PBSで室温で3回、室温で3回ずつ洗浄し、穏やかに振とうしてアクリルアミド溶液を除去します。
- 組織サンプルを0.1 M PBS中、37°Cで15 mLの8% SDSに入れ、2〜5+日間穏やかに揺さぶってクリアリングします。8% SDS 溶液を定期的にリフレッシュし、必要に応じて最大 50 mL の溶液を使用して透明化を早めます。サンプルが視覚的に透明になるか、進行しなくなったら、クリアプロセスを停止します。定期的な参照画像を撮影して、クリアプロセスを経時的に監視します。
- 組織サンプルを滅菌した0.1 M PBSで室温で1日5回、穏やかに振とうしながら洗浄します。
- 内因性蛍光をイメージングする準備ができるまで、サンプルを一時的に0.1 M PBS(さらに0.01%の容量/体積(v / v)NaN3 で長期保存)暗所に保管します。
- 組織を5 mLのイメージングメディアRI-2に入れます( 材料の表を参照)。暗所で室温で一晩インキュベートし、免疫染色前に透明化プロセスの完全性を確認します。参照画像を撮影して、組織の透明性を監視します。
5. CLARITYサンプルのブロッキングと免疫染色
注:これらのステップは、CUBICクリア組織のブロッキングおよび免疫染色と似ていますが、ブロッキング、洗浄、および染色溶液に異なる製剤を使用します。
- リンパ組織サンプルを0.1 M PBSで3回、室温で30分間、穏やかに振とうしながら洗浄します。
- 共焦点顕微鏡を使用してイメージングするには、0.5 mmの組織スライサーとマトリックスを使用して、組織を厚さ0.5〜1 mmのスライスに切断します。LSFMを実行するには、組織サンプル全体をブロックします。
- 5 mLのCLARITYブロッキング溶液( 材料表を参照)でサンプルを4°Cで一晩、振とうしながらブロッキングします。
- サンプルをブロッキング溶液(分子種特異的FcRを含まない)で5 mLの一次抗体( 材料表を参照)で室温で3日間、振とうしながら染色します(オプション:抗体の凝集を最小限に抑えるために、使用前に抗体を2,300 x g で5分間遠心分離します)。
- 染色したサンプルを室温の洗浄液で5回、合計5時間以上振とうします( 材料の表を参照)。
- サンプルをブロッキング溶液(種特異的FcRを含まない)で室温で3日間振とうしながら、5 mLの二次抗体( 材料表を参照)で染色します(オプション:抗体の凝集を最小限に抑えるために、使用前に抗体を2,300 x g で5分間遠心分離します)。プロトコールの全体の長さを短縮するには、一次抗体を蛍光色素と結合させて使用することで、二次抗体とのインキュベーションの必要性をなくします。
- 染色したサンプルを室温の洗浄液で5回洗浄し、合計で5時間以上振とうします。
- 各組織サンプルに5 mLのDAPI染色溶液( 材料表を参照)でサンプルを染色し、室温で10分間インキュベートします。暗所でサンプルを4°Cに保ってDAPI染色液に浸し、後でイメージングします。
- リンパ組織サンプルを洗浄液で室温で3回、毎回30分間振とうしながら洗浄します。
- 組織を5 mLのイメージング培地RI-2(R.I. = 1.46)に入れ、サンプルを埋取する前に、暗所で室温で一晩インキュベートします(プロトコールステップ6および7を参照)。
6. 共焦点顕微鏡法のための透明化された組織サンプルのマウントとイメージング
- 接着性シリコーンアイソレータのプロテクター層の片側をはがします。
- 顕微鏡カバーガラス(22 mm x 40 mm、厚さ0.25 mm)をシリコーンアイソレータの剥離面に貼り付けて、サンプルの耐液スペースを形成します。
- 接着性シリコーンアイソレータのプロテクター層の反対側をはがします。
- イメージング用サンプルをシリコンアイソレーターの中央に置き、液面がアイソレーターの端まで高くなるまで、CUBIC Reagent-2またはイメージングメディアRI-2を適宜加えます。
- シリコンアイソレータ内の気泡の捕捉を最小限に抑えるには、EM鉗子を使用して、2番目のカバーガラスを片側から整列させ、穏やかに重ねます。余分な液体を拭き取ります。鉗子の裏側を使用して、サンプルウェルの周りのカバーガラスをそっと押して接着剤をシールします。マウントされたサンプルは、暗闇の中で水平に保管してください。
注:サンプルは、マウントされてから数週間から数か月後にイメージングできます。ただし、画像品質は一般的に時間の経過とともに低下します。 - 取り付けられたスライドを顕微鏡ステージに置き、白色光と低倍率の対物レンズ(2〜10倍)を使用してサンプルを配置します。
- 選択した個々の蛍光色素に基づいて蛍光取得プロファイルを設定します。
注:個別の蛍光色素チャネルを個別に取得することをお勧めします。これにより、取得時間が長くなりますが、スペクトルのオーバーラップと非特異的蛍光シグナルの取得が減少します。一般的な蛍光色素プロファイルには、DAPI(450 nm)、Alexa488、Alexa594、Alexa647(または関連する組み合わせ)が含まれ、画像取得中のスペクトルのオーバーラップを最小限に抑えることができます。 - 関心領域を画像化するための適切な倍率対物レンズを選択します。単一細胞の解像度で大容量または全組織イメージングを行うには、低倍率の対物レンズ(2〜10倍)を使用し、透明化された組織の細胞内詳細を高解像度で視覚化するには、高倍率の対物レンズ(20〜63倍)を使用します。対物レンズ、イメージング媒体、および組織の屈折率をできるだけ一致させて、画像取得中の光学的歪みの導入を最小限に抑えます。
- Zスタック取得のステップサイズを選択します。低倍率の対物レンズ(2-10倍)の場合、ステップサイズを~3-5 μmに選択して、複数の連続Zスライス中の個々の細胞からの蛍光を検出し、3Dモデリングを行いながら、合計取得時間と全体のファイルサイズを削減します。高倍率の対物レンズ(20-63倍)の場合は、個々のZスライス間の細胞内情報の損失を最小限に抑えるために、~1μm以下のステップサイズを選択します。
- 視野をズームして、XおよびY次元で画像化する組織の全領域を、空いている領域をできるだけ少なくして視覚化します。イメージングする対象領域全体を含むZステージの上下の取り込み座標を設定します。
- Zスタックイメージを取得します。画像を保存してエクスポートし、任意の画像解析ソフトウェアを使用して後処理します。特定のソフトウェアスイートでは、ファイルを特定のファイルタイプ(.tiff、.ome-tiff、.jpegなど)に変換します。変換は、顕微鏡画像取得ソフトウェアまたは画像解析フリーウェア(ImageJ/Fijiなど)を使用して行うことができます。
7. LSFMチャンバーまたはキュベットへのサンプルのマウントとイメージング
- イメージングチャンバーにCUBIC Reagent-2またはRI-2を充填します。これは、使用する特定のプロトコルによって異なります。液体を移送する際の気泡の形成を避けてください。ピペットで余分な気泡を取り除きます。
- サンプルをイメージングチャンバーに浸し、サンプルの動きを制限します。
注:使用する顕微鏡によっては、アガロースへのサンプルの埋め込み、フックまたはヤマアラシアダプターからのサンプルの懸濁、サンプルホルダーの3Dプリント、プラスチック皿への接着剤によるサンプルの取り付けなどが含まれます。 - 対物レンズをイメージング溶液に置き、サンプルに焦点を合わせます。マウントされたサンプルをイメージングチャンバーに数時間または一晩放置して、キュベット内の溶液と組織が完全に平衡化できるようにします。
- 関心領域の Z スタックを取得します (画像取得については、手順 6.7 から 6.11 を参照してください)。
注:このアプローチにより、1 cm3 を超える組織体積を単一細胞の分解能でイメージングできます。
8. Imaris画像解析ソフトウェアによる表面再構成と細胞定量
注:これらの手順はImaris画像分析ソフトウェアに固有のものですが、他のソフトウェアスイート(ImageJ / Fiji、Aivia、Arivis、Amiraなど)を使用して同様の画像処理手順を実行できます。
- Imaris File Converter を使用して、Z-stack イメージ ファイルをネイティブの Imaris 形式の .ims に変換します。これにより、変換エラーや開いた後の潜在的なソフトウェアの問題を最小限に抑えながら、より迅速なファイル変換が容易になります。
注: 一部の新しい LSFM では、ユーザーがファイルを直接 .ims 形式で保存できます。 - 分析する.imsファイルをImarisソフトウェアの Arena 領域にドラッグします。各カラーチャンネルのコントラストや強度は、 ディスプレイ調整 パネルで調整します。左上の 「新規サーフェスを追加 」アイコンをクリックします。
- 「Next: Source Channel」(右向きの矢印が付いた青いアイコン)をクリックします。構築するサーフェスのソース チャネルを選択します。他のパラメータは変更しないでください。
- [Next: Threshold](右向きの矢印が付いた青いアイコン)をクリックします。
- しきい値(絶対強度)を調整するには、しきい値の線を左または右にドラッグします。 [Split Touching Objects] を有効にし、システムの分割標準として平均セル直径をミクロン単位で入力して、個々のサーフェスの原点として多数のドットを生成します。
- 小さすぎたり明るすぎたりする蛍光シグナルは、染色や顕微鏡のアーティファクトの可能性があるため、含めないでください。許容可能なサイズと蛍光強度を持つドットのみを含め、それに応じて平均細胞径を変更します。
注:平均細胞径は、特定の組織または細胞タイプによって異なりますが、一般的には5〜15μmの間にあります。
- 小さすぎたり明るすぎたりする蛍光シグナルは、染色や顕微鏡のアーティファクトの可能性があるため、含めないでください。許容可能なサイズと蛍光強度を持つドットのみを含め、それに応じて平均細胞径を変更します。
- Next: Classify Surfaces(右向きの矢印が付いた青いアイコン)をクリックします。含めるサーフェスを調整するには、しきい値の線を左または右にドラッグします。表面が生の蛍光シグナルに正確に近似していることを確認するとともに、個々の細胞から蛍光シグナルを分離します。
- [ 完了]をクリックします:すべての作成手順を実行し、ウィザードを終了します (右向きの2つの矢印が付いた緑色のアイコン)。表面は正式に構築されています。
- 左側のパネルにある[統計]というラベルの付いた6番目のアイコンをクリックして、セルの数、この状況では、分析された特定のカラーチャネルの表面の数を確認します。
- Number of Disconnected Components per Time Point、Number of Surfaces per Time Point、Total Number of Disconnected Component、Total Number of Surfaces の 4 つの変数が、そのカラーチャネルのセル数と同じ数であることを確認してください。
結果
ティッシュ・クリアリングでは、保存された組織をケミカルカクテルで処理し、組織構造を維持しながら組織から不透明な生体分子を抽出します。これらの組織透明化ソリューションは、組織の屈折率を周囲のイメージング媒体と一致させることで、光学的歪みを最小限に抑え、組織深部のS/N比を高め、バックグラウンドの自家蛍光を最小限に抑えます。光学組織透明化のための2つの水性プロトコル、CUBIC3 およびCLARITY9を使用して、保存されたHIV/SIV感染hu-マウス、非ヒト霊長類、およびヒト組織サンプルを透明化した後、免疫蛍光染色および共焦点およびライトシート蛍光顕微鏡によるイメージングを行いました。
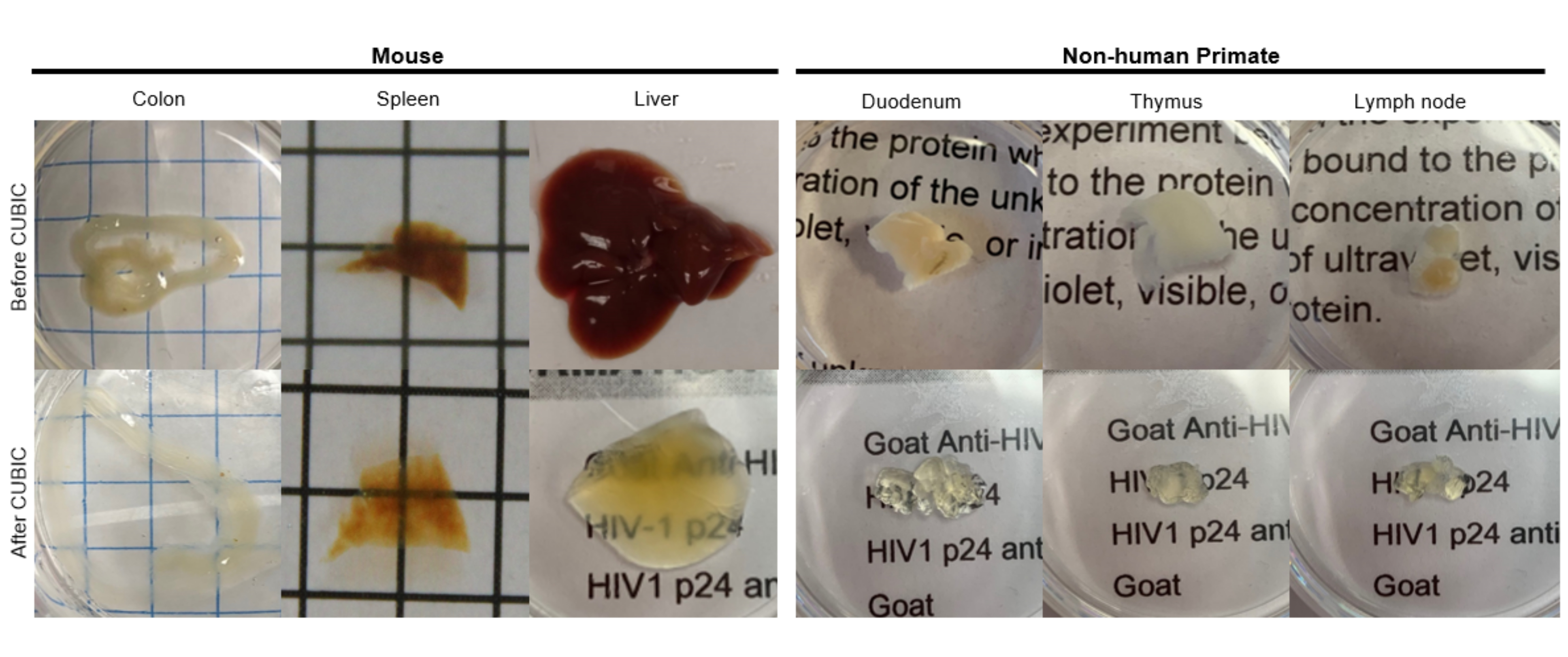
CUBICプロトコールでは、固定組織をPBSで洗浄して固定剤を除去し、ヘムなどの発色団を溶出するアミノアルコールの塩基性緩衝液であるCUBIC Reagent-1に浸漬し、組織の脱色と脱脂を引き起こしました(図1、上)。CUBIC Reagent-1による処理の3日間で、より小さな組織量(~mm3)で脱色することができますが、より大きな組織量(~cm3)や大量のヘムを含む組織(肝臓、脾臓、心臓など)は、より長いインキュベーション時間と溶液量(>1ヶ月および~50mL)を必要とし、2〜3日ごとに溶液を頻繁に交換する必要があります。脱色後、組織を洗浄し、屈折率が約1.48〜1.49のショ糖含有溶液であるCUBIC Reagent-2に入れました。これは、組織の屈折率と一致し、光の透過率を高めます。透明化した組織を免疫染色し、CUBIC Reagent-2溶液にマウントした後、共焦点顕微鏡またはライトシート顕微鏡でイメージングしました。CUBICクリアリング手順の影響を、さまざまなサイズと濃度の発色団のいくつかのhu-mouseおよびNHP組織について画像化しました(図2)。光学的クリアリングにより、組織は肉眼で視覚的に透明になり、紙のシート上のグリッド線とテキストが組織を「透過」して見えるようになりました。脾臓、肝臓、骨髄、心臓などの発色団に富む組織は、完全に脱色しないかもしれませんが、免疫染色やイメージングには適しています(図2 および 図5)。
CLARITYプロトコルでは、固定組織をPBSで洗浄して固定剤を除去した後、熱開始剤を含む40%アクリルアミド溶液で4°Cで一晩インキュベートし、サンプル中のタンパク質とアクリルアミドのモノマーとの間に共有結合を形成しました(図1、下)。翌日、組織を室温まで平衡化し、37°Cの水浴で加温した後、アクリルアミド重合を開始し、サンプルをハイドロゲルに迅速に包み込みました。サンプルを8%SDS溶液で2〜5日間処理し、不透明な脂質を除去しました。蛍光染色の直前に、サンプルを 90% の非イオン性密度グラジエント培地を含む CLARITY (イメージングメディア RI-2) の屈折率マッチング溶液 (RIMS) に浸しました。ヘムを多量に含有する組織の場合、脱脂ステップ9,11,12の最後に脱色ステップを追加することができる。CUBICとCLARITYのクリアリングの進行を、同じヒト脾臓サンプルの異なる切片で比較しました(図3)。CLARITYクリアリングは、溶液を包み込む可視のポリアクリルアミドゲルを生成し、追加の脱色ステップが追加されない限り、CUBICクリアリングと比較して通常、脱色が減少します9,12。
続いて、両方のプロトコルで、透明で無傷の組織を免疫染色して、特定の免疫細胞集団を検出しました。サンプルを洗浄し、α-FcR含有試薬でブロックして非特異的抗体の結合を減らし、蛍光色素に直接結合した一次抗体を使用して3日間染色しました。あるいは、サンプルを非標識一次抗体で3日間染色した後、蛍光色素に標識した二次抗体でさらに3日間染色しました。組織を再度洗浄し、その後、核を可視化するためにDAPI染色剤と4°Cで一晩インキュベートしました。サンプルを洗浄し、CUBIC Reagent-2で24〜36時間インキュベートするか、Imaging Media RI-2(CLARITY)で暗所で一晩インキュベートしました。共焦点顕微鏡法では、イメージングの前に、組織を適切なRIMSの顕微鏡スライドに取り付けました(図4)。ライトシート蛍光顕微鏡(LSFM)の場合、サンプルはイメージング前にRIMSと一晩で完全にライセンシングキュベットに浸されました。
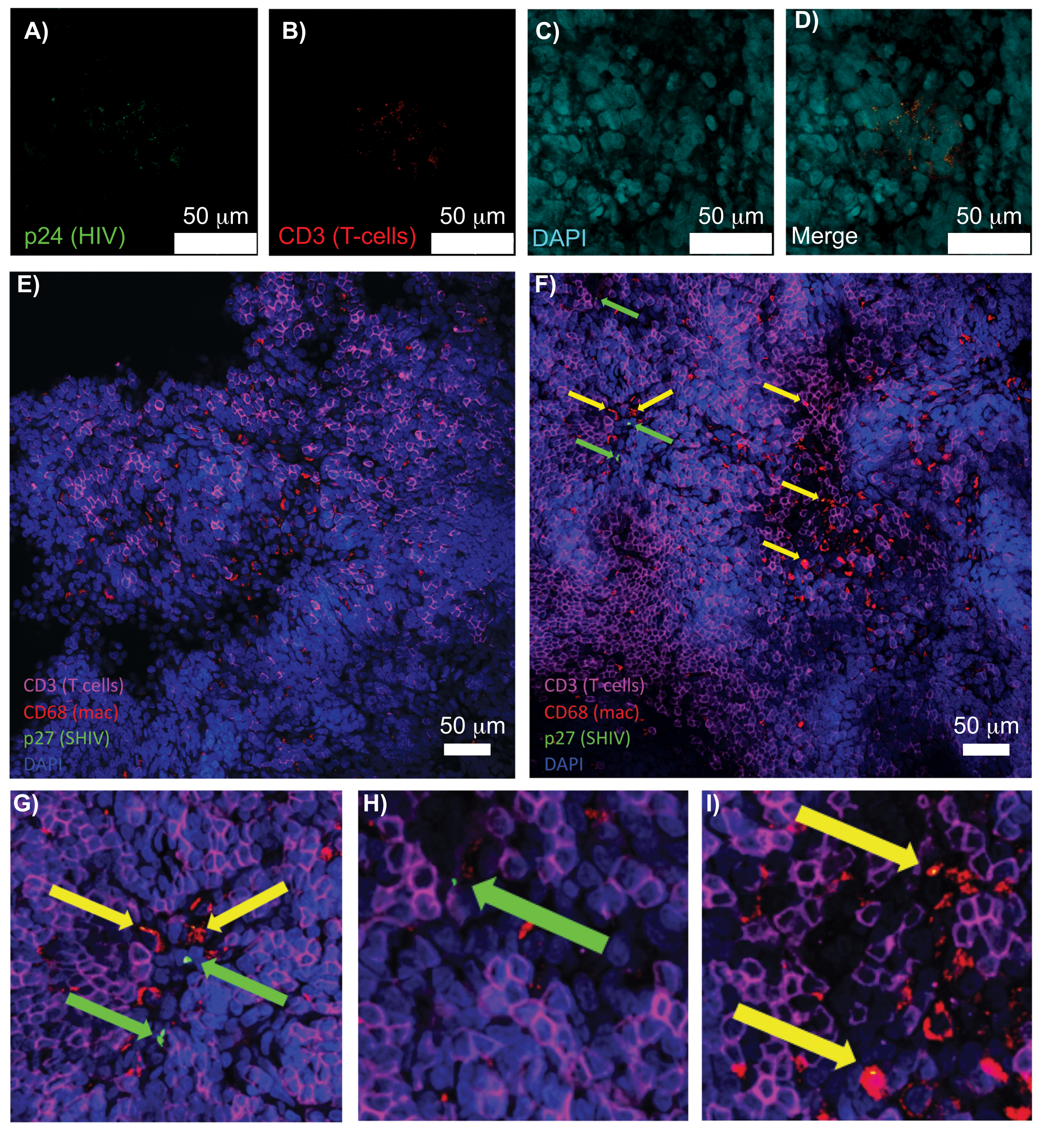
インタクトリンパ組織、透明リンパ組織、免疫染色リンパ組織を共焦点顕微鏡で観察することで、核、免疫細胞マーカー、HIV/SIV CA(キャプシド)タンパク質など、複数の蛍光シグナルを同時に可視化することができました(図5)。ウイルス産生細胞は、免疫細胞マーカーとHIVタンパク質の蛍光共局在によって決定されました。透明化して染色されたHIV感染ヒト脾臓は、HIV p24と共局在する複数のCD3+ T細胞を明らかにし、無傷の組織の領域内にウイルス産生細胞が存在することを示しました(図5A-D)。透明化して免疫染色したSHIV感染NHPリンパ節は、多数のウイルス産生細胞が存在する領域(図5F)に加えて、ウイルスが検出されなかった組織領域(図5E)におけるCD3+ T細胞およびCD68+マクロファージの分布を明らかにしました。これらの結果は、多様な組織源からのウイルス産生細胞が、特定の視野内で他の細胞と区別可能であり、複雑な組織環境内でまれな生物学的事象を検出できることを示しました。
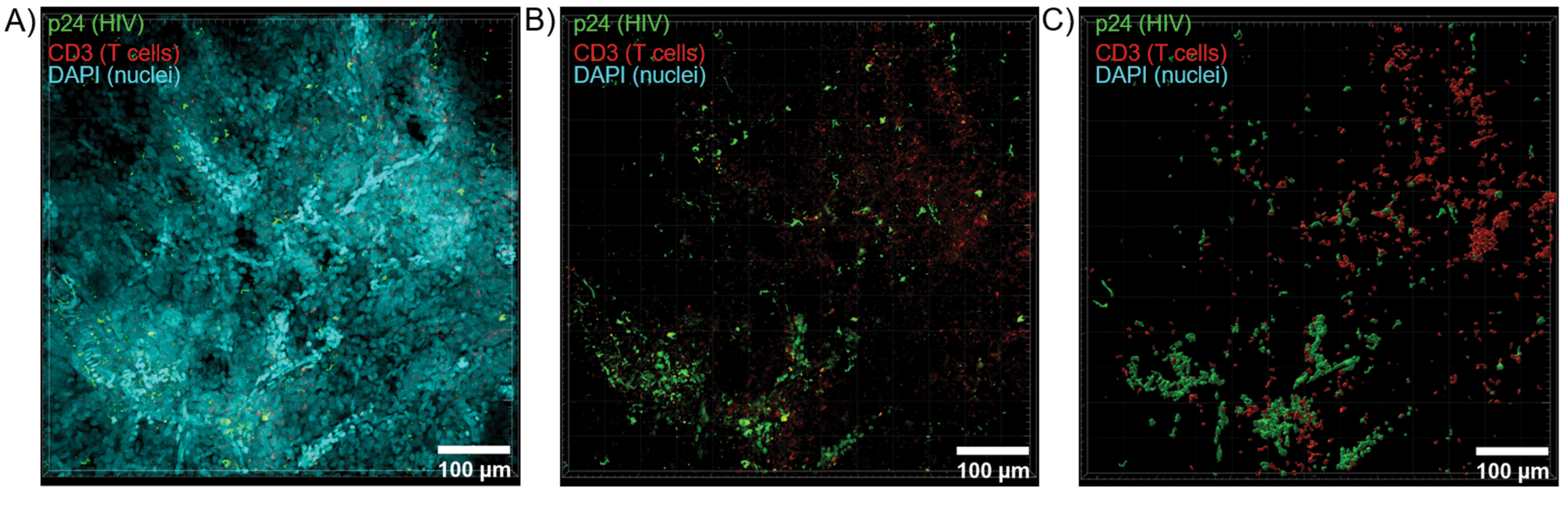
透明化した組織を共焦点顕微鏡で光学的に切片化してZスタックと3D表面モデルを生成したところ、HIV感染時に細胞の不均一性が明らかになりました(図6)。Imarisソフトウェアスイート(図6A)を使用してZスタックをZ投影画像に再結合し、DAPI核チャネルを除去して、組織の全体積にわたるCD3+ T細胞とHIVキャプシドタンパク質(p24)蛍光を明確に可視化しました(図6B)。Z投影蛍光は、Imarisソフトウェアを使用して自動的にセグメント化され、Zスタック全体にわたる蛍光シグナルの空間的可視化と定量化のための再構築された3D表面モデルを生成しました(図6C)。3D表面モデルの解析により、546個のCD3+ T細胞と218個のHIV p24を産生する細胞が明らかになりました。累積的に、透明なHIV感染リンパ組織からの免疫蛍光のZスタック取得により、組織内の細胞組成の3Dモデルの生成と、組織ボリューム内の免疫細胞集団の自動定量が可能になりました。
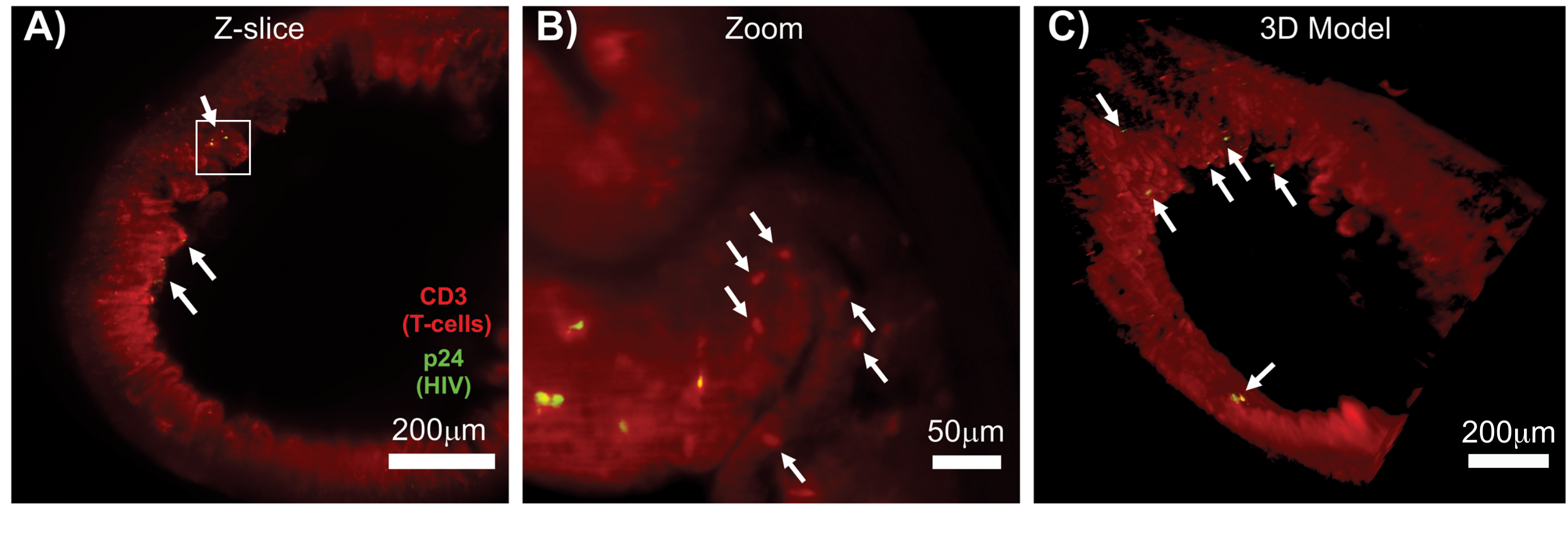
無傷のリンパ組織、透明化されたリンパ組織、および免疫染色されたリンパ組織のLSFMにより、リンパ組織中の免疫細胞およびウイルス産生細胞の分布の免疫蛍光(IF)イメージングが可能になりました(図7)。HIVに感染したhu-mouseの結腸組織をhCD3+ T細胞およびHIV p24に対して免疫染色したところ、感染の証拠がない組織の広い領域に分散したウイルス産生細胞の病巣が明らかになりました(図7A)。ウイルス産生細胞の病巣を拡大して見ると、複数のウイルス産生細胞が潜在的な標的細胞に近接していることが明らかになりました(図7B)。組織自家蛍光(赤色曇色)を用いて、組織全体の構造を可視化し、自家蛍光(赤色の楕円)よりも明るく染色された組織内の特定の免疫細胞集団を区別しました。LSFM Zスタックボリューム全体の3Dモデルは、無傷の組織の領域内のウイルス産生細胞の病巣の空間分布を示し、全体的な組織構造に対するウイルス産生の位置のマッピングを可能にしました(図7C)。驚くべきことに、ウイルス産生細胞の病巣は、ウイルス産生の証拠がないまま、組織の大きな領域に点在することがよくありました。これらの結果により、さまざまな組織内、さまざまな感染時期やさまざまな治療に対する反応におけるウイルス分布と感染細胞密度のパラメーターを定量化できます。

図1:一般的なCUBICおよびCLARITYの組織透明化、免疫染色、イメージングのワークフロー。 CUBIC(上)とCLARITY(下)のクリア時間は、組織のサイズと種類によって大きく異なります。CLARITYのクリアリングには、免疫染色の前に屈折率一致培地との追加のインキュベーションステップが必要であり、組織が透明であることを確認する必要があります。免疫染色には、一次抗体を蛍光色素と結合させる場合は通常3日、蛍光二次抗体が必要な場合は6日かかります。サンプルは、共焦点またはLSFMのいずれかでイメージングできます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:hu-mouseおよびNHP組織サンプルのCUBICクリアリング。 組織サンプルのヘムと脂質の密度が異なるため、各組織タイプの透明化に必要な時間は異なります。例えば、結腸と十二指腸は通常、比較的短い期間(~7日)を必要としますが、脾臓と肝臓は透明になるまでに時間がかかることがあります(~30日)。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:ヒトサンプルの組織透明化方法の縦断的比較。 CUBIC(上面)とCLARITY(下面)は、抗レトロウイルス療法を受けているHIV感染者から脾臓を除去しました。どちらの方法も、免疫染色とイメージングのために32日目までに組織を十分に透明化しました。CUBIC法の脱色ステップでは、脾臓サンプルに含まれるヘムの存在によって引き起こされる自家蛍光が目に見えて減少します。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

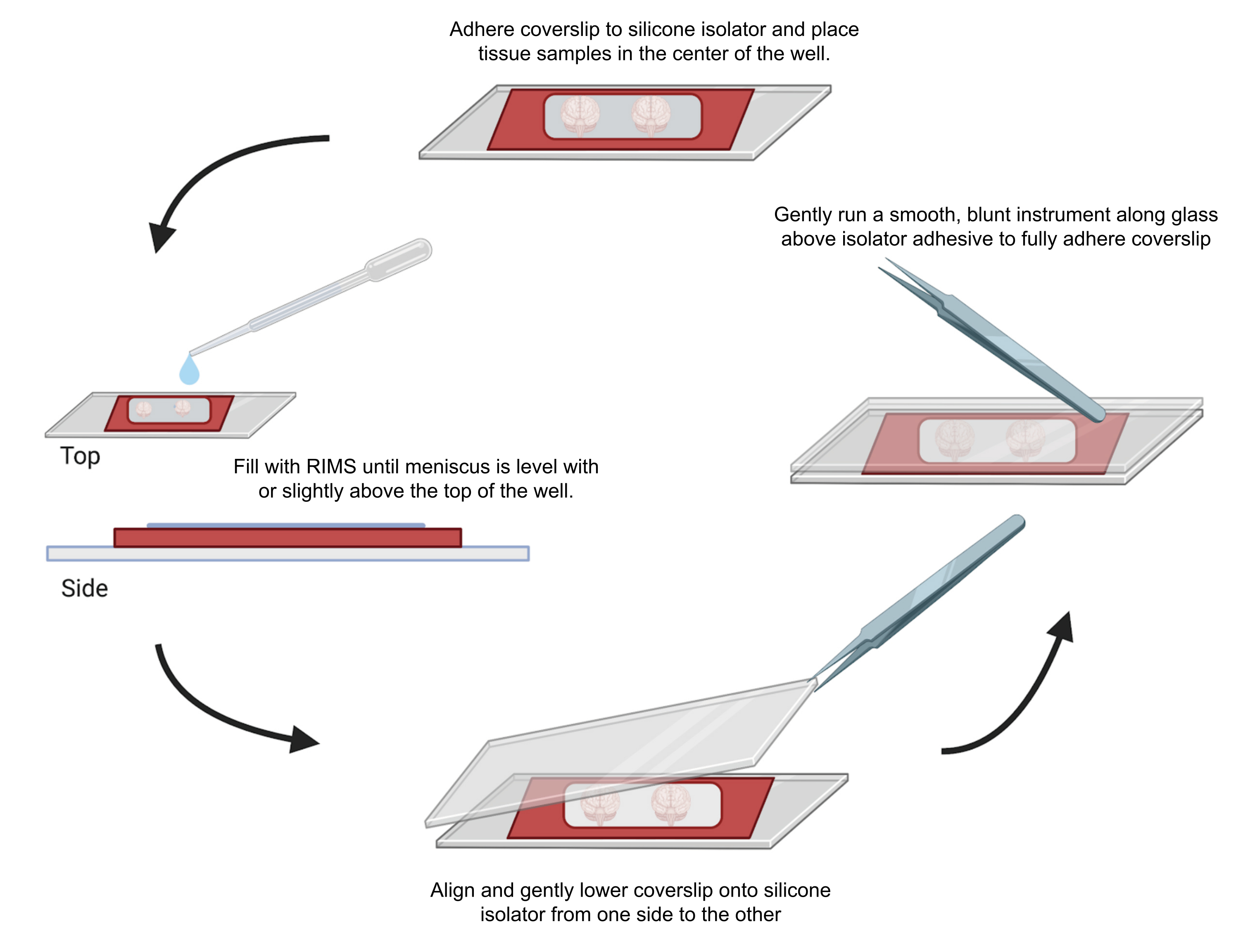
図4:共焦点顕微鏡用のサンプルマウント。 サンプルは、接着剤0.5〜1mmシリコンアイソレーターで分離されたカバースリップの間に取り付けました。シリコーンアイソレーターを最初のカバースリップに接着し、組織をウェルの中央に配置しました(上)。ウェルは、メニスカスがウェルの上部と同じ高さまたはわずかに上になるまでRIMSで満たされました(左)。2枚目のカバースリップは、気泡を避けて片側から反対側に慎重に下げました(底面)。カバースリップは、鈍い器具をウェルの周囲に穏やかに走らせることにより、シリコンアイソレータに完全に接着されました(右)。サンプルは、標準的な共焦点顕微鏡で画像化しました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図5:透明で無傷のヒト脾臓およびNHPリンパ節の共焦点顕微鏡。(A-D)透明化されたHIV感染ヒト組織について、HIV-1 p24(緑)、hCD3+ T細胞(赤)、および核(シアン)について染色しました。(E)感染後8週間のSHIV感染NHPから採取したCUBICクリアリンパ節の共焦点Zスライスを、CD3+ T細胞(マゼンタ)、CD68+マクロファージ(mac/赤)、SHIV p27(緑)、核(青)の免疫染色した。視野にはT細胞、マクロファージ、およびその他の細胞タイプが含まれていますが、SHIV産生細胞の証拠はありません(緑)。 F)同じリンパ節の隣接領域の共焦点Zスライスは、CD3 + T細胞(緑の矢印)とCD68 +マクロファージ(黄色の矢印)を産生するウイルスの存在とともに、細胞密度と数の違いを示しています。(G-I)(F)からp27染色したp27の選択した領域の拡大図。スケールバーは50μmです。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図6:HIVに感染したヒト脾臓からのZスタックボリュームと3D再構築された表面。 (A)HIV-1 p24(緑)、hCD3+ T細胞(赤)、および核(シアン)について染色されたHIV感染ヒト脾臓組織の600μm x 600 μm x 100 μm ZスタックからのZ投影画像。(B)核DAPI染色をしていない同じZプロジェクション画像。(C)Zスタック全容積からのCD3(赤)およびp24(緑)蛍光の再構成3D表面モデル。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図7:HIV感染組織(A)CD3+ T細胞(赤)およびHIV p24(緑)の免疫染色されたHIV感染huマウスの結腸のZスライス(1,000μm x 1,000μm)の表面体積のLSFMおよび3D再構成。鈍い赤い霞は組織の自家蛍光を表し、はっきりとした赤い点はT細胞を示します。絨毛は、ウイルスを含まない広い領域に分散した活発なウイルス産生のいくつかの焦点(白い矢印)が中央の内腔を指して周辺の周りに見られます。ボックスはパネルBのおおよその関心領域を示す(B)個々のウイルスが非感染T細胞(赤)に近接してhCD3+ T細胞(黄色)を産生することを示す組織のズーム領域。画像を回転させて近くのZスライスに変更し、1つのZ面にp24陽性細胞の焦点を示しました。背景の赤色の自家蛍光は、特定のhCD3+ T細胞染色(赤のプンクタ、白い矢印)に加えて、一般的な組織構造を示しています。(C)Imarisソフトウェアで生成された全容積(1,000μm×1,000μm×200μm)の3D表面モデルを回転させて、腸の明確な場所にHIV感染の病巣(黄色)を示す。白い矢印は、ボリューム内の個々の焦点を示します。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
目的のリンパ組織は、染色やイメージングに悪影響を及ぼす可能性のある組織壊死(暗色または黒色組織)を避けるために、死後迅速に採取し、直ちに冷却済みの固定緩衝液に入れる必要があります。目的の組織を採取した後、すぐに組織を氷冷した4%〜8%パラホルムアルデヒド(PFA)に一晩浸して固定します。これにより、サンプルに関連する潜在的な病原体も不活性化されます。4% PFA は LM サンプルの固定に最適ですが、8% PFA は LM と EM の両方の組織を適切に保存できます。これらの手順に従い、暗所で4°Cの固定液にサンプルを保存すると、LMイメージング用の組織を数年間効果的に保存できます。1つの注意点は、固定剤での長期保存は、染色アーティファクト、特に目的のタンパク質への隣接タンパク質の架橋によって引き起こされる抗原マスキングの導入につながる可能性があり、エピトープ13,14に対する染色抗体のアクセス性を閉塞する可能性があることです。組織に内因的に発現した蛍光タンパク質が含まれている場合は、プロトコル全体を通じて可能な限り組織を光にさらさないように対策を講じてください。通常、内因性蛍光タンパク質は固定後6〜12か月間蛍光を保持しますが、個々の組織サンプルは、より長い期間またはより短い期間で変化する可能性があります。タンパク質の分解により内因性蛍光が失われた場合、目的のタンパク質に特異的な一次抗体を使用して蛍光タンパク質を検出できることがよくあります。灌流は、クリアする前に組織を迅速に固定するための別のオプションです12;しかし、HIVなどの病原体を扱う際の懸念から、できるだけ安全にサンプルを調製するために、組織剖検とそれに続く氷冷固定剤への浸漬のルートが選択されました。
記載されている水ベースのクリアリングプロトコルの利点の1つは、一般的に有機ベースのプロトコルよりも穏やかであり、肝臓などのより壊れやすい組織を損傷する可能性があることです。水ベースの清澄化プロトコルは、有機ベースの清算プロトコルと比較して、一般に完全なサンプル清澄化を達成するのに長い時間(数週間対数日)を必要とします。CLARITYおよびCUBICプロトコルは、灌流を使用してより迅速に実施し、げっ歯類内のすべての臓器を同時に除去することができます11,12;しかし、これはNHPと人間の剖検にとって実行可能な選択肢ではありませんでした。CLARITYで処理されたサンプルは、ある程度の体積拡大を示す傾向がありますが、CUBICはサンプル量への影響が減少していることを示しました9。一般的にはより迅速ですが、多くの有機ベースの組織クリアプロトコルは組織を収縮させ15、これによりリンパ節や脾臓などの細胞密集組織内での単一細胞または細胞内の詳細の検出がより困難になる可能性があります。クリアリングによる拡張により、イメージングの分解能が効果的に向上し、組織の元のサイズでは観察しにくい側面を観察することが容易になります。あるいは、組織の縮小によりサンプルの全体的なサイズが効果的に縮小され、解剖せずに臓器全体のイメージングが可能になります。CLARITYプロトコルとCUBICプロトコルの両方の利点は、組織中の内因性に発現した蛍光タンパク質からの蛍光を保持しながら、免疫蛍光染色11,12に順応性を保つことです。免疫染色は、水性または有機組織透明化法を用いて実施できます。しかし、個人的な経験から、水ベースのプロトコールを使用した抗体適合性は、有機ベースのプロトコールと比較して高い割合であることが示されました。研究者は、画像化された組織と対処される生物学的な問題(例えば、全臓器イメージングと特定の関心領域イメージング)に基づいて、どの組織クリアリング法を使用するかを検討する必要があります。すべての大容量イメージングの問題に対して堅牢なターンキー分析を可能にする普遍的な組織透明化技術はなく、利用可能な方法は生物学的アプリケーションに応じて明確な長所と短所を示します。
抗体染色を行う際には、多くの側面を考慮する必要があります。CLARITYサンプルはアクリルアミドハイドロゲルに埋め込まれているため、インキュベーションに時間がかかる傾向があります12。抗体のインキュベーションに必要な時間は、各サンプルの量と厚さによっても異なります。ここに記載されているほとんどのサンプルの厚さは2~3ミリメートルで、組織全体で完全に染色するには3日で十分でした。ターゲットがマウスの脳全体をイメージングすることである場合、抗体のインキュベーション時間は1週間以上かかることがあります12。免疫蛍光イメージングのための水系組織透明化法と有機組織透明化法のどちらを選択するかは、抗体適合性に左右されます。一般に、CUBICまたはCLARITYの場合、培養細胞や組織で機能する抗体のヒット率は~70%です。水系組織透明化法と有機組織透明化法のいずれを使用する場合でも、使用する特定の方法とすべての抗体の適合性と有効性を評価する必要があります。このプロトコールのセクションで示したように、CUBICおよびCLARITYで処理したサンプルの免疫染色は、クリアリングが終了した後に行われます。それどころか、このステップは、一部の有機ベースのプロトコルのクリア手順の前に行われ、その後にポストフィックスが行われます。
組織は、屈折率に一致するイメージング媒体に完全に浸されることが非常に重要です。そうしないと、イメージング中に球面収差が発生し、画像取得中にキャプチャされた光が歪んでしまいます。共焦点とLSFMの両方のサンプルをマウントする際には、気泡がサンプルに出入りする光の経路を乱す可能性があるため、イメージング媒体からすべての気泡を取り除くように注意する必要があります。気泡は、最終サンプルをマウントする前にピペットで手動で除去できます。共焦点顕微鏡でより厚いサンプルをイメージングする場合、複数のシリコーンスペーサーを互いに重ねて、厚さ0.5mmを超える組織に対応できます。推奨事項の1つは、RIMS内のすべての組織を数時間から一晩中平衡化し、追加のサンプル移動なしで顕微鏡に装着することです。組織とイメージング媒体の完全な平衡化により、イメージング中に収差を生じさせる可能性のある屈折率の不整合を持つ溶液の混合を防ぐことができます。透明化された組織サンプルをマウントする場合、すべての顕微鏡ですべてのサンプルをイメージングするための単一のターンキーマウント方法がないことを覚えておくことが重要です。このプロトコルでは、1つのコンテキストで最適に機能したサンプルのマウントオプションについて説明しますが、使用する個々の顕微鏡や対処する生物学的な問題に応じて、サンプルのマウントには多くのアプローチがあります。これらのアプローチには、アガロースへのサンプルの埋め込み、フックまたは屈折率が一致したプラスチックラインからサンプルを懸濁する、ヤマアラシアダプターの使用、サンプルホルダーの3Dプリント、またはサンプルを接着剤でプラスチック皿に取り付けることが含まれますが、これらに限定されません。
共焦点顕微鏡は、組織量~1 mm3-1 cm3のイメージングに適しています。共焦点顕微鏡では、2-10倍の対物レンズを使用して、最初に関心領域を特定し、単一細胞の分解能でより大きな体積または全組織のZスタックを取得します。20-63x対物レンズに切り替えて、細胞内情報を含む特定の関心領域の高解像度画像を取得します。CUBICおよびCLARITYでクリアされた組織のイメージングに最適な対物レンズは、組織およびイメージング溶液の屈折率に正確に適合したCLARITY/Scale固有の対物レンズです。このタイプの対物レンズが利用できない場合は、空気対物レンズではなく、グリセロールまたは油浸対物レンズ(LD LCI Plan-Apochromat 25 x 0.8 NA Imm Corr DIC M27 マルチイマージョン対物レンズ:作動距離 = 0.57 mm)でサンプルをイメージングするのが最適です。これにより、画像キャプチャ中の屈折率の不一致による光学的歪みの発生が最小限に抑えられます。20-25x対物レンズは、複雑な組織環境における個々の細胞からの染色詳細の取得と、大量の画像取得のバランスをとることができます。重要なことは、ほとんどの共焦点顕微鏡には、イメージングボリュームの3Dタイリングを可能にするモジュールが含まれていることです。このタイプの画像取得は、細胞内情報を含む大量のZスタックを生成するのが理想的です。
LSFMイメージングは、大量の組織(>1 cm3)や臓器全体のコンテキスト内で、特定の細胞集団を3Dで視覚化することができます。過去10年間、LSFMと組み合わせた組織除去は、主にげっ歯類内の脳の接続性の理解に焦点を当てていました。しかし、より最近のアプリケーションには、腫瘍転移ランドスケープ16の視覚化、解剖学的コンパートメント内の細胞分布9,17、および病原体分散18が含まれる。培養細胞と比較して、組織内のほとんどの生物学的イベントは不均一であり、LSFMは、これらのイベントの空間的組織の不均一性(ウイルス複製、免疫シグナル伝達、細胞分布など)を視覚化および定量するのに特に優れています。
共焦点またはLSFMを介して取得した3Dデータセットは、多数の画像解析プラットフォームで後処理できます。Imarisソフトウェアスイートは、サーフェスの構築、3Dアニメーションの生成、および細胞の定量化に使用できます。しかし、効率的な画像後処理と解析を可能にする画像解析システムは数多くあります。ImageJ/Fijiフリーウェア19 は、ほとんどのラボでアクセス可能な魅力的な代替画像処理プラットフォームですが、あらゆる形式の画像分析と視覚化に優れた万能の分析ソフトウェアはありません。多くの画像解析ソフトウェアスイートは、共有使用施設を通じて利用できない場合、法外に高価になる可能性があります。最後に、LSFMまたは大規模なタイル型共焦点3Dデータセットの重要な側面は、データ管理です。これらのイメージングプラットフォームは、データの視覚化と定量化のためにハイエンドのコンピューターワークステーションを必要とする大量のファイル(>1 Tb)を生成することができます。最終的に、このイメージングワークフローは、組織全体内の空間的に異なる細胞集団の取得と定量を効率化でき、ほとんどの組織源や生物学的システムに広く適用できます。
開示事項
著者には開示すべき利益相反はありません。
謝辞
イリノイ大学アーバナ・シャンペーン校ゲノム生物学研究所のコア施設に、共焦点およびライトシート蛍光顕微鏡を使用していただき、ありがとうございます。ヒト組織サンプルの「The Last Gift」コホートの素晴らしい方々に感謝します。このコホートは、I147821、DA051915、AI131385、P30 AI036214の助成金によって資金提供されました。Nancy Haigwood氏とAnn Hessell氏、SHIV感染NHP組織サンプルの提供に感謝します。
資料
| Name | Company | Catalog Number | Comments |
| Acrylamide Solution (in 0.1 M PBS, 40 mL in total) | |||
| 40% Acrylamide: 4 mL | Bio-Rad | 1610144 | |
| VA-044 Thermal Initiator: 0.1g | Fujifilm | 011-19365 | |
| CLARITY Blocking solution (in 0.1 M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Tween-20: 5 µL | Fisher Scientific | BP337-500 | |
| CLARITY wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Tween-20: 50 µL | Fisher Scientific | BP337-500 | |
| CUBIC Blocking solution (in 0.1M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Triton X-100: 5 µL | VWR | M143-1L | |
| CUBIC Reagent-1 (in 0.1M PBS, 50 mL in total) | |||
| N, N, N’, N’-tetrakis (2-hydroxypropyl) ethylenediamine: 12.5 g | Aldrich | 122262 | |
| Triton X-100: 7.5 g | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC Reagent-2 (in 0.1M PBS, 50 mL in total) | |||
| Sucrose: 25 g | Sigma | S1888-500G | |
| Sodium azide (in powder form): 10 g | Sigma | 71289-50G | |
| Sodium azide stock solution (in DI H2O, 50 mL in total) | Sigma | 71289-50G | |
| Triethanolamine: 5 g | Sigma | 90270-500mL | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| DAPI staining solution (0.5 µg/mL) | |||
| DAPI stock solution: 1 µL | |||
| Wash solution: 10 mL | |||
| DAPI stock solution (5 mg/mL) | |||
| DAPI powder: 5 mg | Sigma-Aldrich | D9542-1MG | |
| DMSO (100%): 1 mL | ThermoFisher | D12345 | |
| Imaging Media RI-2 (in dH2O) | |||
| 90% Histodenz | Sigma | D2158-100G | |
| 0.01% Sodium azide | Sigma | 71289-50G | |
| 0.02 Sodium Phosphate Buffer, pH 7.5 | Sigma-Aldrich | S9638-250G | |
| 0.1% Tween-20 | Fisher Scientific | BP337-500 | |
| Primary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Goat anti-HIV p24: 10 µL (1:200) | Creative Diagnostics | DPATB-H81692 | |
| Mouse anti-human CD68: 10 µL(1:200) | Dako | M0876 | |
| Rabbit anti-human CD3: 10 µL (1:200) | Dako | A0452 | |
| 8% SDS Solution (in 0.1 M PBS, 50 mL in total) | |||
| SDS powder: 4 g | Sigma-Aldrich | L3771-500G | |
| Secondary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Donkey anti-goat conjugated with AlexaFluor647: 2 µL | Invitrogen | A21447 | |
| Donkey anti-mouse conjugated with AlexaFluor594: 2 µL | Invitrogen | A21203 | |
| Donkey anti-rabbit conjugated with AlexaFluor488: 2 µL | Invitrogen | A21206 |
参考文献
- Spalteholz, W., Barker, L. F., Mall, F. P. Hand-Atlas of Human Anatomy. , J.B. Lippincott Co. Philadelphia. Second edition in English (1907).
- Jacob, T., Gray, J. W., Troxell, M., Vu, T. Q. Multiplexed imaging reveals heterogeneity of PI3K/MAPK network signaling in breast lesions of known PIK3CA genotype. Breast Cancer Research and Treatment. 159 (3), 575-583 (2016).
- Kieffer, C., Ladinsky, M. S., Ninh, A., Galimidi, R. P., Bjorkman, P. J. Longitudinal imaging of HIV-1 spread in humanized mice with parallel 3d immunofluorescence and electron tomography. eLife. 6, 23282(2017).
- Chung, K., Deisseroth, K. CLARITY for mapping the nervous system. Nature Methods. 10 (6), 508-513 (2013).
- Compton, A. A., Schwartz, O. They might be giants: Does syncytium formation sink or spread HIV infection. PLoS Pathogens. 13 (2), 2-8 (2017).
- Symeonides, M., et al. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses. 7 (12), 6590-6603 (2015).
- Sharova, N., Swingler, C., Sharkey, M., Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. The EMBO Journal. 24 (13), 2481-2489 (2005).
- Colby, D. J., et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nature Medicine. 24 (7), 923-926 (2018).
- Ladinsky, M. S., et al. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. eLife. 8, 46916(2019).
- Buettner, M., Bode, U. Lymph node dissection--understanding the immunological function of lymph nodes. Clinical and Experimental Immunology. 169 (3), 205-212 (2012).
- Treweek, J. B., et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nature Protocols. 10, 1860-1896 (2015).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159, 911-924 (2014).
- Sompuram, S. R., Vani, K., Bogen, S. A. A molecular model of antigen retrieval using a peptide array. American Journal of Clinical Pathology. 125 (1), 91-98 (2006).
- Scalia, C. R., et al. Antigen masking during fixation and embedding, dissected. The journal of histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 65 (1), 5-20 (2017).
- Jing, D., et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Research. 28 (8), 803-818 (2018).
- Guldner, I. H., et al. An Integrative platform for three-dimensional quantitative analysis of spatially heterogeneous metastasis landscapes. Scientific Reports. 6, 24201(2016).
- Muntifering, M., et al. Clearing for deep tissue imaging. Current Protocols in Cytometry. 86 (1), 38(2018).
- DePas, W. H., et al. Exposing the three-dimensional biogeography and metabolic states of pathogens in cystic fibrosis sputum via hydrogel embedding, clearing, and rRNA labeling. mBio. 7 (5), 00796(2018).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image. Nature Methods. 9 (7), 676-682 (2012).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved