
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Simultane Quantifizierung von T-Zell-Rezeptor-Excision Kreis (TRECs) und K-Löschen Rekombination Excision Kreis (KRECs) durch Echtzeit-PCR
In diesem Artikel
Zusammenfassung
Here, we describe a method for simultaneous quantification of T-cell receptor excision circles (TRECs) and K-deleting recombination excision circles (KRECs). The TREC/KREC assay can be used as marker of thymic and bone marrow output.
Zusammenfassung
T-cell receptor excision circles (TRECs) and K-deleting recombination excision circles (KRECs) are circularized DNA elements formed during recombination process that creates T- and B-cell receptors. Because TRECs and KRECs are unable to replicate, they are diluted after each cell division, and therefore persist in the cell. Their quantity in peripheral blood can be considered as an estimation of thymic and bone marrow output. By combining well established and commonly used TREC assay with a modified version of KREC assay, we have developed a duplex quantitative real-time PCR that allows quantification of both newly-produced T and B lymphocytes in a single assay. The number of TRECs and KRECs are obtained using a standard curve prepared by serially diluting TREC and KREC signal joints cloned in a bacterial plasmid, together with a fragment of T-cell receptor alpha constant gene that serves as reference gene. Results are reported as number of TRECs and KRECs/106 cells or per ml of blood. The quantification of these DNA fragments have been proven useful for monitoring immune reconstitution following bone marrow transplantation in both children and adults, for improved characterization of immune deficiencies, or for better understanding of certain immunomodulating drug activity.
Einleitung
T-Zell-Rezeptor-Exzision Kreise (TRECs) und K-Löschen Rekombination Exzision Kreise (KRECs) sind kleine zirkularisiertes DNA-Elemente, die in einem Anteil von T- und B-Zellen jeweils während einer genomischen DNA-Rekombination herausgeschnitten werden, was zu dem Bildung einer stark diversifizierten Repertoire von T- und B-Zell-Rezeptoren. Sie haben keine Funktion, sondern weil sie stabil sind und nicht repliziert werden, werden sie nach jeder Zellteilung verdünnt, also nur in einer der beiden Tochter persistierenden Zellen. Daher kann ihre Ebenen im peripheren Blut als eine Schätzung des Thymus und des Knochenmarks Ausgangs angenommen werden.
Während die TREC-Assay wurde weitgehend in den letzten 15 Jahren um das Ausmaß der Thymusfunktion auszuwerten, um 1 der KREC Assay, die ursprünglich entwickelt wurde B-Zell-Proliferation und ihren Beitrag zur B-Zell-Homöostase in Gesundheit und Krankheit, 2 messen wurde erst kürzlich als Marker der Knochen m vorgeschlagenenarrow Ausgang. 3,4 Hier beschreiben wir die Methode, die wir für die simultane Quantifizierung von beiden TRECs und KRECs entwickelt. 4
Mit diesem kombinierten Verfahren wird die Variabilität DNA Quantifizierung durch Echtzeit-PCR verbunden sind, durch die Verwendung eines einzigartigen Standardkurve durch Verdünnung einer Triple-Insert enthaltenden Plasmid Fragmente TRECs, KRECs und T-Zell-Rezeptor alpha konstant erhalten (TCRAC) beseitigt Gens in einer 1: 1: 1-Verhältnis. Dies ermöglicht eine genauere Bewertung der TREC und KREC Kopienzahl. Ferner ist die gleichzeitige Quantifizierung der zwei Targets in der gleichen Reaktion ermöglicht Reagenz Kostenreduzierung.
Die vorgeschlagene TREC / KREC Assay kann nützlich sein, um das Ausmaß der T- und B-Zell-neo-Produktion bei Kindern oder Erwachsenen mit schwerer kombinierter Immundefizienz (SCID), 4 Common Variable Immunodeficiency, 5 Autoimmunerkrankungen, 6-8 und HIV-Infektionen zu messen Ferner. 9 zeigt, kann es verwendet werdenÜberwachung der Immun Erholung nach Transplantation hämatopoetischer Stammzellen, 10 Enzymersatz, 11 und antivirale 9 oder immunmodulierende Therapien. 6-8 Schließlich weil SCID-Patienten werden mit TREC-Test trotz der zugrunde liegenden genetischen Defekte erkannt und Agammaglobulinämie Patienten können mit KREC Quantifizierung identifiziert werden Die TREC / KREC Assay kann auch verwendet werden, um Immun in Neugeborenen-Screening-Programme erkennen. 12 In diesem Fall muss der Test an aus kleinen Flecken von Blut abgetupft extrahierten DNA durchgeführt, und auf Filterpapier getrocknet werden müssen hochempfindlich und spezifisch für sein die Zielkrankheiten, sowie in hohem Maße reproduzierbar und kostengünstig.
Die Einführung KREC Quantifizierung im Test sollte Aufführungen von Neugeborenen-Screening für Immundefekte, die routinemäßig in den einigen Teilen der USA (WI, MA, CA) seit 2008 durchgeführt wurde, zu verbessern, wenn Wisconsin war der erste, um introduce die Analyse TRECs in seiner postnatale Screening-Programm. 13
Access restricted. Please log in or start a trial to view this content.
Protokoll
HINWEIS: Ethics Statement: Dieses Protokoll folgt den Richtlinien unserer Institution, die Spedali Civili di Brescia
1. Herstellung eines "Triple-Insert" Plasmid
- Auswahl und Herstellung von geeigneten Ausgangsmaterials:
- Abrufen einer Zellen enthaltenden Probe, sehr wahrscheinlich TRECs und KRECs durch PCR nachweisbar sind, wie peripherem Blut eines jungen, gesunden Individuum in EDTA-Röhrchen entnommen.
HINWEIS: Ursprünglich haben wir die Methode mit Thymozyten für TRECs und mononuklearen Zellen aus Tonsillen Fragmente für KRECs entwickelt hatte, aber gesunden Probanden haben in der Regel einen Betrag von TRECs und KRECs ausreichend, um deren Nachweis durch PCR zu ermöglichen. Daher sollten peripheren Blut eine gute Alternative, leichter zu erhalten und damit zu arbeiten. In Anbetracht, dass TRECs insbesondere mit dem Alter bei gesunden Erwachsenen, peripheren Blut verringern von einem jungen Erwachsenen, wenn nicht von einem Kinder, wird empfohlen. - Separate peripheren Blut mononucLear Zellen (PBMC) von einem Standard-Dichtegradiententrennung Methode.
- Abrufen einer Zellen enthaltenden Probe, sehr wahrscheinlich TRECs und KRECs durch PCR nachweisbar sind, wie peripherem Blut eines jungen, gesunden Individuum in EDTA-Röhrchen entnommen.
- DNA-Extraktion:
- Extrahieren von DNA aus PBMC unter Verwendung eines kommerziellen DNA Blood Mini Kit. Folgen Sie den Anweisungen des Herstellers mit den wenigen unten beschriebenen Änderungen.
- Verwendung eines Schüttlers-Inkubator bei 1.400 rpm Inkubieren der Proben bei 70 ° C (statt 56 ° C) für 10 min.
- Hinzufügen des Elutionspuffers bei 70 ° C auf die Säule erwärmt, für 5 min bei 70 ° C und Eluieren der DNA durch Zentrifugation gemäß den Anweisungen des Herstellers. Bestimmen Sie die extrahierte DNA-Konzentration durch photometrische Bestimmung bei 260/280 nm.
- Extrahieren von DNA aus PBMC unter Verwendung eines kommerziellen DNA Blood Mini Kit. Folgen Sie den Anweisungen des Herstellers mit den wenigen unten beschriebenen Änderungen.
- Herstellung eines Plasmids, das die Einsätze der TREC und KREC Signalverbindungen (SJ) und TCRAC Referenzgens:
- Verstärken die aus PBMC extrahiert mit für TRECs, KRECs und TCRAC Primer (die TREC-SJ, KREC-SJ und TCRAC Fragmente zu erhalten) DNA (siehe Tabelle 1).
ANMERKUNG: Am 5'-Ende, die KREC- und TCRAC Spezifische Primer enthalten HindIII und SpeI-Restriktionsstellen (in Kleinbuchstaben in der Tabelle 1 gezeigt Sequenzen) mit einer entsprechenden Anzahl von flankierenden zusätzliche Nukleotide (in Kursivschrift angegeben); Beide Funktionen sind nicht in den kanonischen KREC und TCRAC Sequenzen.
- Verstärken die aus PBMC extrahiert mit für TRECs, KRECs und TCRAC Primer (die TREC-SJ, KREC-SJ und TCRAC Fragmente zu erhalten) DNA (siehe Tabelle 1).
| TRECs | vorwärts | 5'-AAA GAG GGC AGC CCT CTC CAA GGC-3 ' |
| umkehren | 5'-GGC TGA TCT TGT CTG ACA TTT GC-3 ' | |
| KRECs | vorwärts | 5'CCC aag ctt TCA GCG CCC ATT ACG TTT CT-3 ' |
| umkehren | 5'CCC aag ctt GTG AGG GAC ACG CAG CC-3 ' | |
| TCRAC | vorwärts | 5'-G ac Tag TAT GAG ACC GTG ACTTGC CAG-3 ' |
| umkehren | 5'-G ac Tag TGC TGT TGT TGA AGG CGT TTG C-3 ' |
Tabelle 1. Primer für die Klonierungsverfahren verwendet. Am 5'-Ende, in Kleinbuchstaben sind entsprechende Restriktionsenzymstellen gezeigt, Nukleotide, wohingegen in Kursivschrift dargestellt sind hinzugefügt flankierenden Nukleotiden.
- Führen PCR-Reaktionen in einem Endvolumen von 25 & mgr; l, bestehend aus 1x Puffer II, 1,5 mM MgCl 2, dNTP-Mix (jeweils 200 uM), Primer mit einer Endkonzentration von 900 nM, AmpliTaq DNA Polymerase (2,5 U / 25 ul), und 100 ng der DNA.
- Mit den folgenden PCR-Parameter: erster Schritt bei 95 ° C für 10 min, gefolgt von 40 Zyklen bei 95 ° C für 30 sec; 60 ° C für 30 sec; 72 ° C für 30 sec, und schließlich 1 Zyklus 72 ° C für 10 min. PCR-Produkt Längen sollte: 380 bp für TRECs, 166 bp für KREC, 381 bp für TCRAC.
- Legen Sie die PCR-Produkt von TREC-SJ in der TA-Akzeptor-Stelle des pCR2.1-TOPO Vektor. Wandeln Sie die Plasmid-DNA in XL1-Blue chemisch kompetente Zellen durch Hitzeschock nach dem Protokoll mit den Zellen zur Verfügung gestellt. Unter den Kolonien, die wegen der Ampicillin-Resistenz zu wachsen, zu identifizieren, die die Durchführung des Einsatzes, die wegen der verloren β-Galactosidase Ergänzung weiß erscheinen wird, und impfen sie in einen Master-Gericht. Schließlich identifizieren die Kolonien, die das TREC Fragment durch PCR.
- Erweitern Sie eine der identifizierten Kolonien und reinigen die Plasmid-DNA mit einem Plasmid Miniprep Kit, und folgen Sie den Anweisungen des Herstellers. Verdauen die Plasmid-DNA und die TCRAC amplifizierte Produkt mit SpeI Restriktionsenzym und ligieren die TCRAC Fragment in den SpeI-Schnittstelle.
- Wandeln Sie die Plasmid-DNA in XL1-Blue-Zellen und Identifizierung der Kolonien, die das TCRAC Fragment durch PCR. Erweitern Sie eine der identifizierten Kolonien und zu reinigendie Plasmid-DNA unter Verwendung des Plasmid-Miniprep-Kit.
- Verdauen die Plasmid-DNA und die KREC amplifizierte Produkt mit HindIII und klonen die KREC-SJ-Fragment in die HindIII-Schnittstelle. Wandeln Sie die Plasmid-DNA in XL1-Blue-Zellen und Identifizierung der Kolonien, die das dritte Fragment durch PCR.
- Zu überprüfen, durch direkte Sequenzierung die Gegenwart der drei Einsätze und die Abwesenheit von Mutationen. Die Karte des abschließenden Dreieinsatz Plasmid ist in Abbildung 1 dargestellt.

Abbildung 1. Dreifach-Einsatz Plasmidkarte. Dreifach-Einsatz Plasmidkarte, die die Position von TREC, KREC, TCRAC Sequenzen und Restriktionsenzymstellen. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
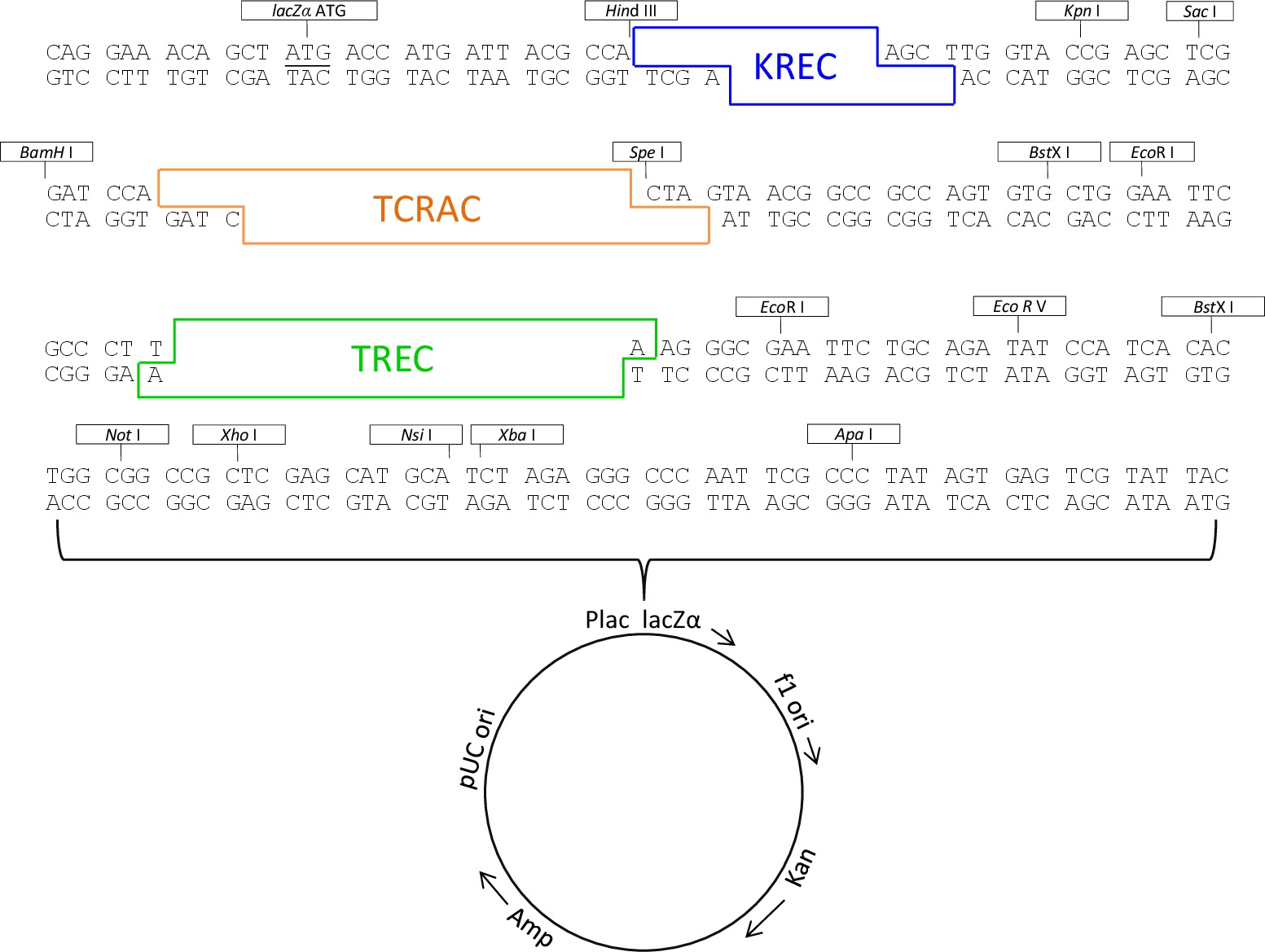{kind=link}
- Erweitern eines der identifizierten Kolonien, die Plasmid-DNA zu reinigen, unter Verwendung eines Plasmids Midiprep kit. Bestimmen Sie die Plasmid-DNA-Konzentration durch photometrische Bestimmung bei 260/280 nm und die DNA-Qualität durch Gelelektrophorese. Speicherung geeigneten Aliquots des Plasmids bei -80 ° C (z. B. jeweils 50 ul).
2. Standard Curve Vorbereitung
HINWEIS: Bereiten Sie alle Verdünnungen in 0,1 × TE-Puffer in einem Ort speziell für Rückhaltung von DNA-Verschleppung konzipiert.
- Berechnen Sie die Masse der Plasmid-Kopienzahl von Interesse (siehe Tabelle 2), unter Berücksichtigung, dass die Plasmid-Größe ist 4846 bp und dass die durchschnittliche Masse pro bp ist 1,096 × 10 -21 g / bp und deshalb: m p = ( 4846 bp) x (1,096 × 10 -21 g / bp) = 5,311.216 x 10 -21 g = 5,311 x 10 -18 g.
| Kopieren # | x 5,311 x 10 -18 g | Masse von Plasmid-DNA (g) |
| 1 x 10 6 | 5,311 x 10 -12 | |
| 1 x 10 & sup5; | 5,311 x 10 -13 | |
| 1 x 10 & sup4; | 5,311 x 10 -14 | |
| 1 x 10 3 | 5,311 × 10 -15 | |
| 1 x 10 2 | 5,311 × 10 -16 |
Tabelle 2. Messe von Plasmid für jede Standardkurve Verdünnungspunkt benötigt.
- Berechnen der Konzentrationen von Plasmid-DNA, die durch Unterteilen der benötigten Plasmids Massen durch das Volumen (5 ul), die in jeder Reaktion pipettiert werden (siehe Tabelle 3).
| Masse von Plasmid-DNA (g) | ÷ 5 ul | Endkonzentration von Plasmid-DNA (g / & mgr; l) |
| 5,311 x 10 -12 | 1,062 x 10 -12 | |
| 5,311 x 10 -13 | 1,062 x 10 -13 | |
| 5,311 x 10 -14 | 1,062 x 10 -14 | |
| 5,311 × 10 -15 | 1,062 x 10 -15 | |
| 5,311 × 10 -16 | 1,062 × 10 -16 |
Tabelle 3. Berechnung von Plasmid-Konzentrationen für jede Verdünnung Punkt benötigt.
- Tauwetter ein Aliquot der Plasmid. Linearisieren 2 ug der Plasmid-DNA mit XhoI und Bestimmung seiner Konzentration by spektrophotometrische Bestimmung bei 260/280 nm.
- Bereiten Sie eine richtige Verdünnung der linearisierte Plasmid-DNA, um eine komfortable Arbeitsstammlösung zu erreichen, um aus (zB 100 ng / & mgr; = 0,1 ug / ul = 1 x 10 -7 g / ul) zu starten. Speichert den Rest der linearisierte Plasmid DNA bei -80 ° C.
- Führen Sie eine geeignete Anzahl von 1:10 oder 1: 100 Verdünnungen, um das Plasmid an der praktikabler Konzentration von 1 x 10 -10 g / & mgr; l zu bringen.
- Verwenden Sie die Formel C 1 V 1 = C 2 V 2, das Verdünnungsvolumen berechnen (V 2 - V 1) benötigt, um den ersten Standard-Kurvenpunkt der Serie vorzubereiten (1 x 10 6 Kopien, entsprechend 1,062 x 10 -12 g / 5 & mgr; l; siehe Tabelle 4).
| Initial konz. (G / & mgr; l) | Die Plasmid-DNA vol (ul) | Diluent vol (ul) | Schluss Vol. (Ul) | Schluss konz. (G / & mgr; l) | Schlusskopienzahl von Plasmid-DNA / 5 ul |
| C 1 | V 1 | V 2 -V 1 | V 2 | C 2 | |
| 1 x 10 -7 | 5 & mgr; | 45 & mgr; | 50 & mgr; | 1 x 10 -8 | N / A |
| 1 x 10 -8 | 5 & mgr; | 495 & mgr; | 500 & mgr; | 1 x 10 -10 | N / A |
| 1 x 10 -10 | 5 & mgr; | 465 & mgr; | 470 & mgr; | 1,062 x 10 -12 | 1 x 10 6 |
| 1,062 x 10 -12 | 50 & mgr; | 450 & mgr; | 500 & mgr; | 1.062 x 10 -13 | 1 x 10 & sup5; |
| 1,062 x 10 -13 | 50 & mgr; | 450 & mgr; | 500 & mgr; | 1,062 x 10 -14 | 1 x 10 & sup4; |
| 1,062 x 10 -14 | 50 & mgr; | 450 & mgr; | 500 & mgr; | 1,062 x 10 -15 | 1 x 10 3 |
| 1,062 x 10 -15 | 50 & mgr; | 450 & mgr; | 500 & mgr; | 1,062 x 10- 16 | 1 x 10 2 |
Tabelle 4. Dilution Berechnungen.
- Fahren Sie mit einer regelmäßigen Reihe von 1:10 Verdünnungen, um die verbleibenden Punkte Standardkurve zu erhalten.
HINWEIS: Vergessen Sie nicht, jedes Plasmid Verdünnung kurz bevor Sie den nächsten Wirbel.
HINWEIS: Die höchste Verdünnung des standard Kurve (10 Kopien / 5 & mgr; l) wird kurz vor der Durchführung des Assays hergestellt werden, weil eine solche geringe Menge an Plasmid-DNA ist nicht stabil genug, um für eine zukünftige Verwendung gespeichert werden. - Bewahren Sie die Triple-Plasmid Verdünnung Punkte bei -80 ° C in getrennten Röhren bis zur Verwendung. Die verdünnte Triple-Plasmid-DNA ist für mindestens 6 Monate sehr stabil.
3. DNA-Extraktion von Zielstichproben
- Separate PBMC aus peripherem Blut in EDTA-Röhrchen mit dem Dichtegradiententrennung Verfahren gesammelt.
HINWEIS: Alternativ können andere geeignete Ausgangsmaterial (beispielsweise sortiert Lymphozytensubpopulationen, Vollblut) als Zielprobe. - Extrahieren Sie die DNA aus PBMC von Zielstichproben, wie in Schritt 1.2 berichtet.
4. Real-time PCR zur Quantifizierung der TRECs und KRECs
- Plattenherstellung und das Laden:
- Vereinbaren Sie einen PCR-Platte, die mindestens zwei Wiederholungen für jede Probenanalysiert werden: Standardkurve (A → F 1 → 4), Positivkontrolle (CTRL +; G1 → 4) und Kontrolle ohne Matrize (NTC; H 1 → 4).
HINWEIS: ein typisches Schema beachten in der Tabelle 5, aber unterschiedlichen Formaten erstellt werden können. Beachten Sie, dass TRECs und KRECs zusammen in den gleichen Quellen, die sich von denen der TCRAC verstärkt werden.
- Vereinbaren Sie einen PCR-Platte, die mindestens zwei Wiederholungen für jede Probenanalysiert werden: Standardkurve (A → F 1 → 4), Positivkontrolle (CTRL +; G1 → 4) und Kontrolle ohne Matrize (NTC; H 1 → 4).
| TRECs + KRECs ein | TCRAC b | TRECs + KRECs | TCRAC | TRECs + KRECs | TCRAC | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| A | 10 6 Kopien | 10 6 Kopien | 10 6 Kopien | 10 6 Kopien | 1 | 1 | 1 | 1 | 9 | 9 | 9 | 9 |
| B | 10 5 Kopien | 10 5 Kopien | 10 5 Kopien | 10 5 Kopien | 2 | 2 | 2 | 2 | 10 | 10 | 10 | 10 |
| C | 10 4 Kopien | 10 4 Kopien | 10 4 Kopien | 10 4 Kopien | 3 | 3 | 3 | 3 | 11 | 11 | 11 | 11 |
| D | 10 3 Kopien | 10 3 Kopien | 10 3 Kopien | 10 3 Kopien | 4 | 4 | 4 | 4 | 12 | 12 | 12 | 12 |
| E | 10 2 Kopien | 10 2 Kopien | 10 2 Kopien | 10 2 Kopien | 5 | 5 | 5 | 5 | 13 | 13 | 13 | 13 |
| F | 10 Exemplare | 10 Exemplare | 10 Exemplare | 10 Exemplare | 6 | 6 | 6 | 6 | 14 | 14 | 14 | 14 |
| G | STRG + | STRG + | STRG + | STRG + | 7 | 7 | 7 | 7 | 15 | 15 | 15 | 15 |
| H | NTC | NTC | NTC | NTC | 8 | 8 | 8 | 8 | 16 | 16 | 16 | 16 |
Tabelle 5. Beispiel Echtzeit-PCR-Platte.
- Berechnen Sie die Gesamtzahl der Bohrungen erforderlich (enthalten 1-2 zusätzliche Bohrlöcher auf mögliche Pipettierfehler zu berücksichtigen) und Vorbereitung des Master-Mix für TRECs / KRECs und TCRAC in getrennten Röhren (siehe Tabelle 6).
HINWEIS:. Die Primer und Sonden, die spezifisch für TRECs, KRECs und TCRAC sind in Tabelle 7 genannten Der Primer und die Sonde Endkonzentrationen 900 nM und 200 nM.
- Berechnen Sie die Gesamtzahl der Bohrungen erforderlich (enthalten 1-2 zusätzliche Bohrlöcher auf mögliche Pipettierfehler zu berücksichtigen) und Vorbereitung des Master-Mix für TRECs / KRECs und TCRAC in getrennten Röhren (siehe Tabelle 6).
| TRECs / KRECs | TCRAC | ||
| H 2 O | 2 & mgr; | H 2 O | 4,75 & mgr; |
| KRECs 20 pmol / & mgr; | 1,125 & mgr; | TCRAC 20 pmol / & mgr; | 1,125 ul |
| KRECs rev 20 pmol / ul | 1,125 & mgr; | TCRAC rev 20 pmol / ul | 1,125 & mgr; |
| KRECs Sonde 10 pmol / ul | 0,5 ul | TCRAC Sonde 10 pmol / ul | 0,5 ul |
| TRECs 20 pmol / ul | 1,125 & mgr; | 2x Universal-TaqMan PCR Master Mix | 12,5 ul |
| TRECs rev 20 pmol / ul | 1,125 & mgr; | ||
| TRECs Sonde 10 pmol / ul | 0,5 ul | ||
| 2x Universal-TaqMan PCR Master Mix | 12,5 ul | ||
Tabelle 6. Volumen der Reagenzien für die angegebenen Bohrungen benötigt.
| TRECs | vorwärts | 5'-CAC ATC CCT TTC AAC CAT GCT-3 ' |
| umkehren | 5'-TGC AGG TGC CTA TGC ATC A-3 ' | |
| Sonde | 5'-FAM-ACA CCT CTG GTT GTA TTT AAG GTG CCC ACT-TAMRA-3 ' | |
| KRECs | vorwärts | 5'-TCC CTT AGT GGC ATT ATT TGT ATC ACT-3 |
| umkehren | 5'-AGG AGC CAG CTC TTA CCC TAG AGT-3 ' | |
| Sonde | 5'-HEX-TCT GCA CGG GCA GCA GGT TGG-TAMRA-3 | |
| TCRAC | vorwärts | 5'-TGG CCT CCT AAC GAT CCT CTT-3 ' |
| umkehren | 5'-GGA TTT AGA TCT GTC CAG CTG GTA CAC-3 | |
| Sonde | 5'-FAM-TCC CAC AGA TAT CCA GAA CCC TGA CCC-TAMRA-3 ' |
Tabelle 7 Sequenz der Primer und Sonden für die Echtzeit-PCR-Test.
- In 20 ul jeder Mischung (TRECs / KRECs und TCRAC). Vor dem Verlassen des Reagenz Vorbereitungsraum, dichten die Reaktionsplatte mit einem optischen Klebstoff Abdeckung.
- In einer Nukleinsäureextraktion Raum, heben Sie die Klebeabdeckung und mit 5 ul der genomischen DNA-Probe (400 bis 500 ng) in die Vertiefungen. Mit 5 & mgr; l genomischer DNA (dh von PBMC aus Nabelschnurblut erhalten wurde, hergestellt), die mit CTRL + Brunnen.
HINWEIS: Als Alternative zu Nabelschnurblut verwenden DNA aus der gleichen bekannten, positiven peripheren Blutprobe oder Pool von Proben oder bereiten eine "künstliche" positive Standard, indem man eine bestimmte Menge an Triple-Einsatz Plasmid mit genomischer DNA aus TREC- und KREC-negative Zelllinien. - In 5 ul Wasser zu den NTC Brunnen. Verschließen Sie die Reaktionsplatte wieder mit dem optischen Kleber Abdeckung.
- Sorgen Sie für eine Sicherung Eindämmung von Plasmid-DNA-Verschleppung; auftauen jeder Verdünnung point der Plasmid-DNA-Standardkurve nur vor dem Gebrauch und bereiten die höchste Verdünnung (10 Kopien / 5 ul). Heben Sie die Klebeabdeckung nur von A → H 1 → 4 Brunnen und mit 5 ul jeder Punkt Verdünnung von Plasmid-DNA-Standardkurve. Verschließen Sie die Klebeabdeckung wieder.
- Überprüfen Sie die Abwesenheit von Blasen auf dem Boden der Vertiefungen, wodurch sichergestellt wird, dass die Reagenzien an der Unterseite positioniert ist.
- Führen Sie den Test auf einer Echtzeit-PCR-System. Das Standardprotokoll besteht aus einer ersten Stufe bei 50 ° C für 2 min, einer anfänglichen Erhitzen bei 95 ° C für 10 min, gefolgt von 45 Zyklen der Denaturierung bei 95 ° C für 15 sec, und einer kombinierten Primer / Sonde-Glühen und Dehnung 60 ° C für 1 min.
- Speichern Sie die Daten am Ende der PCR-Programm.
HINWEIS: Um DNA Kreuzkontaminationen zu vermeiden, denken Sie daran: die üblichen guten Laborpraxis Empfehlungen für quantitative PCR folgen beim Einrichten der Echtzeit-PCR-Assay: Verwenden Sie separate Bereiche / Räume für die Nukleinsäureextraktion,Reagenzienvorbereitung, Plasmid-Manipulation, Amplifikationsreaktion; benutzen Pipettensätze; ändern Handschuhe / Mäntel als angemessen.
- Ergebnisse und Berechnung:
HINWEIS: Die folgenden Schritte werden auf unseren Erfahrungen mit dem mit der in der Tabelle aufgeführten Materialien in Echtzeit-PCR-Instrument, aber in der Regel zur Verfügung gestellten Software basieren, sollten sie mit anderen Echtzeit-PCR-Analyse-Plattformen und Software gelten. Beachten Sie, dass, auch wenn mit der gleichen Software, in der Regel gibt es mehr als einen Weg, um die erforderlichen Befehle (zB Symbole, Verknüpfungen) auszuführen.- Öffnen Sie die gespeicherte Ergebnisdatei in der Software und klicken Sie auf der Registerkarte "Ergebnisse". Am oberen Rand des Fensters, klicken Sie im Menü "Analyse" und wählen Sie "Analyse-Einstellungen". Lassen Sie die Software bestimmen die Schwelle automatisch, indem Sie "Alle", Detektoren und "Auto Ct" (Ct = Schwellenzyklus. Lassen Sie die Software-Reihe "Automatische Baseline"; für die Hintergrund Beseitigung standardmäßig.
- Klicken Sie auf der Registerkarte mit dem Namen "Amplification Plot" und, an der äußersten rechten Teil des Fensters, wählen Sie eine der drei "Detektoren" im entsprechenden Dropdown-Menü (zB Starten mit TRECs). Direkt unter, wählen Sie "Manuell Ct", um einen Schwellenwert manuell einstellen; gehen Sie zum unteren Teil des Fensters und wählen Sie die Brunnen, die den Standardkurve Verdünnung Punkte des gewählten Detektor, um die Verstärkung Plots zeigen. Dazu klicken Sie einfach und ziehen Sie mit der Maus über den entsprechenden Zellen der Tabelle.
- Um die Schwelle manuell einzustellen, ziehen Sie den roten Grenzlinie auf und ab, und klicken Sie dann auf "Analysieren", um Ergebnisse erneut analysieren. Um zu überprüfen, wie die Einstellung der Schwelle auf die Ergebnisse auswirkt, wählen Sie die Registerkarte "Report", und sehen Sie die daraus resultierende Ct der jeweiligen Standardkurve Verdünnung Punkten. Beim Bewegen der Schwelle, versuchen Sie, mit den folgenden Empfehlungen nachzukommen, um eine optimale Platzierung zu bekommen.
- Gehen Sie auf der Registerkarte "Amplification Plot" und überprüfen Sie, ob der Schwellenwert in einem Bereich der exponentiellen Amplifikation ausreichend über dem Hintergrundrauschen, aber unterhalb der Plateauphase. Wenn die Amplifikationsplot nicht im logarithmischen Maßstab gezeigt, rechtsklicken Sie auf dem Grundstück, die "Diagrammeinstellungen" und stellen Einstellung Nachlauf Y-Achse als "log", stellen Sie den Schwellenwert auf halbem Weg in den linearen Teil des Rückrufs Grundstück, und beobachten Sie die Verstärkungskurven annähernd parallel zueinander.
- Klicken Sie auf der Registerkarte "Report", um die Ergebnisse zu sehen Ct. Stellen Sie sicher, dass die Schwelle Platzierung zu Ergebnissen geführt, die die Genauigkeit der zwei Wiederholungen jeder Standardkurve Verdünnung Punkte zu maximieren.
- Prüfen Sie, ob der Schwellenwert an der Stelle, die am besten trägt der extremen Größenordnungen der Standardkurve Verdünnung Punkte (optimale Empfindlichkeit) gestellt. Wiederholen Sie Schritt 4.2.2 und verbleibenden Teilschritte für die verbleibenden zwei Detektoren (KRECs und TCRAC).
- Gehen Sie auf die Registerkarte Bericht, ziehen Sie die Maus wieder in der Tabelle im unteren Bereich des Fensters, um die Zellen, die den Vertiefungen der Standardkurve für alle Detektoren (TRECs, KRECs, TCRAC) zu wählen. Stellen Sie sicher, dass die resultierende Ct der drei Ziele ist zu den gleichen Verdünnung Punkten sehr ähnlich (zB Unterschied nicht mehr als 0,5 Ct). Nachstellen Schwellenwerte bei Bedarf und erneut prüfen.
Hinweis: Da die Dreifach-Einsatz Plasmid enthält eine einzelne-Kopie jedes Ziel und damit das Verstärkungsverhältnis theoretisch in der Nähe von 1: 1: 1. In einigen Fällen kann es erforderlich sein, von Hand auch die Grundlinie für einen oder mehrere der Detektoren durch, um bessere Ergebnisse zu erzielen. Genau daran erinnern, das Fenster "Analyse-Einstellungen", wählen Sie den Detektor für die der Anpassung erforderlich ist, stellen Sie "Manual Baseline", und legen Sie benutzerdefinierte Werte für die Start- und End-Cycles (in der Regel 3 bis 15). Klicken Sie dann auf "OK & erneut analysieren" und überprüfen Sie vorherige Punkte.
- Übernehmen Sie die experimentellen Sitzung erst nach Überprüfung der folgenden:
- Klicken Sie auf der Registerkarte "Report", gehen Sie auf den unteren Teil des Fensters, wählen NTC Brunnen und überprüfen, ob die entsprechenden Ct "Undet" (dh keine Verstärkung in NTC Brunnen vorhanden).
- Wählen Sie die Registerkarte "Standardkurve" und suchen Sie auf der rechten Seite des Plot-Fenster; Wählen Detektoren aus der Drop-down-Menü und sehen, ob die gemeldete Standardkurve Steigung im Bereich zwischen -3,55 und -3,32 (entspricht Wirkungsgrade von 91 PCR - 100%)
- Stellen Sie sicher, dass der Bestimmungskoeffizient (R 2) ist höher als 0,998 (Abbildung 2), durch das Lesen seines Wertes direkt unterhalb der Steigung und Achsenabschnitt (in der gleichen Registerkarte "Standardkurve", nachdem Sie jeden Detektor).
HINWEIS: Achten Sie auf die extreme Verdünnung Punkte, weil ihre Fehlausrichtung kann die Neigung beeinflussen leichter (sie eine hohe "leve habenWut "Auswirkungen auf die Regressionsgerade). Insbesondere im Hinterkopf behalten, dass die höchste Plasmid Verdünnung kann früher als erwartet beeinträchtigen. , Wann immer dies möglich ist es jedoch sinnvoll, nicht um sie zu verwerfen, da ein vernünftiger Ansatz sein kann, um 100 als Grenze der quantitativen Nachweis berücksichtigen: mit anderen Worten, für Proben zwischen 10 und 100, kann das Ergebnis als <100 oder "positive gemeldet werden , aber nicht quantifizierbar ", während, wenn außerhalb der Kurvenbereich (<10) kann es als 0 oder gemeldet werden" nicht nachweisbar ".
HINWEIS: Es kann sinnvoll sein, ein Archiv der Ct-Werte der bisherigen Standardkurve Verdünnungen zu halten, um einen Durchschnittswert für jede Verdünnung Punkt zu berechnen sein, als eine Art optimal "Referenz" Bereich oder interne Qualitätskontrolle für zukünftige Experimente gehalten werden (zB Berechnung der Mittelwert und SD zu Shewhart Regelkarten zu bauen). Analog kann es hilfreich sein, ein Archiv der vergangenen Ct der positiven Kontrolle zu halten.

Abbildung 2. Standardkurven für TRECs, KRECs und TCRAC. Stücke der Standardkurvenpunkte und Log-Regressionslinie Schätzungen für TRECs (A), KRECs (B), und TCRAC (C) werden errichtet, um die Einhaltung der idealen exponentiellen überprüfen Verstärkungsrate (Steigung = 3,32), die mit einem Wirkungsgrad von 100% entsprechen würde. Ct: Schwellenzyklus; R 2:. Regressions Bestimmtheitsmaß Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
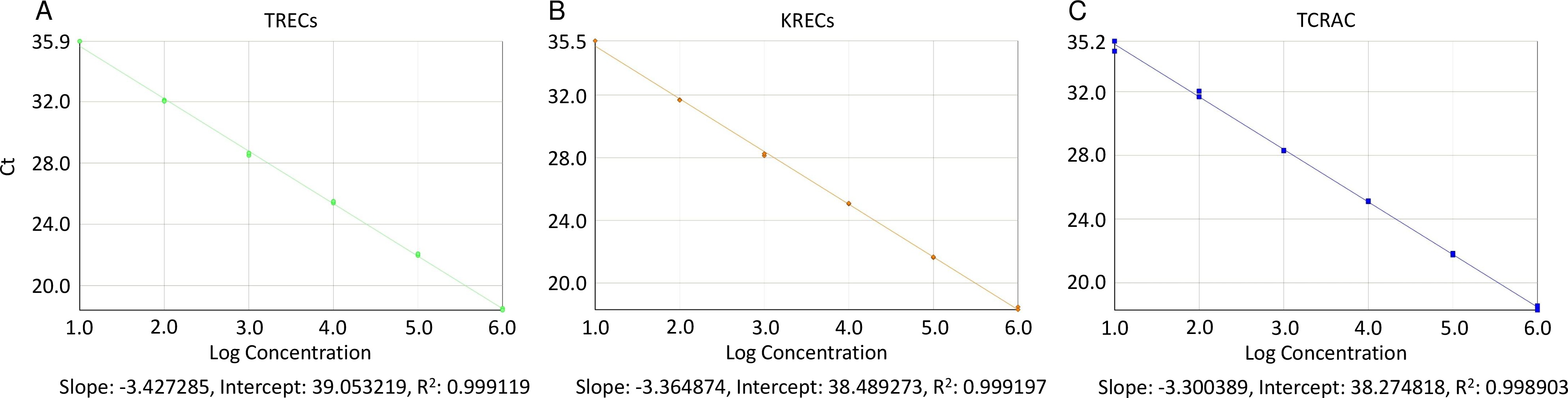{kind=link}
- Klicken Sie auf der Registerkarte "Report" und stellen Sie sicher, dass die Ct der Positivkontrolle (in den entsprechenden Zellen der Tabelle unter der Grafik gefunden) steht im Einklang mit den Ergebnissen der bisherigen Tests auf die samir bekannt positive Kontrolle.
HINWEIS: Die Registerkarte "Report" zeigt die Menge an TRECs, KRECs und TCRAC der Proben getestet, dass die Software durch Interpolation aus den jeweiligen Standardkurve berechnet, unter Verwendung der CT-Werte nach der Schwelle und der Basislinie geeignet eingestellt worden sind (a neue Analyse mit einer neuen Schwelle / Basis würden diese Ergebnisse ändern). Um zu sehen, wo die Interpolation auf der Standardkurve erfolgt, wählen Sie die Registerkarte "Standardkurve" und wählen Sie dann die gewünschten Brunnen, und suchen Sie den schwarzen "X" auf der Regressionslinie erscheinen. Ihre y-Achse Wert ist der interpolierte Menge. Wenn Sie immer die gleiche positive Kontrolle können geeignete Referenzbereiche und / oder Qualitätsregelkarten erstellt werden, basierend auf vorherigen Werten auf der Positivkontrolle ermittelt.
- Klicken Sie auf der Registerkarte "Report" und stellen Sie sicher, dass die Ct der Positivkontrolle (in den entsprechenden Zellen der Tabelle unter der Grafik gefunden) steht im Einklang mit den Ergebnissen der bisherigen Tests auf die samir bekannt positive Kontrolle.
- Klicken Sie auf das Menü "Datei", "Speichern" und dann auf "Export" in eine CSV-Datei, um die Mengen aller Vertiefungen in einer te exportierenxt-Datei mit kommagetrennten Werten.
- Importieren the.csv Datei in einem Tabellenkalkulationsprogramm und berechnen Sie die Anzahl der TRECs oder KRECs pro 10 6 PBMC, indem Sie die folgende Formel entsprechend: [(mittlere Menge TRECs oder KRECs) / (Durchschnittsmenge TCRAC / 2)] x 10 6
HINWEIS: Die mittlere Menge TCRAC durch 2 geteilt wird, da in jeder Zelle gibt es zwei TCRAC Genkopien, beispielsweise eine für jedes Chromosom.
HINWEIS: Die Konsistenz der endgültigen Anzahl der TRECs und KRECs der Positivkontrolle kann angesehen werden, aber bedenken Sie, dass Werte nicht perfekt wegen der zusätzlichen Variabilität der Standardkurve Verdünnungspunkten entsprechen. Bei grober Divergenz, muss der Versuch wiederholt werden. Auch hier kann Regelkarten erstellt werden, um einen geeigneten Referenzbereich festlegen. - (Optional, aber empfohlen) Wenn die Ergebnisse der Blutbild sind, berechnen Sie die TRECs oder KRECs pro ml Blut nach folgender Formel:
TRECs oder KRECs pro 1 x 10 6) x (Lymphozyten + Monozytenzahl in 1 ml Blut) / 10 = 6 Kopien / ml.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Der Test wurde in einer repräsentativen Stichprobe von 87 gesunden Kontrollpersonen durchgeführt: 42 Kinder im Alter von 0 bis 17 (männlich / weiblich: 25/17) und 45 Erwachsene im Alter von 24 bis 60 (Männer / Frauen: 29/16). Die Ergebnisse wurden als TRECs und KRECs pro 10 6 PBMC und dann den TRECs und KRECs pro ml Blut wurden berechnet erhalten.
Die Anzahl der TRECs nimmt mit dem Alter aufgrund Thymusinvolution, 4 insbesondere in einem sehr scharfen Mode 0-3 - 4 J...
Access restricted. Please log in or start a trial to view this content.
Diskussion
TREC and KREC quantification can be considered a good estimate of recent thymic and bone marrow output provided that some caveats are taken into account. Even though an absolute quantification method employing standard curve requires more reagents and more space on the real-time PCR reaction plate, it ensures highly accurate quantitative results because unknown sample quantities are interpolated from standard curves built upon known amounts of starting material. Moreover this method is better fitted to detect low amount ...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
The authors have nothing to disclose.
Danksagungen
The authors have no acknowledgements.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| Histopaque-1077 | Sigma Aldrich SRL | 10771-500 ML | density gradient separation method |
| QIAamp DNA Blood Mini Kit (250) | QIAGEN | 51106 | DNA extraction |
| Unmodified DNA Oligonucleotides HPSF 0.01 mmol | Eurofins MWG Operon/Carlo Erba Reagents S.r.l | Resuspend the lyophilized product to 100 pmol/µl | |
| AmpliTaq DNA Polymerase: including 10x Buffer II and 25 mM MgCl2 | Applied Biosystems/Life-Technologies | N8080156 | |
| GeneAmp dNTP Blend (100 mM) | Applied Biosystems/Life-Technologies | N8080261 | |
| TOPO TA Cloning Kit for Subcloning | Invitrogen/Life-Technologies | K4500-01 | |
| XL1-Blue Subcloning Grade Competent Cells | Stratagene | 200130 | |
| PureYield Plasmid Miniprep System | Promega | A1223 | |
| SpeI 500U | New England Biolabs | R0133S | |
| HindIII-HF 10,000 U | New England Biolabs | R3104S | |
| PureYield Plasmid Midiprep System | Promega | A2492 | |
| XhoI 5,000 U | New England Biolabs | R0146S | |
| TRIS Utrapure | Sigma Aldrich SRL | T1503 | |
| EDTA | Sigma Aldrich SRL | E5134 | |
| TE buffer (1 mM TRIS and 0.1 mM EDTA) | |||
| TaqMan Universal PCR Master Mix | Applied Biosystems/Life-Technologies | 4364338 | |
| Dual labeled probes HPLC 0.01 mmol | Eurofins MWG Operon/Carlo Erba Reagents S.r.l | Resuspend the lyophilized product to 100 pmol/µl | |
| NanoDrop 2000c spectrophotometer | ThermoFisher | ||
| Applied Biosystems 2720 Thermal Cycler | Applied Biosystems/Life-Technologies | 4359659 | |
| Fast 7500 Real-Time PCR system | Applied Biosystems/Life-Technologies | ||
| SDS Sequence Detection Software 1.4 | Applied Biosystems/Life-Technologies |
Referenzen
- Douek, D. C., et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 396 (6712), 690-695 (1998).
- Zelm, M. C., Szczepanski, T., van der Burg, M., van Dongen, J. J. M. Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J. Exp. Med. 204 (3), 645-655 (2007).
- Fronkova, E., et al. B-cell reconstitution after allogeneic SCT impairs minimal residual disease monitoring in children with ALL. Bone Marrow Transplant. 42 (3), 187-196 (2008).
- Sottini, A., et al. Simultaneous quantification of recent thymic T-cell and bone marrow B-cell emigrants in patients with primary immunodeficiency undergone to stem cell transplantation. Clin. Immunol. 136 (2), 217-227 (2010).
- Serana, F., et al. Thymic and bone marrow output in patients with common variable immunodeficiency. J. Clin. Immunol. 31 (4), 540-549 (2011).
- Zanotti, C., et al. Opposite effects of interferon-β on new B and T cell release from production sites in sclerosis. J. Neuroimmunol. 240-241, 147-150 (2011).
- Zanotti, C., et al. Peripheral accumulation of newly produced T and B lymphocytes in natalizumab-treated multiple sclerosis patients. Clin. Immunol. 145 (1), 19-26 (2012).
- Sottini, A., et al. Pre-existing T- and B-cell defects in one progressive multifocal leukoencephalopathy patient. PLoS One. 7, e34493(2012).
- Quiros-Roldan, E., et al. Effects of combined antiretroviral therapy on B- and T-cell release from production sites in long-term treated HIV-1+ patients. J. Transl. Med. 10, 94(2012).
- Mensen, A., et al. Utilization of TREC and KREC quantification for the monitoring of early T- and B-cell neogenesis in adult patients after allogeneic hematopoietic stem cell transplantation. J. Transl. Med. 11, 188(2013).
- Serana, F., et al. The different extent of B and T cell immune reconstitution after hematopoietic stem cell transplantation and enzyme replacement therapies in SCID patients with adenosine deaminase deficiency. J. Immunol. 185 (12), 7713-7722 (2010).
- Borte, S., et al. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood. 119 (11), 2552-2555 (2012).
- Baker, M. W., et al. Implementing routine testing for severe combined immunodeficiency within Wisconsin's newborn screening program. Public Health Rep. 125 (2), 88-95 (2010).
- Lorenzi, A. R., et al. Determination of thymic function directly from peripheral blood: a validated modification to an established method. J. Immunol. Meth. 339 (2), 185-194 (2008).
- De Boer, R. J., Perelson, A. S. Quantifying T lymphocyte turnover. J. Theor. Biol. 327, 45-87 (2013).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten