
Method Article
3D-визуализация популяций иммунных клеток в ВИЧ-инфицированных тканях с помощью клиринга, иммуноокрашивания, конфокальной и световой флуоресцентной микроскопии
В этой статье
Резюме
Очистка тканей в сочетании с иммунофлуоресцентной микроскопией позволяет визуализировать и количественно оценить популяции иммунных клеток и вирусные белки в интактных тканях. Оптическое срезание очищенных тканей с помощью конфокальной и световой флуоресцентной микроскопии позволяет создавать 3D-модели сложных тканевых сред и выявлять пространственную гетерогенность, проявляющуюся при ВИЧ-инфекции.
Аннотация
Вирус иммунодефицита человека (ВИЧ), возбудитель синдрома приобретенного иммунодефицита (СПИДа), является серьезной глобальной проблемой здравоохранения, в которой инфицировано почти 40 миллионов человек во всем мире и нет широко доступного лекарства. Несмотря на интенсивные усилия, детальное понимание взаимодействия вируса и клетки-хозяина в тканях во время инфекции и в ответ на терапию остается неполным. Чтобы устранить эти ограничения, методы очистки тканей на водной основе CUBIC (Clear, Unobstructed Brain/Body Imaging Cocktails and Computational analysis) и CLARITY (Clear Lipid-exchange Acrylamide-hybridized Rigid Imaging/Immunostaining/in situ-hybridization-compatible Tissue hYdrogel) применяются для визуализации сложных взаимодействий вируса-хозяина и клеток в ВИЧ-инфицированных тканях животных моделей и людей с использованием конфокальной и световой флуоресцентной микроскопии. Оптический срез интактных тканей и анализ изображений позволяют быстро реконструировать пространственную информацию, содержащуюся в целых тканях, и количественно оценить популяции иммунных клеток во время инфекции. Эти методы применимы к большинству источников тканей и различным биологическим вопросам, включая инфекционные заболевания и рак.
Введение
Растущая потребность в количественной пространственной визуализации тканей в биологических исследованиях в последнее время привела к появлению методов очистки тканей для получения изображений большего объема (мм3-см 3) интактных тканей с разрешением одной клетки. Ткани включают в себя сложные организации биомолекул с уникально определенными структурами, составами и функциями. К сожалению, многие биомолекулы, присутствующие в тканях (например, липиды и хромофоры), рассеивают, поглощают или излучают свет при визуализации с помощью световой микроскопии, что затрудняет визуализацию больших объемов. Кроме того, ткани часто демонстрируют показатель преломления, не соответствующий стандартным решениям для визуализации и оптическим линзам, что приводит к оптическим искажениям во время визуализации. Оптимальный подход к визуализации больших объемов тканей с помощью светового микроскопа должен включать согласование показателя преломления тканей, решений для визуализации и объективов, при этом позволяя свету проникать глубоко в ткань без нарушения биологических особенностей интактных тканей во время обработки. Первоначальные попытки уменьшить различия в показателе преломления между тканями и визуализирующими растворами путем очистки от непрозрачных образцов тканей были предприняты немецким анатомом Вернером Шпалтехольцем в конце 1800-хгодов1. Этот метод очистки тканей включал в себя агрессивные химические растворители, которые могут повредить образцы тканей, но, тем не менее, представлял собой первую зарегистрированную визуализацию неповрежденных тканей в больших объемах. Современные методы световой микроскопии в сочетании с вычислительными мощностями для захвата и анализа изображений недавно вновь ввели в моду очищение тканей в качестве метода визуализации больших, неповрежденных образцов тканей с разрешением одной клетки. За последние два десятилетия появились десятки передовых методов очистки тканей, в том числе на органической и водной основе, каждая из которых имеет свои сильные и слабые стороны для конкретных применений.
3D-визуализация тканей может исследовать более сложные биологические взаимодействия, которые не могут быть воспроизведены в клеточной культуре. Например, паттерны клеточной сигнализации2, пространственное распределение различных типов клеток3 и связь мозга4были ранее картированы количественным образом с использованием методов визуализации всей ткани/органа. Здесь описано применение протоколов очистки тканей на водной основе для очистки, иммуноокрашивания и визуализации различных популяций клеток-мишеней ВИЧ в интактных ВИЧ-инфицированных лимфоидных тканях во время активной инфекции. В организме ВИЧ преимущественно инфицирует CD4+ Т-клетки и интегрирует копию своего генома в геномы инфицированных клеток-хозяев. Впоследствии вирус захватывает механизм инфицированной клетки-хозяина для самовоспроизведения, что приводит к распространению вируса, уничтожению клеток-хозяев, иммунной дисфункции и долгосрочному прогрессированию в сторону СПИДа. Важно отметить, что поведение инфицированных Т-клеток в тканях и клеточных культурах заметно различается. Культивируемые CD4+ Т-клетки, инкубированные с ВИЧ, могут продуцировать массивную индуцированную ВИЧ синцитию, которая может включать десятки ядер5, в то время как аналогичные эксперименты с первичными CD4+ Т-клетками, культивируемыми в гидрогелях 3D внеклеточного матрикса (ECM) или образцах тканей ВИЧ-инфицированных гуманизированных мышей (hu-мышей), обычно дают синцитию с 2-5 ядрами6. Понимание местной передачи вируса от клетки к клетке и системного распространения вируса среди ВИЧ-инфицированных людей, вероятно, еще более сложное, поскольку включает транспортировку вируса несколькими типами инфицированных клеток из тканей в кровеносные сосуды и в новые ткани, где свободные вирионы и клетки, продуцирующие вирус, могут получить доступ к большому количеству восприимчивых лимфоцитов7. Эти сценарии в настоящее время невозможно воспроизвести в системах клеточных культур, и ткани животных моделей и человека остаются важным ресурсом для понимания патогенеза вируса в контексте сложного организма с функционирующей иммунной системой.
Современные антиретровирусные препараты (АРТ) значительно увеличивают ожидаемую продолжительность и качество жизни людей с ВИЧ (ЛСГ), подавляя репликацию ВИЧ и останавливая прогрессирование заболевания в сторону СПИДа. К сожалению, АРТ не устраняет латентно инфицированные иммунные клетки, содержащие инсерцию ретровирусного генома, которые находятся в состоянии покоя и не продуцируют активно вирус. Несмотря на то, что вирус не обнаруживается в крови большинства лиц, получающих АРТ, вирусная нагрузка быстро восстанавливается после прерывания АРТ и продолжения прогрессирования заболевания8. Персистирующий характер ВИЧ-инфекции, вызванный латентным резервуаром инфицированных клеток, представляет собой серьезное препятствие для разработки лекарства от ВИЧ. Тканевые резервуары ВИЧ остаются плохо изученными, и крайне важно установить более глубокое понимание этих резервуаров в лимфоидных тканях до, во время и после АРТ, чтобы полностью охарактеризовать патогенез вируса и оценить новые методы лечения, которые эффективно устраняют латентно инфицированные клетки, не продуцирующие активно вирус.
Здесь CUBIC3 и CLARITY9, два ранее адаптированных протокола очистки тканей на водной основе, были применены для визуализации иммунных клеточных популяций в многочисленных интактных лимфоидных тканях ВИЧ-инфицированных мышей с гуманизированной иммунной системой (hu-mice), SIV/SHIV-инфицированных приматов (NHP) и ВИЧ-инфицированных людей. Эти протоколы могут быть адаптированы как к конфокальной, так и к световой флуоресцентной микроскопии в зависимости от целей визуализации (более высокое разрешение против большего объема) и доступных инструментов. Хотя световая микроскопия не может разрешить отдельные вирионы, использование иммунофлуоресценции может идентифицировать участки ткани, содержащие вирус и вируспродуцирующие клетки, которые могут быть дополнительно проанализированы с помощью методов с более высоким разрешением. Представленные здесь методы могут быть адаптированы для визуализации практически любой ткани в организме с разрешением по одной клетке с целью количественной оценки пространственных отношений между конкретными типами клеток в различных условиях во время инфекции и могут быть легко переведены на высокорелевантные образцы пациентов для изучения инфекционных заболеваний или рака.
протокол
Все эксперименты на животных проводились в соответствии с утвержденными протоколами институционального ухода за животными. Все человеческие ткани были получены в соответствии с утвержденными институциональными руководящими принципами этики исследований человека.
1. Забор и фиксация тканей (одинаково для CUBIC и CLARITY)
- Определите и препарируйте лимфоидные ткани, как описано ранее10.
- Иссекайте лимфоидные ткани с помощью препарирующих ножниц и пинцета в течение нескольких минут после вскрытия, когда это безопасно.
- Поместите образцы тканей в свежеприготовленный ледяной фиксирующий буфер, содержащий 8% параформальдегида (PFA), 5% сахарозы в 0,1 М тригидрате какодилата натрия, чтобы обеспечить адекватное сохранение образцов тканей для световой микроскопии (LM), электронной микроскопии (EM) или иммуно-ЭМ. В качестве альтернативы можно зафиксировать образцы на LM с 4% PFA в 0,1 М PBS. Зафиксируйте образцы на ночь перед началом процесса очистки, чтобы обеспечить полную деактивацию вируса.
ВНИМАНИЕ: Параформальдегид токсичен при контакте с кожей и вдыхании, а также является легковоспламеняющимся твердым веществом; Обращайтесь бережно и храните в легковоспламеняющемся шкафу для хранения. Тригидрат какодилата натрия токсичен при проглатывании или вдыхании. - Сделайте эталонный снимок ткани перед началом процесса очищения.
ПРИМЕЧАНИЕ: В этих условиях образцы LM могут храниться не менее 1 года. Для работы с образцами, экспрессирующими эндогенные флуоресцентные белки, на последующих этапах всегда держите образцы в темноте.
2. КУБИЧЕСКАЯ очистка тканей
- Образцы лимфоидной ткани промывают в стерильном 0,1 М PBS три раза с встряхиванием при комнатной температуре в течение 15 мин для обеспечения удаления PFA при каждой смене буфера.
ПРИМЕЧАНИЕ: Утилизируйте жидкости, содержащие PFA, в соответствии с рекомендациями учреждения. - Погрузите образец лимфоидной ткани в реагент CUBIC Reagent-1 (см. Таблицу материалов) при температуре 37 °C на 3 дня с легким встряхиванием. Регулярно делайте эталонные изображения, чтобы отслеживать процесс обесцвечивания с течением времени.
- Замените Реагент-1 на дополнительные 3-4 дня погружения или до полного обесцвечивания тканей. Время, необходимое для очистки, зависит как от объема, так и от типа ткани. Чтобы ускорить процесс обесцвечивания тканей, ежедневно обновляйте реагент CUBIC Reagent-1 и используйте большие объемы.
- Образцы лимфоидной ткани трижды промыть 0,1 М PBS в течение 30 мин при комнатной температуре с легким встряхиванием.
- Погрузите образцы лимфоидной ткани в реагент CUBIC Reagent-2 (см. Таблицу материалов) при температуре 37 °C с легким встряхиванием в течение 2-7 дней или до достижения полной прозрачности. Если образцы не достигают полной прозрачности, повторяйте шаги 2.2-2.5 до тех пор, пока очистка больше не будет продолжаться. Регулярно делайте эталонные изображения, чтобы контролировать процесс очистки с течением времени.
- Образцы лимфоидной ткани трижды промыть 0,1 М PBS в течение 30 мин при комнатной температуре с легким встряхиванием.
- Образцы хранят в реактиве КУБИЧЕСКИЙ КУБ-2 с содержанием азида натрия 0,01% объем/объем (V/V) в темноте (см. Таблицу материалов).
ПРИМЕЧАНИЕ: При использовании этого метода образцы могут храниться не менее 6 месяцев.
ВНИМАНИЕ: Азид натрия очень токсичен и представляет серьезную опасность при вдыхании. Рекомендуется приобретать разбавленные растворы с содержанием 5% азида натрия или менее.
3. Блокирование и иммуноокрашивание кубических образцов
- Образцы лимфоидной ткани промыть три раза по 0,1 М PBS в течение 30 мин при комнатной температуре с легким встряхиванием.
- Чтобы получить изображение с помощью конфокального микроскопа, разрежьте ткань на срезы толщиной ~0,5-1 мм с помощью матрицы для среза ткани. Для проведения флуоресцентной микроскопии световых листов (ЛСФМ) необходимо заблокировать весь участок ткани.
- Заблокируйте образцы 5 мл блокирующего раствора CUBIC на ночь при температуре 4 °C с встряхиванием (см. Таблицу материалов). При работе с NHP или человеческими образцами используйте античеловеческий FcR. При работе с образцами мышей используйте антимышиный FcR в блокирующем растворе.
- Окрашивайте образцы 5 мл первичных антител (см. Таблицу материалов) в блокирующий раствор (без видоспецифичного FcR) в течение 3 дней при комнатной температуре с встряхиванием (Дополнительно: центрифугируйте концентрированный запас антител при 2300 x g в течение 5 минут перед использованием, чтобы уменьшить добавление агрегированного антитела).
- Окрашенный образец промывают при комнатной температуре с встряхиванием в течение не менее 5 часов в общей сложности с не менее чем пятью заменами буфера промывочного раствора (см. Таблицу материалов).
- Окрашивание образцов вторичными антителами (см. Таблицу материалов) в блокирующий раствор (без видоспецифичного FcR) в течение 3 дней при комнатной температуре с встряхиванием (необязательно: центрифугирование антител при концентрации 2300 x g в течение 5 минут перед использованием для минимизации агрегации антител).
- Испачканный образец пять раз промыть промывочным раствором комнатной температуры с встряхиванием в общей сложности не менее 5 часов.
- Окрасьте образцы 5 мл раствора для окрашивания DAPI (см. Таблицу материалов) на каждый образец ткани и инкубируйте в течение 10 минут при комнатной температуре. Оставьте образцы в растворе красителя DAPI в темноте при температуре 4 °C для последующей визуализации.
- Образцы лимфоидной ткани трижды промыть промывочным раствором при комнатной температуре с встряхиванием в течение 30 мин каждый.
- Перед сборкой образца погрузите окрашенный образец в реагент CUBIC Reagent-2 на ночь при комнатной температуре в темноте.
4. Очищение тканей CLARITY
- Образцы лимфоидной ткани промыть в стерильном 0,1 М PBS три раза с встряхиванием при комнатной температуре в течение 15 мин каждый для удаления PFA.
- Поместите образцы тканей в 15 мл свежеприготовленного раствора акриламида и инкубируйте при температуре 4 °C в течение ночи при осторожном перемешивании (см. Таблицу материалов).
ВНИМАНИЕ: Неполимеризованный акриламид является мощным нейротоксином и легко всасывается через кожу. Избегайте любого контакта с кожей и немедленно смойте при контакте. - Дайте образцам тканей нагреться до комнатной температуры.
- ДОПОЛНИТЕЛЬНО: Дегазируйте образцы тканей путем барботирования азота в растворе акриламида в течение 1 минуты. Следите за тем, чтобы использовать низкую скорость потока, чтобы избежать разбрызгивания токсичного неполимеризованного акриламида (~1-2 пузырька/с).
- Поместите образцы тканей в водяную баню при температуре 37 °C на 1-3 часа для полимеризации, переворачивая каждые 15 минут. Удаляйте образцы, как только будет обнаружена заметная полимеризация, о чем свидетельствует вязкая жидкость, появление линий Шлерена при смешивании или образование прозрачной капсулы вокруг ткани.
ПРИМЕЧАНИЕ: Если происходит полная полимеризация раствора акриламида, удалите излишки гидрогеля из образца и продолжайте протокол. - Промойте образцы тканей стерильным 0,1 М PBS три раза по 30 мин каждый при комнатной температуре с легким встряхиванием для удаления раствора акриламида.
- Поместите образцы тканей в 15 мл 8% SDS в 0,1 М PBS при 37 °C с легким покачиванием в течение 2-5+ дней, чтобы обеспечить очищение. Периодически обновляйте 8% раствор SDS и при необходимости используйте до 50 мл раствора для ускорения очистки. Остановите процесс очистки, когда образцы становятся визуально прозрачными или больше не продвигаются. Регулярно делайте эталонные изображения, чтобы контролировать процесс очистки с течением времени.
- Образцы тканей промыть стерильным 0,1 М PBS пять раз в течение 1 суток при комнатной температуре с легким встряхиванием.
- Временно храните образцы в соотношении 0,1 M PBS (плюс 0,01% объема/объема (v/v) NaN3 для более длительного хранения) в темноте до тех пор, пока они не будут готовы к визуализации эндогенной флуоресценции.
- Поместите салфетку в 5 мл среды для визуализации RI-2 (см. Таблицу материалов). Инкубируйте в течение ночи при комнатной температуре в темноте, чтобы убедиться в полноте процесса очищения перед иммуноокрашиванием. Сделайте референсные изображения для контроля прозрачности тканей.
5. Блокирование и иммуноокрашивание образцов CLARITY
ПРИМЕЧАНИЕ: Эти шаги аналогичны блокировке и иммуноокрашиванию очищенных тканей CUBIC, но используют другие составы для блокировки, промывания и окрашивания растворов.
- Образцы лимфоидной ткани трижды промыть 0,1 М PBS в течение 30 мин каждый раз при комнатной температуре с легким встряхиванием.
- Для получения изображения с помощью конфокального микроскопа разрежьте ткань на срезы толщиной ~0,5-1 мм с помощью слайсера и матрицы диаметром 0,5 мм. Для выполнения ЛСФР необходимо заблокировать весь образец ткани.
- Заблокируйте образцы 5 мл раствора, блокирующего CLARITY (см. Таблицу материалов) на ночь при температуре 4 °C с встряхиванием.
- Окрашивайте образцы 5 мл первичных антител (см. Таблицу материалов) в блокирующий раствор (без видоспецифичного FcR) в течение 3 дней при комнатной температуре с встряхиванием (необязательно: центрифугируйте антитела при 2300 x g в течение 5 минут перед использованием для минимизации агрегации антител).
- Испачканный образец пять раз промыть раствором для промывки при комнатной температуре с встряхиванием в общей сложности не менее 5 ч (см. Таблицу материалов).
- Окрашивание образцов 5 мл вторичных антител (см. Таблицу материалов) в блокирующий раствор (без видоспецифичного FcR) в течение 3 дней при комнатной температуре с встряхиванием (необязательно: центрифугирование антител при 2300 x g в течение 5 минут перед использованием для минимизации агрегации антител). Чтобы сократить общую продолжительность протокола, используйте первичные антитела, конъюгированные с флуорофорами, чтобы исключить необходимость инкубации со вторичными антителами.
- Испачканный образец пять раз промыть раствором для промывки при комнатной температуре с встряхиванием в общей сложности не менее 5 ч.
- Окрасьте образцы 5 мл раствора для окрашивания DAPI (см. Таблицу материалов) на каждый образец ткани и инкубируйте в течение 10 минут при комнатной температуре. Оставьте образцы при температуре 4°C в темноте в растворе морилки DAPI для последующей визуализации.
- Образцы лимфоидной ткани трижды промыть раствором для умывания, при комнатной температуре, каждый раз встряхивая в течение 30 минут.
- Поместите ткань в 5 мл среды для визуализации RI-2 (R.I. = 1,46) и инкубируйте в течение ночи при комнатной температуре в темноте перед сбором образца (см. протокольные шаги 6 и 7).
6. Монтаж и визуализация очищенных образцов тканей для конфокальной микроскопии
- Снимите с одной стороны защитный слой клейкого силиконового изолятора.
- Приклейте защитное стекло микроскопа (22 мм x 40 мм, толщина 0,25 мм) к отклеенной стороне силиконового изолятора, чтобы образовать водонепроницаемое пространство для образца.
- Снимите с другой стороны защитный слой клейкого силиконового изолятора.
- Поместите образец для визуализации в центр силиконового изолятора, а затем добавьте CUBIC Reagent-2 или Imaging Media RI-2 в зависимости от ситуации, пока поверхность жидкости не достигнет высоты края изолятора.
- Чтобы свести к минимуму скопление пузырьков воздуха в силиконовом изоляторе, выровняйте и аккуратно наложите слой второго защитного стекла с одной стороны с помощью электромагнитных щипцов. Вытрите лишнюю жидкость. Осторожно прижмите покровное стекло вокруг лунки (лунок) образца (лунок) с помощью тыльной стороны щипцов, чтобы запечатать клей. Храните смонтированные образцы в горизонтальном положении в темное время суток.
ПРИМЕЧАНИЕ: Образцы могут быть сфотографированы через недели или месяцы после монтажа; Однако качество изображения, как правило, со временем снижается. - Поместите установленное предметное стекло на предметный столик микроскопа и найдите образец с помощью белого света и объектива с меньшим увеличением (2-10x).
- Настройте профиль регистрации флуоресценции на основе отдельных выбранных флуорофоров.
ПРИМЕЧАНИЕ: Рекомендуется индивидуально приобретать отдельные флуорофорные каналы. Это приводит к увеличению времени регистрации, но снижает спектральное перекрытие и регистрацию неспецифического флуоресцентного сигнала. Общий профиль флуорофора может включать DAPI (450 нм), Alexa488, Alexa594 и Alexa647 (или связанные комбинации) для минимизации спектрального перекрытия во время получения изображения. - Выберите подходящий объектив увеличения для изображения интересующих областей. Используйте объективы с меньшим увеличением (2-10x) для визуализации больших объемов или целых тканей с разрешением одной клетки и используйте объективы с более высоким увеличением (20-63x) для визуализации субклеточных деталей в очищенных тканях с более высоким разрешением. Максимально точное соответствие показателя преломления объективов, носителей изображения и тканей сводит к минимуму появление оптических искажений при получении изображения.
- Выберите размер шага для получения Z-стека. Для объективов с меньшим увеличением (2–10x) выберите размер шага ~3–5 мкм, чтобы обнаружить флуоресценцию отдельной ячейки в нескольких непрерывных Z-срезах для 3D-моделирования при одновременном уменьшении общего времени сбора данных и общего размера файла. Для объективов с большим увеличением (20-63x) выберите размер шага ~1 мкм или меньше, чтобы свести к минимуму потерю субклеточной информации между отдельными Z-срезами.
- Увеличьте поле зрения, чтобы визуализировать всю область ткани, которую необходимо изобразить, в размерах X и Y с как можно меньшим количеством незанятой области. Задайте верхнюю и нижнюю координаты захвата Z-ступени, которые охватывают всю интересующую область для изображения.
- Получение образов Z-стека. Сохраните и экспортируйте файл для постобработки с помощью любого программного обеспечения для анализа изображений. Для определенных наборов программного обеспечения преобразуйте файлы в определенные типы файлов (например, .tiff, .ome-tiff, .jpeg и т. д.). Выполните преобразование с помощью любого программного обеспечения для получения изображений микроскопа или бесплатного программного обеспечения для анализа изображений (например, ImageJ/Fiji).
7. Монтаж и визуализация образцов в камере или кювете LSFM
- Заполните камеру визуализации CUBIC Reagent-2 или RI-2 в зависимости от конкретного используемого протокола. Избегайте образования пузырьков во время перекачивания жидкости. Удалите лишние пузырьки с помощью пипетки.
- Погрузите образец в камеру визуализации и ограничьте движение образца.
ПРИМЕЧАНИЕ: В зависимости от конкретного используемого микроскопа, это может включать в себя встраивание образца в агарозу, подвешивание образца к крючку или адаптеру для дикобраза, 3D-печать держателя образца или прикрепление образца с помощью клея к пластиковой тарелке. - Поместите объектив в решение для визуализации, сфокусировавшись на образце. Оставьте установленный образец в камере визуализации на несколько часов или на ночь, чтобы обеспечить полное равновесие растворов и тканей в кювете.
- Получите Z-стек интересующей области (см. шаги 6.7-6.11 для получения изображений).
ПРИМЕЧАНИЕ: Этот подход позволяет визуализировать объемы тканей более 1 см3 с разрешением для одной клетки.
8. Реконструкция поверхности и количественное определение клеток с помощью программного обеспечения для анализа изображений Imaris
ПРИМЕЧАНИЕ: Эти шаги относятся к программному обеспечению для анализа изображений Imiris, но аналогичные шаги обработки изображений могут быть выполнены с использованием других программных пакетов (например, ImageJ/Fiji, Aivia, Arivis, Amira и т. д.).
- Используйте конвертер файлов Imaris для преобразования файла изображения Z-стека в родной формат Imaris .ims. Это будет способствовать более быстрому преобразованию файлов, сводя к минимуму ошибки преобразования и потенциальные проблемы с программным обеспечением после открытия.
ПРИМЕЧАНИЕ: Некоторые новые LSFM позволяют пользователю сохранять файлы непосредственно в формате .ims. - Перетащите файл .ims для анализа в область «Арена » программного обеспечения Imis. Отрегулируйте контрастность или интенсивность каждого цветового канала на панели «Настройка дисплея ». Нажмите на иконку « Добавить новые поверхности» в левом верхнем углу.
- Нажмите на «Далее: Исходный канал » (синий значок со стрелкой, указывающей вправо). Выберите исходный канал поверхности для построения. Остальные параметры не изменяйте.
- Нажмите на Далее: Порог (синий значок со стрелкой, указывающей вправо).
- Чтобы настроить порог (абсолютную интенсивность), перетащите линию порога влево или вправо. Включите функцию Split Touching Objects и введите средний диаметр ячейки в микронах в качестве стандарта разделения для системы, чтобы получить множество точек в качестве начала координат для каждой отдельной поверхности.
- Не включайте слишком маленькие или слишком яркие флуоресцентные сигналы, так как они могут представлять собой потенциальное окрашивание или артефакты микроскопа. Включайте только те точки, которые имеют приемлемые размеры и интенсивность флуоресценции, соответственно изменяя средний диаметр ячейки.
ПРИМЕЧАНИЕ: Средний диаметр клеток будет варьироваться для конкретных тканей или типов клеток, но обычно будет находиться в пределах 5-15 мкм.
- Не включайте слишком маленькие или слишком яркие флуоресцентные сигналы, так как они могут представлять собой потенциальное окрашивание или артефакты микроскопа. Включайте только те точки, которые имеют приемлемые размеры и интенсивность флуоресценции, соответственно изменяя средний диаметр ячейки.
- Нажмите на кнопку «Далее: Классифицировать поверхности» (синий значок со стрелкой, указывающей вправо). Отрегулируйте поверхности, которые будут включены, перетащив линию порога влево или вправо. Убедитесь, что поверхности точно аппроксимируют исходный сигнал флуоресценции, отделяя при этом сигнал флуоресценции от отдельных ячеек.
- Нажмите на кнопку «Готово: выполнить все шаги создания» и «Завершить работу мастера » (зеленый значок с двумя стрелками, указывающими вправо). Поверхность официально построена.
- Нажмите на шестую иконку с надписью «Статистика» на левой панели, чтобы увидеть количество ячеек, в данном случае количество поверхностей для конкретного анализируемого цветового канала.
- Убедитесь, что четыре переменные «Число разъединенных компонентов на момент времени», «Количество поверхностей на момент времени», «Общее число отключенных компонентов» и «Общее число поверхностей » имеют одно и то же число, которое является количеством ячеек этого цветового канала.
Результаты
Очистка тканей включает в себя обработку законсервированных тканей химическими коктейлями для извлечения непрозрачных биомолекул из ткани при сохранении архитектуры ткани. Эти решения для очистки тканей сопоставляют показатель преломления ткани с окружающей средой визуализации, чтобы свести к минимуму оптические искажения, улучшить соотношение сигнал/шум в глубине тканей и свести к минимуму фоновую автофлуоресценцию. Два протокола на водной основе для оптической очистки тканей, CUBIC3 и CLARITY9, были использованы для очистки консервированных ВИЧ/SIV-инфицированных мышей, приматов и образцов тканей человека перед иммунофлуоресцентным окрашиванием и визуализацией с помощью конфокальной и световой флуоресцентной микроскопии.
Для протокола CUBIC неподвижные ткани промывали PBS для удаления фиксаторов и погружали в CUBIC Reagent-1, основной буферный раствор аминоспиртов, который элюирует хромофоры, такие как гем, что приводит к обесцвечиванию и делипидации ткани (рис. 1, вверху). Меньшие объемы тканей (~мм3) могут быть обесцвечены после 3 дней лечения CUBIC Reagent-1, но большие объемы тканей (~см3) или ткани с большим количеством гема (такие как печень, селезенка или сердце) требуют более длительного времени инкубации и объемов раствора (>1 месяц и ~50 мл), а также частой замены раствора каждые 2-3 дня. После обесцвечивания ткани промывали и помещали в CUBIC Reagent-2, содержащий сахарозу раствор с показателем преломления примерно 1,48-1,49, который соответствует показателю преломления ткани и увеличивает пропускание света. Очищенные ткани подвергали иммуноокрашиванию и помещали в раствор реагента CUBIC Reagent-2 перед визуализацией с помощью конфокального или светового листового микроскопа. Эффекты процедуры очистки CUBIC были визуализированы для нескольких тканей hu-mouse и NHP различных размеров и концентраций хромофоров (рис. 2). Оптическое очищение сделало ткани видимыми прозрачными для невооруженного глаза, что позволило увидеть линии сетки и текст на листах бумаги «сквозь» ткань. Богатые хромофорами ткани, такие как селезенка, печень, костный мозг и сердце, могут не полностью обесцвечиваться, но остаются пригодными для иммуноокрашивания и визуализации (Рисунок 2 и Рисунок 5).
Для протокола CLARITY фиксированные ткани промывали PBS для удаления фиксаторов, а затем инкубировали в течение ночи при 4 °C в 40% растворе акриламида с термическим инициатором для образования ковалентных связей между белками в образце и мономерами акриламида (рис. 1, внизу). На следующий день, после того как ткань была уравновешена до комнатной температуры, а затем нагрета на водяной бане при температуре 37 °C, была начата полимеризация акриламида, которая быстро окутала образец гидрогелем. Образец обрабатывали раствором 8% SDS в течение 2-5 дней для удаления помутневших липидов. Непосредственно перед флуоресцентным окрашиванием образец погружали в раствор для согласования показателей преломления (RIMS) для CLARITY (Imaging Media RI-2), содержащий 90% неионогенной градиентной среды. Для тканей, содержащих большое количество гема, стадия обесцвечивания может быть добавлена в конце стадии делипидации 9,11,12. Прогрессирование CUBIC и CLARITY clearing сравнивали на разных срезах одного и того же образца селезенки человека (рис. 3). При очистке CLARITY образуется видимый полиакриламидный гель, который обволакивает раствор и, как правило, демонстрирует меньшую обесцвечиваемость по сравнению с CUBIC clearing, если не добавляется дополнительная стадия обесцвечивания 9,12.
Впоследствии в обоих протоколах очищенные, неповрежденные ткани подвергались иммуноокрашиванию для обнаружения специфических иммунных клеточных популяций. Образцы промывали, блокировали реагентом, содержащим α-FcR, для уменьшения связывания неспецифических антител и окрашивали в течение 3 дней при использовании первичного антитела, непосредственно конъюгированного с флуорофором. В качестве альтернативы образцы окрашивали в течение 3 дней неконъюгированным первичным антителом, а затем еще 3 дня вторичным антителом, конъюгированным с флуорофором. Ткани снова промывали, а затем инкубировали с окрашиванием DAPI в течение ночи при температуре 4 °C для ядерной визуализации. Образцы промывали и инкубировали либо в реагенте CUBIC в течение 24-36 часов, либо в среде Imaging Media RI-2 (CLARITY) в течение ночи в темноте. Для конфокальной микроскопии ткани наносили на предметное стекло микроскопа в соответствующем RIMS перед визуализацией (рис. 4). Для флуоресцентной микроскопии световых листов (LSFM) образцы были полностью погружены с помощью RIMS в кювету для визуализации в течение ночи перед визуализацией.
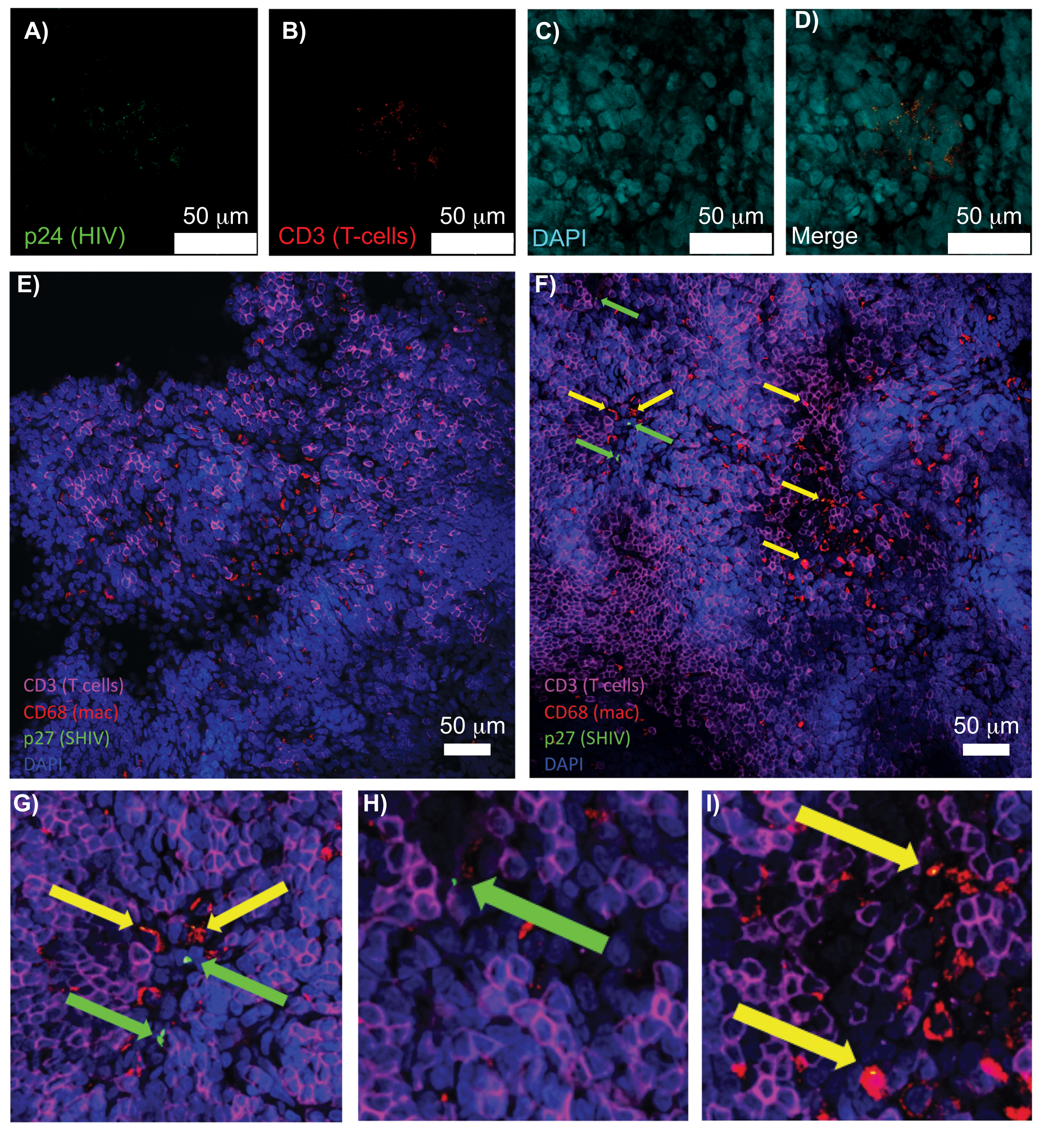
Конфокальная микроскопия интактных, очищенных и иммуноокрашенных лимфоидных тканей позволила одновременно визуализировать несколько флуоресцентных сигналов, включая ядра, маркеры иммунных клеток и белки ВИЧ/SIV CA (капсид) (рис. 5). Вирус-продуцирующие клетки определяли путем флуоресцентной колокализации маркеров иммунных клеток и белков ВИЧ. Очищенная и окрашенная ВИЧ-инфицированная селезенка человека выявила множественные CD3+ Т-клетки, колокализованные с ВИЧ p24, что указывает на присутствие вируспродуцирующих клеток в области интактной ткани (рисунок 5A-D). Очищенные и иммуноокрашенные инфицированные SHIV-инфицированные лимфатические узлы NHP выявили распределение CD3+ Т-клеток и макрофагов CD68+ в областях тканей, где вирус не обнаружен (рисунок 5E), в дополнение к областям с многочисленными вирусопродуцирующими клетками (рисунок 5F). Эти результаты показали, что вирус-продуцирующие клетки из различных тканевых источников отличались от других клеток в пределах данного поля зрения и позволяли обнаруживать редкие биологические события в сложной тканевой среде.
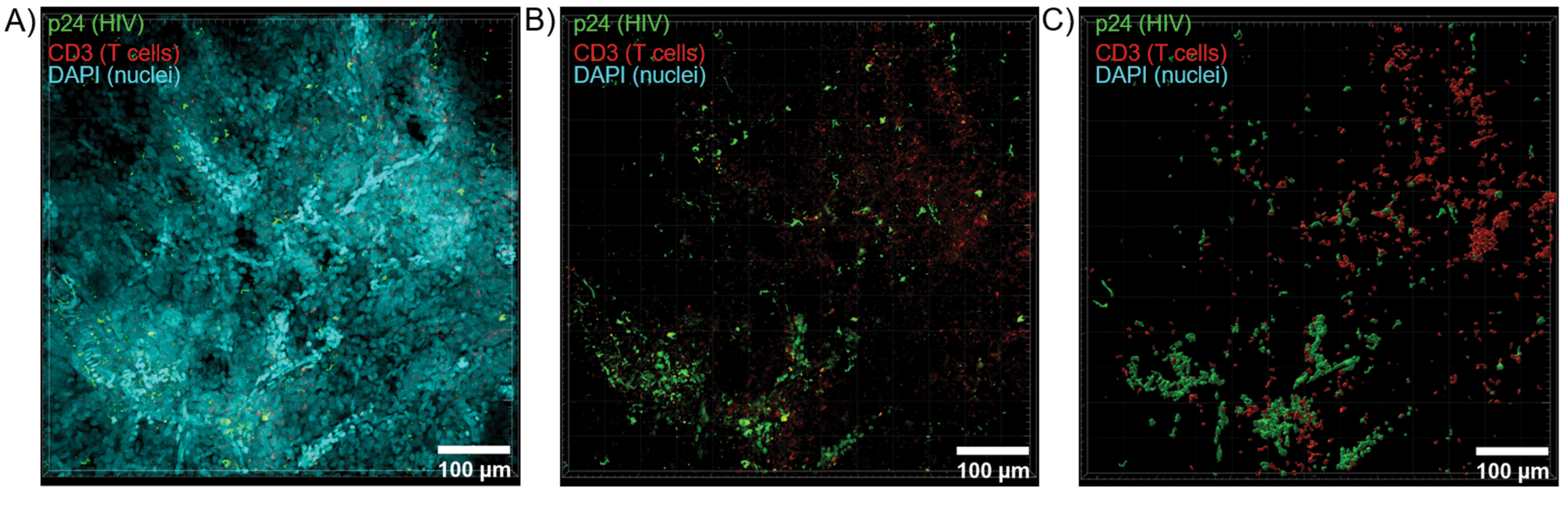
Оптическое срезирование очищенных тканей с помощью конфокального микроскопа было применено для создания Z-стеков и 3D-моделей поверхности, которые выявили клеточную гетерогенность, проявляющуюся во время ВИЧ-инфекции (рис. 6). Z-стеки рекомбинировали в Z-проекционное изображение с помощью программного комплекса Imaris (рис. 6A) и удаляли ядерный канал DAPI для четкой визуализации флуоресценции CD3+ Т-клеток и капсидного белка ВИЧ (p24) во всех объемах ткани (рис. 6B). Флуоресценция Z-проекции была автоматически сегментирована с помощью программного обеспечения Imaris для создания реконструированной 3D-модели поверхности для пространственной визуализации и количественной оценки сигнала флуоресценции по всему Z-стеку (рис. 6C). Анализ 3D-модели поверхности выявил 546 CD3+ Т-клеток и 218 клеток, продуцирующих ВИЧ p24. В совокупности получение иммунофлуоресценции с помощью Z-стека из очищенных ВИЧ-инфицированных лимфоидных тканей позволило создать 3D-модели клеточного состава в ткани и автоматизировать количественное определение популяций иммунных клеток в тканевых объемах.
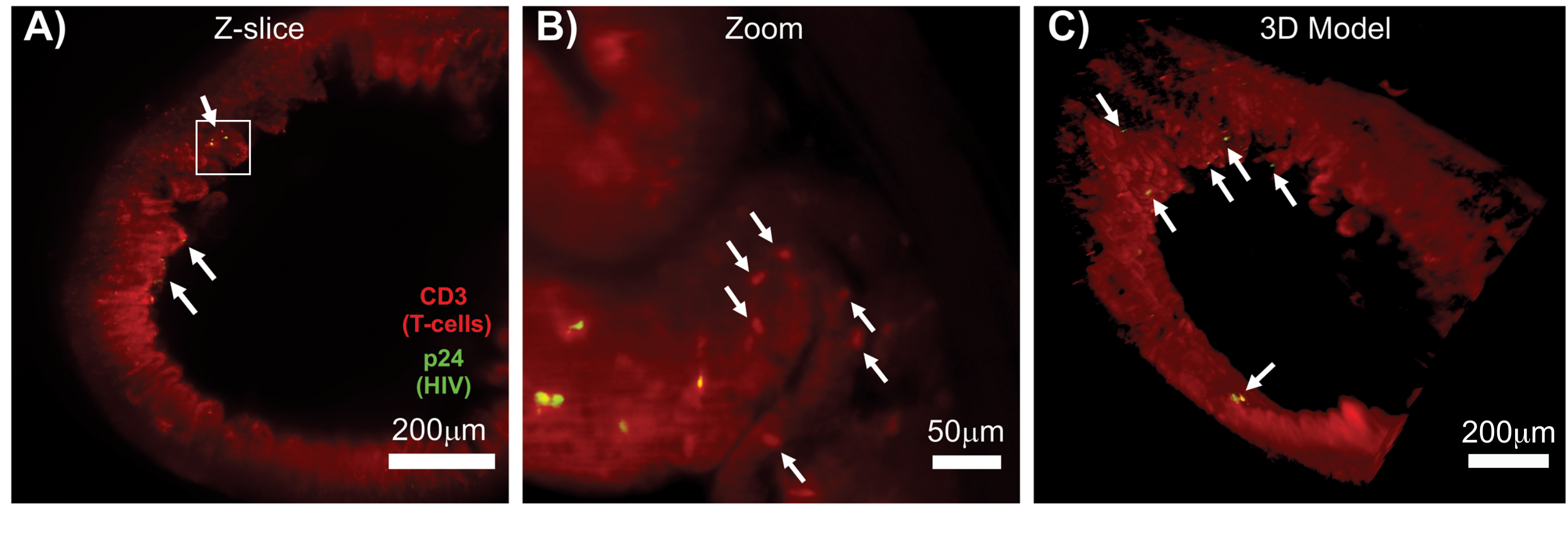
LSFM интактных, очищенных и иммуноокрашенных лимфоидных тканей позволила получить более объемную иммунофлуоресцентную (ИФ) визуализацию распределения иммунных клеток и вирус-продуцирующих клеток в лимфоидных тканях (рис. 7). Иммуноокрашивание ткани толстой кишки ВИЧ-инфицированной мыши на hCD3+ Т-клетки и ВИЧ p24 выявило очаги вирус-продуцирующих клеток, рассеянных среди больших участков ткани без признаков инфекции (рис. 7A). Увеличенное изображение очагов вирус-продуцирующих клеток выявило несколько вирус-продуцирующих клеток в непосредственной близости от потенциальных клеток-мишеней (рис. 7B). Тканевая аутофлуоресценция (красная дымка) использовалась для визуализации всей архитектуры ткани при одновременном выделении специфических популяций иммунных клеток в ткани, которые окрашивались ярче, чем автофлуоресценция (красные овалы). 3D-модель всего объема Z-стека LSFM показала пространственное распределение очагов вирус-продуцирующих клеток в области интактной ткани и позволила составить карту мест производства вируса относительно общей архитектуры ткани (рис. 7C). Удивительно, но очаги вирус-продуцирующих клеток часто были разбросаны между большими участками ткани без каких-либо признаков производства вируса. Эти результаты могут позволить количественно оценить параметры распространения вируса и плотность инфицированных клеток в различных тканях и в разное время инфекции или ответа на различные методы лечения.

Рисунок 1: Рабочий процесс типичной очистки тканей CUBIC и CLARITY, иммуноокрашивания и визуализации. Время очистки CUBIC (вверху) и CLARITY (внизу) может варьироваться в широких пределах в зависимости от размера и типа ткани. Для очистки CLARITY требуется дополнительная стадия инкубации со средой, соответствующей индексу преломления, перед иммуноокрашиванием, чтобы убедиться, что ткань прозрачна. Иммуноокрашивание обычно занимает 3 дня, если первичные антитела конъюгированы с флуорофорами, и 6 дней, если требуются флуоресцентные вторичные антитела. Образцы могут быть визуализированы с помощью конфокального или LSFM-сигнала. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

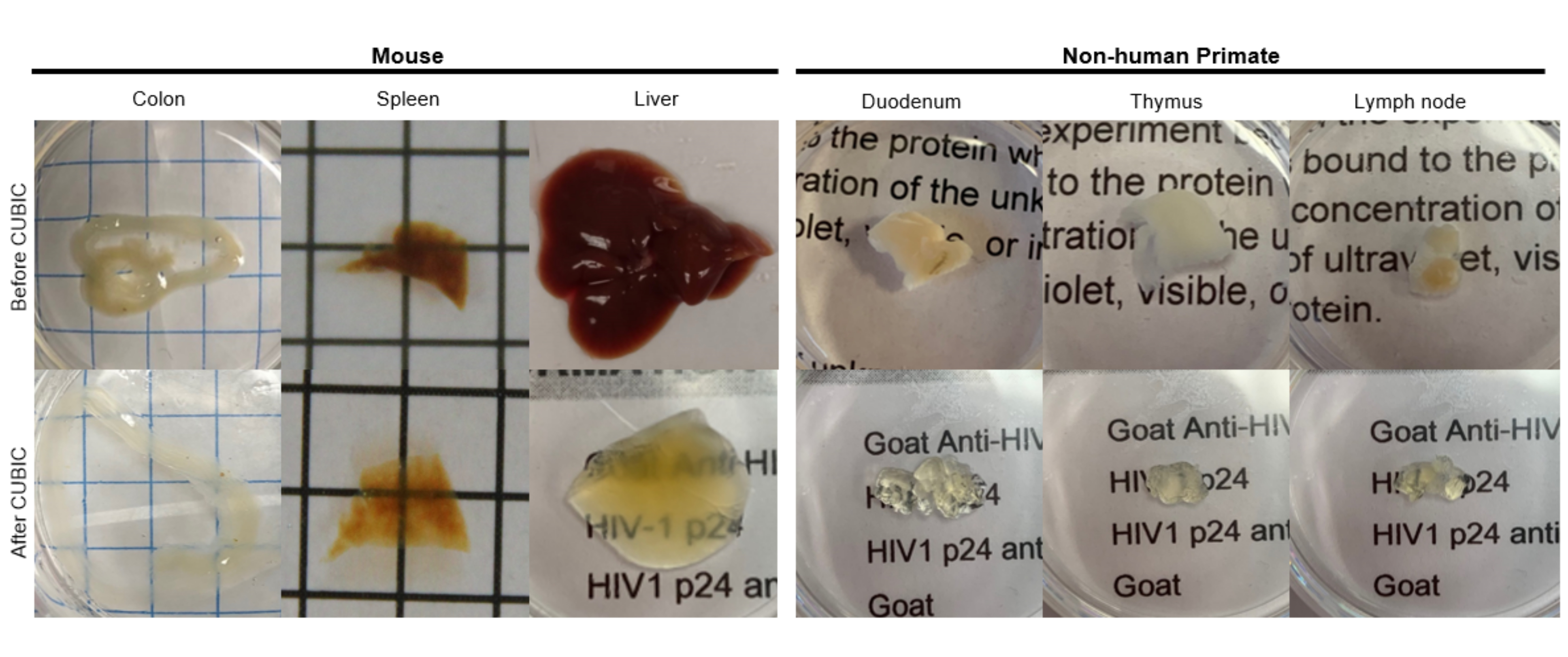
Рисунок 2: CUBIC Очистка образцов тканей hu-mouse и NHP. В зависимости от различной плотности гема и липидов в образцах тканей, время, необходимое для очистки каждого типа ткани, варьируется. Например, толстой кишке и двенадцатиперстной кишке обычно требуются относительно короткие периоды (~7 дней), в то время как селезенке и печени может потребоваться больше времени, чтобы стать прозрачными (~30 дней). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 3: Продольное сравнение методов очищения тканей на образцах человека. CUBIC (верхние панели) и CLARITY (нижние панели) очищали селезенку от ВИЧ-инфицированного человека, получавшего антиретровирусную терапию. Оба метода адекватно очищали ткани к 32-му дню для иммуноокрашивания и визуализации. Стадия обесцвечивания по методу CUBIC заметно снижает автофлуоресценцию, вызванную присутствием гема, содержащегося в образцах селезенки. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

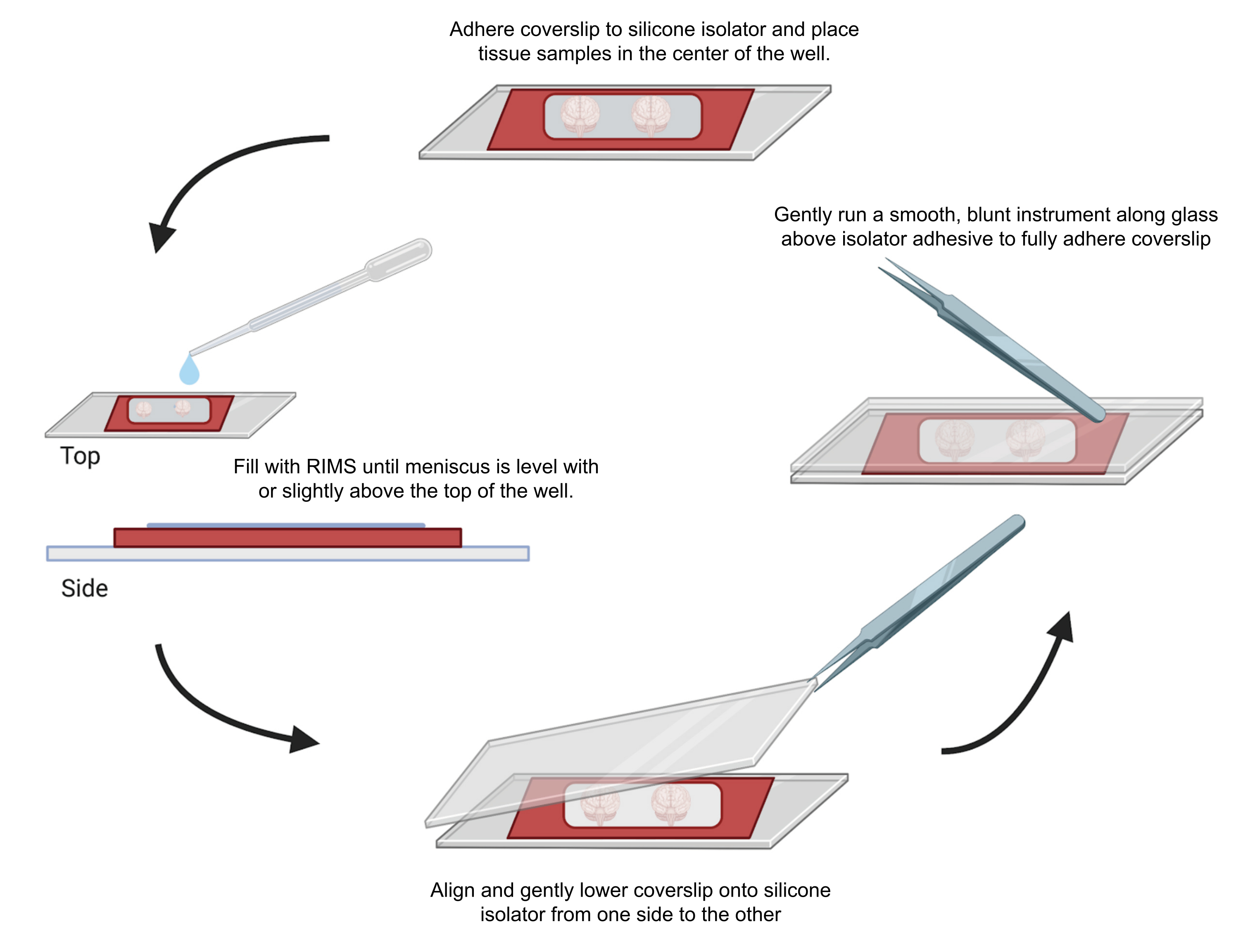
Рисунок 4: Монтаж образца для конфокальной микроскопии. Образцы монтировались между покровными стеклами, разделенными клеевыми силиконовыми изоляторами толщиной 0,5-1 мм. Силиконовые изоляторы были прикреплены к первому покровному стеколу, а салфетка была помещена в центр лунки (сверху). Колодец заполнялся RIMS до тех пор, пока мениск не оказывался на уровне или немного выше верхней части колодца (слева). Второй покровный стекло был аккуратно опущен на место с одной стороны на другую, не допуская образования пузырей (дно). Покровные стекла полностью приклеивались к силиконовому изолятору путем аккуратного проведения тупым инструментом по периметру колодца (справа). Образцы были визуализированы в стандартном конфокальном микроскопе. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 5: Конфокальная микроскопия очищенной, неповрежденной селезенки человека и лимфатических узлов NHP. (А-Г) Очищенные ткани ВИЧ-инфицированного человека окрашивали на ВИЧ-1 p24 (зеленый), hCD3+ Т-клетки (красный) и ядра (голубой). (E) Конфокальный Z-срез CUBIC очищен от инфицированного SHIV NHP через 8 недель после инфицирования иммуноокрашенным на CD3+ Т-клетки (пурпурный), CD68+ макрофаги (mac/красный), SHIV p27 (зеленый) и ядра (синий). Поле зрения содержит Т-клетки, макрофаги и другие типы клеток, но нет доказательств наличия клеток, продуцирующих SHIV (зеленый цвет). Е) Конфокальный Z-срез соседнего участка того же лимфатического узла, показывающий различия в плотности и количестве клеток, а также наличие вирус-продуцирующих CD3+ Т-клеток (зеленые стрелки) и макрофагов CD68+ (желтые стрелки). (Г-И) Увеличенный вид выбранных областей окрашивания p27 от (F). Масштабные линейки имеют размер 50 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 6: Объем Z-стека и 3D-реконструированная поверхность из селезенки ВИЧ-инфицированного человека. (A) Z-проекционное изображение из Z-стека размером 600 мкм x 600 мкм x 100 мкм ВИЧ-инфицированной ткани селезенки человека, окрашенной на HIV-1 p24 (зеленый), hCD3+ Т-клетки (красный) и ядра (голубой). (B) То же Z-проекционное изображение без ядерного окрашивания DAPI. (C) Реконструирована 3D модель поверхности CD3 (красный) и p24 (зеленый) флуоресценции из всего объема Z-стека. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 7: LSFM и 3D-реконструкция поверхностных объемов ВИЧ-инфицированных тканей (A) Z-среза (1000 мкм x 1000 мкм) толстой кишки ВИЧ-инфицированной мыши hu-мыши, иммуноокрашенной на CD3+ Т-клетки (красный) и ВИЧ p24 (зеленый). Тусклая красная дымка представляет собой тканевую аутофлуоресценцию, в то время как отчетливая красная точка указывает на Т-клетки. По периферии видны ворсинки, направленные к центральному просвету, с несколькими очагами активного производства вируса (белые стрелки), рассредоточенными по большим участкам, не содержащим вируса. Прямоугольником обозначена приблизительная область интереса для панели B. (B) Увеличенная область ткани, показывающая отдельные вирус-продуцирующие hCD3+ Т-клетки (желтые) в непосредственной близости от неинфицированных Т-клеток (красные). Изображение было повернуто и изменено на ближайший Z-срез, чтобы показать фокус p24-положительных клеток в одной Z-плоскости. На заднем плане красная автофлуоресценция показывает общую тканевую архитектуру в дополнение к специфическому окрашиванию hCD3+ Т-клеток (красные точки; белые стрелки). (C) 3D-модель поверхности всего объема (1 000 мкм x 1 000 мкм x 200 мкм), созданная с помощью программного обеспечения Imis, повернутая, чтобы показать очаги ВИЧ-инфекции (желтый цвет) в различных местах кишечника. Белыми стрелками обозначены отдельные очаги в пределах объема. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}
Обсуждение
Представляющие интерес лимфоидные ткани должны быть собраны быстро после вскрытия и немедленно помещены в предварительно охлажденные фиксирующие буферы, чтобы избежать некроза тканей (темной или черной ткани), который может негативно повлиять на окрашивание и визуализацию. После сбора желаемых тканей немедленно погрузите ткани в ледяной 4%-8% параформальдегид (PFA) на ночь для фиксации, который также инактивирует потенциальные патогены, связанные с образцами. 4% PFA является оптимальным для фиксации образцов LM, в то время как 8% PFA может адекватно сохранить ткани как для LM, так и для EM. Следование этим процедурам и хранение образцов в фиксаторе при температуре 4 °C в темноте может эффективно сохранить ткани для LM-визуализации в течение нескольких лет. Одним из предостережений является то, что более длительное хранение в фиксаторе может привести к введению окрашивающих артефактов, особенно к маскировке антигена, которая вызвана сшивкой соседних белков с интересующим белком, что может окклюзировать доступность окрашивающих антител к эпитопу13,14. Если ткани содержат эндогенно экспрессируемые флуоресцентные белки, примите меры, чтобы по возможности не подвергать ткани воздействию света, на протяжении всего протокола. Как правило, эндогенные флуоресцентные белки сохраняют флуоресценцию в течение 6-12 месяцев после фиксации, но отдельные образцы тканей могут меняться в течение более или менее длительных периодов времени. Если эндогенная флуоресценция теряется из-за деградации белка, флуоресцентные белки часто могут быть обнаружены с помощью первичного антитела, специфичного для интересующего белка. Перфузия – еще один вариант быстрой фиксации тканей до выведения12; Однако из-за опасений при работе с такими патогенами, как ВИЧ, был выбран путь вскрытия тканей с последующим погружением в ледяной фиксатор, чтобы максимально безопасно подготовить образцы.
Одним из преимуществ описанных протоколов очистки на водной основе является то, что они, как правило, мягче, чем протоколы на основе органических веществ, которые иногда могут повредить более хрупкие ткани, такие как печень. Протоколы очистки на водной основе, как правило, требуют больше времени для достижения полной очистки проб (недели вместо дней) по сравнению с протоколами очистки на основе органических веществ. Протоколы CLARITY и CUBIC могут быть проведены более быстро с использованием перфузии для одновременного выведения всех органов у грызуна11,12; однако это был неосуществимый вариант для НПЗ и вскрытия человека. Образцы, обработанные с CLARITY, как правило, демонстрируют некоторое увеличение объема, в то время как CUBIC показал уменьшенное влияние на объем образца9. Несмотря на то, что в целом это происходит быстрее, многие протоколы очистки тканей на основе органических веществ вызывают усадку тканей15, что может затруднить обнаружение отдельных клеток или субклеточных деталей в плотных тканях, таких как лимфатические узлы и селезенка. Расширение, вызванное очищением, может эффективно увеличить разрешение визуализации, облегчая наблюдение за аспектами, которые было бы трудно наблюдать при исходном размере ткани. В качестве альтернативы, усадка ткани может эффективно уменьшить общий размер образца, что может сделать возможным визуализацию всего органа без вскрытия. Преимущество протокола CLARITY и CUBIC заключается в том, что они сохраняют флуоресценцию эндогенно экспрессируемых флуоресцентных белков в ткани, оставаясь при этом поддающимися иммунофлуоресцентному окрашиванию11,12. Иммуноокрашивание может проводиться с использованием водных или органических методов очищения тканей; Тем не менее, личный опыт показал более высокую долю совместимости антител при использовании протоколов на водной основе по сравнению с протоколами на основе органических веществ. Исследователям необходимо решить, какой метод очистки тканей следует использовать, основываясь на визуализируемых тканях (тканях) и затронутых биологических вопросах (например, визуализация всего органа или визуализация конкретной области интереса). Не существует универсального метода очистки тканей, который позволяет проводить надежный анализ «под ключ» для всех вопросов визуализации больших объемов, а доступные методы демонстрируют явные преимущества и недостатки в зависимости от биологического применения.
При проведении окрашивания антителами необходимо учитывать множество аспектов. Поскольку образцы CLARITY встроены в акриламидный гидрогель, они, как правило, требуют больше времени для инкубации. Время, необходимое для инкубации антител, также зависит от объема и толщины каждого образца. Большинство описанных здесь образцов имели толщину ~2-3 миллиметра, и 3 дней было достаточно для полного окрашивания всей ткани. Если целью является изображение всего мозга мыши, время инкубации антител может занять 1 неделюили дольше. Выбор метода очистки водных и органических тканей для иммунофлуоресцентной визуализации может зависеть от совместимости антител. В целом, для CUBIC или CLARITY частота попадания антител, которые работают в культивируемых клетках и тканях, составляет ~70%. Независимо от того, используется ли метод очистки водных или органических тканей, необходимо оценить совместимость и эффективность всех антител с конкретным используемым методом. Как показано в этом разделе протокола, иммуноокрашивание образцов, обработанных CUBIC и CLARITY, происходит после завершения очистки. Напротив, этот шаг происходит перед процедурой очистки для некоторых протоколов на основе органических веществ, за которой следует постфиксинг.
Крайне важно, чтобы ткани были полностью погружены в среду визуализации, которая соответствует их показателю преломления. Если этого не сделать, возникнут сферические аберрации при получении изображения и исказится свет, полученный во время получения изображения. Необходимо соблюдать осторожность при удалении всех пузырьков воздуха из среды визуализации при монтаже образцов как для конфокальных образцов, так и для LSFM, так как пузырьки могут нарушить путь света к образцу или от него. Пузырьки могут быть удалены вручную с помощью пипетки перед окончательным монтажом образца. Для визуализации более толстых образцов с помощью конфокального микроскопа несколько силиконовых спейсеров могут быть наложены друг на друга, чтобы вместить ткани толщиной более 0,5 мм. Одна из рекомендаций заключается в том, чтобы уравновесить все ткани в RIMS на несколько часов или на ночь, пока они установлены на микроскопе, без дополнительного перемещения образца. Полное равновесие тканей и сред визуализации предотвратит смешивание растворов с несоответствующими показателями преломления, которые могут привести к аберрациям во время визуализации. Важно помнить, что при монтаже очищенных образцов тканей не существует единого готового метода монтажа, позволяющего получить изображение всех образцов во всех микроскопах. В этом протоколе обсуждаются варианты монтажа образцов, которые оптимально работают в одном контексте, но существует множество подходов к монтажу образцов в зависимости от используемого микроскопа и решаемого биологического вопроса. Эти подходы могут включать, помимо прочего, встраивание образца в агарозу, подвешивание образца к крючку или пластиковой линии, согласованной с показателем преломления, использование адаптера для дикобраза, 3D-печать держателя образца или прикрепление образца с помощью клея к пластиковой чашке.
Конфокальные микроскопы хорошо подходят для визуализации объемов тканей ~1 мм3-1 см3. Для конфокальных микроскопов используйте объектив с 2-10-кратным увеличением для первоначального определения местоположения интересующих областей и получения Z-стеков большего объема или целой ткани с разрешением для одной клетки. Переключитесь на объективы 20–63x для получения изображений с более высоким разрешением определенных областей интереса с субклеточной информацией. Идеальным объективом для визуализации тканей с функцией CUBIC и CLARITY является объектив CLARITY/Scale, который точно соответствует показателю преломления ткани и решению для визуализации. Если этот тип объектива недоступен, то оптимальным является получение изображений образцов с помощью глицеринового или масляного иммерсионного объектива (например, мультииммерсионного объектива LD LCI Plan-Apochromat 25 x 0,8 NA Imm Corr DIC M27: рабочее расстояние = 0,57 мм), а не воздушного объектива. Это сведет к минимуму внесение оптических искажений из-за несовпадающих показателей преломления во время захвата изображения. 20-25-кратные объективы могут сбалансировать получение большего объема изображений с получением деталей окрашивания отдельных клеток в сложной тканевой среде. Важно отметить, что большинство конфокальных микроскопов содержат модули, которые позволяют 3D-мозаику объемов изображений. Этот тип получения изображений в идеале может генерировать Z-стеки большего объема, содержащие субклеточную информацию.
Визуализация LSFM может позволить 3D-визуализацию конкретных клеточных популяций в контексте больших объемов ткани (>1 см3 ) и даже целых органов. В течение последних 10 лет очистка тканей в сочетании с LSFM в основном была сосредоточена на понимании мозговых связей у грызунов; Тем не менее, более поздние приложения включают визуализацию метастатических ландшафтов опухолей16, распределения клеток в анатомических компартментах 9,17 и дисперсии патогенов18. По сравнению с культивируемыми клетками, большинство биологических событий в тканях являются неоднородными, и LSFM может быть особенно хороша для визуализации и количественной оценки пространственной тканевой гетерогенности этих событий (например, репликация вируса, иммунная сигнализация, распределение клеток и т. д.).
3D-наборы данных, полученные с помощью конфокального или LSFM, могут быть обработаны с помощью многочисленных платформ анализа изображений. Программный комплекс Imaris может быть использован для построения поверхностей, создания 3D-анимации и количественной оценки клеток; Тем не менее, существует множество систем анализа изображений, которые обеспечивают эффективную постобработку и анализ изображений. ImageJ/Fiji freeware19 — это привлекательная альтернативная платформа для обработки изображений, доступная для большинства лабораторий, но не существует универсального программного обеспечения для анализа, которое преуспело бы во всех формах анализа и визуализации изображений. Многие пакеты программного обеспечения для анализа изображений могут быть непомерно дорогими, если они не доступны через объекты общего пользования. Наконец, важнейшим аспектом LSFM или больших мозаичных конфокальных 3D-наборов данных является управление данными. Эти платформы обработки изображений могут генерировать большие файлы (>1 Тбайт), для которых требуются более мощные компьютерные рабочие станции для визуализации и количественной оценки данных. В конечном счете, этот рабочий процесс визуализации может упростить получение и количественную оценку пространственно различных клеточных популяций в целых тканях и широко применим к большинству источников тканей и биологических систем.
Раскрытие информации
Авторы не имеют конфликта интересов, который можно было бы раскрыть.
Благодарности
Выражаем благодарность Институту геномной биологии Университета штата Иллинойс в Урбане-Шампейне за использование конфокальных и световых флуоресцентных микроскопов. Благодарим замечательных людей из когорты «Последний подарок» за образцы человеческих тканей, которая была профинансирована за счет следующих грантов: I147821, DA051915, AI131385 и P30 AI036214. Благодарим Нэнси Хейгвуд и Энн Хесселл за образцы тканей NHP, инфицированных SHIV.
Материалы
| Name | Company | Catalog Number | Comments |
| Acrylamide Solution (in 0.1 M PBS, 40 mL in total) | |||
| 40% Acrylamide: 4 mL | Bio-Rad | 1610144 | |
| VA-044 Thermal Initiator: 0.1g | Fujifilm | 011-19365 | |
| CLARITY Blocking solution (in 0.1 M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Tween-20: 5 µL | Fisher Scientific | BP337-500 | |
| CLARITY wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Tween-20: 50 µL | Fisher Scientific | BP337-500 | |
| CUBIC Blocking solution (in 0.1M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Triton X-100: 5 µL | VWR | M143-1L | |
| CUBIC Reagent-1 (in 0.1M PBS, 50 mL in total) | |||
| N, N, N’, N’-tetrakis (2-hydroxypropyl) ethylenediamine: 12.5 g | Aldrich | 122262 | |
| Triton X-100: 7.5 g | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC Reagent-2 (in 0.1M PBS, 50 mL in total) | |||
| Sucrose: 25 g | Sigma | S1888-500G | |
| Sodium azide (in powder form): 10 g | Sigma | 71289-50G | |
| Sodium azide stock solution (in DI H2O, 50 mL in total) | Sigma | 71289-50G | |
| Triethanolamine: 5 g | Sigma | 90270-500mL | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| DAPI staining solution (0.5 µg/mL) | |||
| DAPI stock solution: 1 µL | |||
| Wash solution: 10 mL | |||
| DAPI stock solution (5 mg/mL) | |||
| DAPI powder: 5 mg | Sigma-Aldrich | D9542-1MG | |
| DMSO (100%): 1 mL | ThermoFisher | D12345 | |
| Imaging Media RI-2 (in dH2O) | |||
| 90% Histodenz | Sigma | D2158-100G | |
| 0.01% Sodium azide | Sigma | 71289-50G | |
| 0.02 Sodium Phosphate Buffer, pH 7.5 | Sigma-Aldrich | S9638-250G | |
| 0.1% Tween-20 | Fisher Scientific | BP337-500 | |
| Primary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Goat anti-HIV p24: 10 µL (1:200) | Creative Diagnostics | DPATB-H81692 | |
| Mouse anti-human CD68: 10 µL(1:200) | Dako | M0876 | |
| Rabbit anti-human CD3: 10 µL (1:200) | Dako | A0452 | |
| 8% SDS Solution (in 0.1 M PBS, 50 mL in total) | |||
| SDS powder: 4 g | Sigma-Aldrich | L3771-500G | |
| Secondary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Donkey anti-goat conjugated with AlexaFluor647: 2 µL | Invitrogen | A21447 | |
| Donkey anti-mouse conjugated with AlexaFluor594: 2 µL | Invitrogen | A21203 | |
| Donkey anti-rabbit conjugated with AlexaFluor488: 2 µL | Invitrogen | A21206 |
Ссылки
- Spalteholz, W., Barker, L. F., Mall, F. P. . Hand-Atlas of Human Anatomy. , (1907).
- Jacob, T., Gray, J. W., Troxell, M., Vu, T. Q. Multiplexed imaging reveals heterogeneity of PI3K/MAPK network signaling in breast lesions of known PIK3CA genotype. Breast Cancer Research and Treatment. 159 (3), 575-583 (2016).
- Kieffer, C., Ladinsky, M. S., Ninh, A., Galimidi, R. P., Bjorkman, P. J. Longitudinal imaging of HIV-1 spread in humanized mice with parallel 3d immunofluorescence and electron tomography. eLife. 6, 23282 (2017).
- Chung, K., Deisseroth, K. CLARITY for mapping the nervous system. Nature Methods. 10 (6), 508-513 (2013).
- Compton, A. A., Schwartz, O. They might be giants: Does syncytium formation sink or spread HIV infection. PLoS Pathogens. 13 (2), 2-8 (2017).
- Symeonides, M., et al. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses. 7 (12), 6590-6603 (2015).
- Sharova, N., Swingler, C., Sharkey, M., Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. The EMBO Journal. 24 (13), 2481-2489 (2005).
- Colby, D. J., et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nature Medicine. 24 (7), 923-926 (2018).
- Ladinsky, M. S., et al. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. eLife. 8, 46916 (2019).
- Buettner, M., Bode, U. Lymph node dissection--understanding the immunological function of lymph nodes. Clinical and Experimental Immunology. 169 (3), 205-212 (2012).
- Treweek, J. B., et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nature Protocols. 10, 1860-1896 (2015).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159, 911-924 (2014).
- Sompuram, S. R., Vani, K., Bogen, S. A. A molecular model of antigen retrieval using a peptide array. American Journal of Clinical Pathology. 125 (1), 91-98 (2006).
- Scalia, C. R., et al. Antigen masking during fixation and embedding, dissected. The journal of histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 65 (1), 5-20 (2017).
- Jing, D., et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Research. 28 (8), 803-818 (2018).
- Guldner, I. H., et al. An Integrative platform for three-dimensional quantitative analysis of spatially heterogeneous metastasis landscapes. Scientific Reports. 6, 24201 (2016).
- Muntifering, M., et al. Clearing for deep tissue imaging. Current Protocols in Cytometry. 86 (1), 38 (2018).
- DePas, W. H., et al. Exposing the three-dimensional biogeography and metabolic states of pathogens in cystic fibrosis sputum via hydrogel embedding, clearing, and rRNA labeling. mBio. 7 (5), 00796 (2018).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image. Nature Methods. 9 (7), 676-682 (2012).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены