
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Spannklemme Fluorometrie in
In diesem Artikel
Zusammenfassung
Dieser Artikel beschreibt eine Verbesserung der konventionellen Voltage-Clamp Fluorometry (VCF), bei der fluoreszierende unnatürliche Aminosäuren (fUAA) anstelle von Maleinimidfarbstoffen verwendet werden, um strukturelle Umlagerungen in Ionenkanälen zu untersuchen. Das Verfahren umfasst Xenopus- Oozyten-DNA-Injektion, RNA / fUAA-Coinjektion und gleichzeitige Strom- und Fluoreszenzmessungen.
Zusammenfassung
Voltage-Clamp Fluorometry (VCF) war die Technik der Wahl, um die Struktur und die Funktion von elektrogenen Membranproteinen zu untersuchen, bei denen Echtzeitmessungen von Fluoreszenz und Strömen gleichzeitig über lokale Umlagerungen und globale Funktion berichten. Während hochauflösende Strukturtechniken wie Kryo-Elektronenmikroskopie oder Röntgenkristallographie statische Bilder der interessierenden Proteine liefern, liefert VCF dynamische Strukturdaten, die es uns ermöglichen, die strukturellen Umlagerungen (Fluoreszenz) mit dynamischen Funktionsdaten (Elektrophysiologie) zu verknüpfen. Bis vor kurzem beschränkte die Thiol-reaktive Chemie, die für die ortsgerichtete fluoreszierende Markierung der Proteine verwendet wurde, den Anwendungsbereich des Ansatzes, da alle zugänglichen Cysteine, einschließlich endogener, markiert werden. Es war also erforderlich, Proteine aus endogenen Cysteinen zu konstruieren. Die Etikettierung war auch auf Websites beschränkt, die von der extrazellulären zugänglich warenSeite. Dies änderte sich mit der Verwendung von fluoreszierenden unnatürlichen Aminosäuren (fUAA), um spezifisch eine kleine fluoreszierende Sonde als Reaktion auf die Stop-Codon-Suppression unter Verwendung eines orthogonalen tRNA- und tRNA-Synthetasepaar 2 zu integrieren. Die VCF-Verbesserung erfordert nur ein zweistufiges Injektionsverfahren der DNA-Injektion (tRNA / Synthetase-Paar), gefolgt von einer RNA / fUAA-Co-Injektion. Nun ist die Etikettierung sowohl intrazellulärer als auch begrabener Standorte möglich, und die Verwendung von VCF hat sich erheblich erweitert. Die VCF-Technik wird dadurch attraktiv für das Studium einer breiten Palette von Proteinen und - noch wichtiger - ermöglicht die Untersuchung zahlreicher zytosolischer Regulationsmechanismen.
Einleitung
Über 200 unnatürliche Aminosäuren verschiedener chemischer und physikalischer Eigenschaften wurden in Proteine in E. coli , Hefe und Säugetierzellen genetisch eingearbeitet 3 . Die unnatürliche Aminosäure wird als Reaktion auf ein spezifisches Stopcodon über ein orthogonal konstruiertes tRNA / Synthetasepaar eingebaut. Der genetische Ansatz zur Veränderung von Proteinen hat wertvolle Einblicke in die Proteinstruktur und -funktion gegeben. Hier stellen wir ein Protokoll zur Verwendung von Spannungs-Klammer-Fluorometrie (VCF) in Kombination mit einer fluoreszierenden UAA vor.
In VCF ermöglicht die gleichzeitige Beobachtung von Funktionsdaten und strukturellen Umordnungen, die um die Fluoreszenzsonde (~ 5 Å) lokalisiert sind, eine dynamische Information mit Millisekundenauflösung 1 zu erhalten. Die fluoreszierenden Sonden verändern ihren Abschreckzustand bei lokalisierter Bewegung des Proteins. Eine Bewegung von nur 1-2 Å reicht aus, um zu signifikanten Veränderungen in der Fluoreszenz zu führenIntensität 4 Nach der Identifizierung der interessanten Stelle im Zielprotein wird die Stelle durch Punktmutation mutiert. Klassisch war der Rest zu einem Cystein mutiert worden, während jetzt ein Bernstein-Stop-Codon (TAG) für den genetischen fUAA-Einbau eingeführt wird. Das Protein wird dann in vitro transkribiert.
Während andere Expressionssysteme ( z. B. Säugetierzellen) 5 , 6 , 7 verwendet werden können, sind Xenopus- Oozyten aufgrund ihrer größeren Größe für Strukturfunktionsstudien vorzuziehen, was zu einer leichteren Manipulation und einer höheren Fluoreszenzintensität (mehr Fluorophoren) führt, Zu-Rausch-Verhältnis Darüber hinaus haben Xenopus- Oozyten einen geringen Hintergrund aus endogenen Proteinen 2 , 8 , und die dunkle Pigmentierung auf dem Tierpol schützt vor der Hintergrundfluoreszenz von tEr zytosol Die Xenopus- Oozyten werden chirurgisch entfernt und DNA, die für das für die fUAA spezifische orthogonale tRNA / tRNA-Synthetase-Paar kodiert, wird in den Kern der Oozyten injiziert. Nach einer 6-24 h Inkubationszeit wird die Protein-RNA mit dem fUAA in das Cytosol der Oozyten co-injiziert, gefolgt von einer 2-3-tägigen Inkubationszeit. Um eine Beschädigung der fUAA (Photobleichung) zu verhindern, müssen die Verfahren einschließlich Anap unter rotem Licht durchgeführt werden, um eine Fluorophor-Erregung zu vermeiden.
Oozyten werden auf einem aufgeschnittenen Oozytenspannungsklemmenaufbau untersucht, der auf einem aufrechten Fluoreszenzmikroskop montiert ist und elektrische Strom- und Fluoreszenzänderungen gleichzeitig aufgezeichnet werden 9 , 10 . Alternativ können zwei Elektrodenspannungsklemmen 1 oder Patch-Clamp-Konfigurationen 11 verwendet werden. Die Fluoreszenz wird durch entsprechende Wellenlängen mit niedrigem RMS-Rauschen angeregt Emission, die unter Verwendung einer Photodiode aufgezeichnet wurde, die mit einem Verstärker mit hoher Verstärkung verbunden ist.
Es gibt mehrere Vorteile der Verwendung von fluoreszierenden unnatürlichen Aminosäuren (fUAAs) in der Spannungskammer-Fluorometrie. Einer ist Zugang zur cytosolischen Seite der Membranproteine; Hier befinden sich viele regulatorische Prozesse ( zB Ca 2+ - oder Nukleotidbindungsstellen, schnelle und geschlossene Inaktivierung von spannungsgesteuerten Ionenkanälen, Porenöffnung, Modulkopplung). Alle diese Prozesse sind nun für die fluoreszierende Markierung zugänglich.
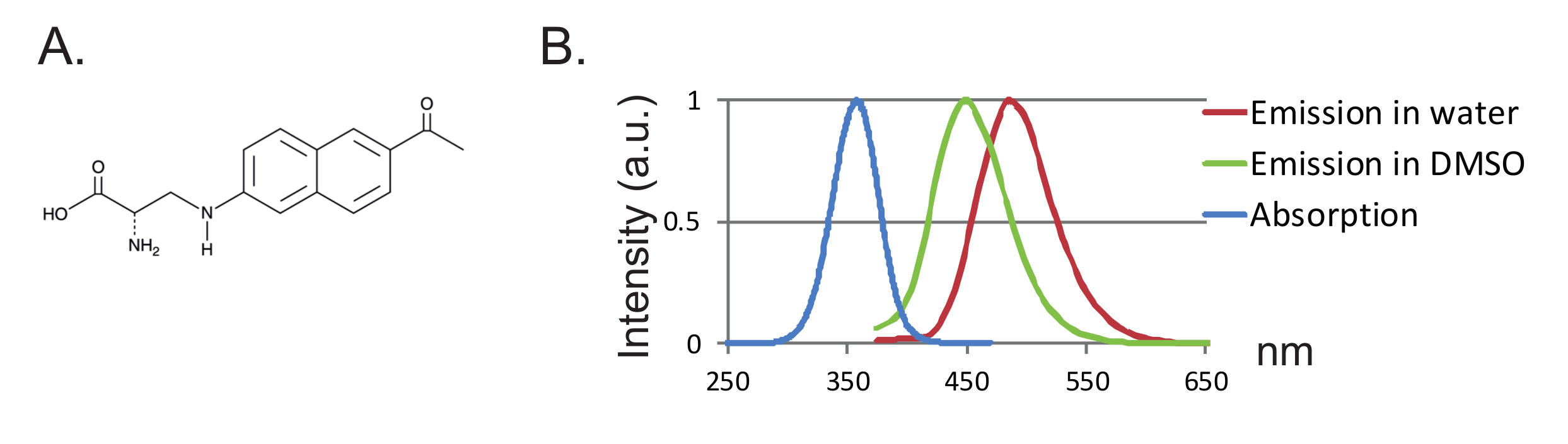
Ein weiterer Vorteil ist die geringe Größe der Sonde, die zu einer geringeren Störung des Proteins führt. Bisher wurden zwei orthogonale tRNA / tRNA-Synthetasepaare für fUAAs 12 , 13 entwickelt, wobei 3- (6-Acetylnaphthalin-2-ylamino) -2-aminopropansäure (Anap) die einzige FUAA ist, die in Xenopus- Oozyten verwendet wurde 2 ,"Xref"> 8 Anap ist ein umweltfreundlicher Fluorophor mit einem Molekulargewicht von 272,3 g / mol und ist nur etwas größer als Tryptophan 12 ( Fig. 1A, 1B ). Aufgrund ihrer geringen Größe werden wahrscheinlich weniger sterische Effekte durch das Fluorophor eingeführt, verglichen mit herkömmlichen Fluorophoren, die über einen Linker (typischerweise mehr als 500 g / mol) befestigt sind. Darüber hinaus befindet sich im Falle von Anap das Fluorophor näher an dem Proteinrückgrat als die an Cysteine gebundenen, und folglich untersucht Anap mehr lokalisierte Umlagerungen. Schließlich ist die Entfernung von endogenen Cysteinen in konventionellem VCF, um eine ortsspezifische Markierung zu gewährleisten, in UAA-VCF nicht mehr erforderlich und verlässt daher (i) die Proteine in (fast) ihrem nativen Zustand und (ii) erlaubt es, VCF anzuwenden Um eine breitere Palette von Proteinen zu untersuchen, in denen die Funktion durch Cysteinsubstitution verändert werden kann.

Abbildung 1 : Anap- und Fluoreszenz-Spektren. ( A ) Chemische Struktur von Anap. ( B ) Normalisiertes Absorptionsspektrum und Emissionsspektren für 1 nM Anap, was die Empfindlichkeit der Anap-Fluoreszenz gegenüber der Lösungsmittel-Hydrophobie zeigt. Emissionsspektren wurden durch Anregung bei 350 nm erhalten. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Ein Nachteil bei der Verwendung von fluoreszierenden UAAs besteht darin, dass eine heterogene Population von Proteinen aus der Stop-Codon-Durchlesung, der Translationsneubinierung, den C-terminalen trunkierten Proteinen oder dem Übersprechen mit endogener Aminoacylierung resultieren kann, wenn die Menge an aminoacylierten tRNAs knapp ist. Solche Leck-Expression sollte immer in Abwesenheit von fUAA und dem tRNA / tRNA-Synthetase-Paar überprüft werden. Wir haben uns mit der Frage befasstLationaler Reinitiation und deren Umgehung für N-terminale Einführungsstellen zuvor 14 . Wenn jedoch die fUAA-, tRNA- und tRNA-Synthetase in gesättigten Mengen vorliegen, bleibt nur eine geringe Wahrscheinlichkeit der Leck-Expression bestehen.
Der wesentliche prozedurale Unterschied zwischen fUAA-VCF und konventionellem VCF ist die Injektion und Handhabung der Oozyten; Die Injektion von DNA, die für die tRNA und die tRNA-Synthetase (pAnap) kodiert, folgt der Einführung von Anap, die entweder mit der Protein-mRNA co-injiziert wird oder alternativ der Inkubationslösung als Acetoxymethyl (AM) -ester zugesetzt wird.
Protokoll
Froschmanipulationen wurden in Übereinstimmung mit den kanadischen Richtlinien durchgeführt und wurden vom Ethikkomitee (CDEA, Protokoll Nr. 15-042) der Universität von Montréal genehmigt.
1. mRNA Vorbereitung für fUAA Incorporation
- Wählen Sie eine interessante Stelle im Protein, bei der Konformationsänderungen auftreten werden. Wählen Sie eine Aminosäure in dieser Region, um die fUAA zu substituieren.
HINWEIS: Die Wahl der Position basiert auf den strukturellen Umbuchungen, die erwartet werden. Wenn eine hochauflösende Struktur existiert und eine Hypothese der erwarteten Bewegungen, sollte die Anap so platziert werden, dass sich die chemische Umgebung ändern wird; Dies könnte entweder eine Änderung der Dielektrizitätskonstante (hydrophobe versus hydrophile Umgebung) oder eher eine Abschreckung durch eine andere Aminosäure sein. Die besten Quencher sind Tryptophane. Anap sollte in Kontakt mit dem Quencher in einem Zustand (Überlappung der van-der-Waals Radien) und frei davon indas andere. Wenn keine hochauflösenden Strukturen oder Modelle existieren, müsste man die interessierende Region scannen. In jedem Fall ist es ratsam, mehrere nahe gelegene Orte auszuwählen, um die Wahrscheinlichkeit des Erhaltens von Expressions- und Fluoreszenzsignal zu erhöhen. Um sterische Effekte während der Proteinreifung und / oder -funktion zu minimieren, kann man große und aromatische Aminosäuren (Phe, Trp, Tyr) ersetzen. Die Autoren haben jedoch erfahren, dass das Scannen eines interessierenden Bereichs für die fUAA-Insertion unabhängig von der substituierten Aminosäure produktiver ist. - Legen Sie ein Amber Stop Codon (TAG) an der ausgewählten Stelle mit site-directed Mutagenese 15 . Stellen Sie sicher, dass das Protein von Interesse nicht auf einem Bernstein-Stop-Codon (TAG) endet. Wenn ja, mutieren Sie zu einem anderen (Ocker oder Opal Stop Codon). Amplifizieren, isolieren und sequenzieren die DNA. Erhalten Sie Protein-mRNA mit in vitro Transkription 16 und speichern Sie die mRNA bei 20 ° C oder 80 ° C.
- Chirurgisch erhalten Stage V oder VI Oozyten aus Xenopus laevis Frösche und defolliculieren mit Kollagenase wie zuvor beschrieben 17 .
- Anästhesieren von Fröschen mit einem geeigneten Anästhetikum nach dem zugelassenen Tierprotokoll (hier: 3-Aminobenzoesäureethylester). Wenn sie nicht auf eine sanfte Prise auf eine Zehenspitze reagieren (Verlust des Rückzugsreflexes), dann werden sie für eine Operation in geeigneter Weise betäubt.
- Sofort die Frösche aus der Narkosemittel entfernen und die Haut mit frischem Wasser gründlich ausspülen. Diese Spülung wird verhindern, dass das Tier in tieferen Niveaus der Anästhesie fällt, indem es unabsorbierte Chemikalien von der Hautoberfläche entfernt.
- Eierstockknoten von einer Seite chirurgisch entfernen und die Knoten sorgfältig mit zwei Zangen öffnen. Inkubieren und rühren die Oozyten in "Standard-Oozyten-Lösung" (SOS), die 1% (w / v) Collagenase für 20-30 min enthältZu defollulieren Waschen Sie dreimal mit SOS-Lösung.
- Wählen Sie große und gesunde Oozyten einzeln und inkubieren sie in Barths Lösung, ergänzt mit Antibiotika (100 U / ml Penicilin, 100 μg / ml Streptomycin, 10 mg / 100 ml Kanamycin) und 5% Pferdeserum bei 18 ° C für mindestens 4 h vor der Injektion .
ANMERKUNG: Nach 2 - 4 Operationen mit einer Verzögerung von 4 Monaten dazwischen wird Xenopus laevis durch verlängerte (> 1 h) Inkubation mit 3-Aminobenzoesäureethylester euthanasiert.
- Für die nukleare Injektion von DNA, bereiten Sie eine lange und dünne Injektion Spitze, um in der Lage, den Kern zu erreichen und um eine Beschädigung der Oozyte zu vermeiden. Füllen Sie die Injektionsspitze mit Öl und montieren Sie sie auf dem Nanoinjektor Gerät.
- Installieren Sie den Nano-Injektor unter einem Stereomikroskop und verwenden Sie Pinzette, um das Ende der Spitze zu brechen. Öl auswerfen, bis keine Luftblasen im Ende der Spitze eingefangen sind.
- 1 μl 0,1 μg /# 181; L pAnap in nukleasefreiem Wasser, enthaltend NaOH (1% 1 N NaOH) auf einem Stück Parafilm unter einem Stereoskop und füllen die Injektionsspitze mit der DNA.
- Übertragen Sie 40 Oozyten auf eine mesh-beschichtete Injektionsschale mit Barth-Lösung, ergänzt mit Antibiotika.
HINWEIS: Um die mesh-beschichtete Injektionsschale herzustellen, schneiden Sie ein entsprechendes Stück von 800 μm Nylon-Mesh aus, um eine Polystyrol-Petrischale zu füllen. Fügen Sie Chloroform in die Mitte und legen Sie dann das Netz auf die Oberseite. Halten Sie das Netz flach, bis der Plastik sitzt. - Wenn sich der Oozytenkern im tierischen (dunklen) Pol befindet, zielen Sie die Injektionsspitze in die Mitte des Tierpols und schlagen so, dass die Spitze in der Nähe des Zentrums der Tierhalbkugel (oder 2-3x der Tiefe im Vergleich zur RNA-Injektion) liegt ). Spritzen Sie 9,2 nL pAnap in den Kern jeder Oozyte ein. Die dünne Spitze und das kleine Einspritzvolumen können zu einer unregelmäßigen Injektion oder einer blockierten Spitze führen. Gelegentlich prüfen, ob die Injektion durch Einspritzen in die Luft arbeitet.
HINWEIS: Ob die DNA richtig in den Kern injiziert wird, ist unsicher. Erwarten Sie daher 10 - 40% der Oozyten, um das tRNA / Synthetase-Paar nicht zu exprimieren. Siehe Diskussion zur weiteren Ausarbeitung. - Inkubieren Sie die Oozyten in 2 ml Barth-Lösung, ergänzt mit Antibiotika und 5% Pferdeserum (HS) bei 18 ° C für 6-24 h, um eine robuste Expression von anap-spezifischen tRNAs und tRNA-Synthetasen zu ermöglichen.
HINWEIS: Die DNA-Inkubationszeit kann einige Tage vor der RNA-Injektion dauern, aber sie erhöht die Expression nicht. - Bereiten Sie den Nano-Injektor für die RNA-Injektion vor (wie in Schritt 2.2, aber die Injektionsspitze muss nicht so dünn sein wie bei der DNA-Injektion). Arbeit nur unter rotem Licht von diesem Punkt zu verhindern, dass Photobleaching der Anap.
- Mischen Sie 1 μl 1 mM Anap mit 1 μl 1-2 μg / μL mRNA direkt auf ein Stück Parafilm und füllen Sie die Injektionsspitze mit der gemischten Lösung. Unkraut knapp unterhalb der Membran im pflanzlichen(Hellen) Pol und injizieren 46 nL in jeder pAnap-injizierten Oozyte.
HINWEIS: Die erforderliche mRNA-Konzentration hängt von dem Protein von Interesse ab. - Inkubieren Sie die Oozyten, die vor Licht geschützt sind, in einer Schachtel oder in Aluminiumfolie gewickelt, in Barths Lösung ergänzte Antibiotika und 5% Pferdeserum bei 18 ° C für 2-3 Tage. Tauschen Sie täglich mit frischer Barth-Lösung aus und entfernen Sie tote Oozyten, um eine Kontamination zu vermeiden.
- Installieren Sie die aufgeschnittene Oozytenspannungsklemme wie oben beschrieben 18 .
- Montieren Sie das elektrophysiologische Aufnahmesystem auf einem aufrechten Fluoreszenzmikroskop, indem Sie die Aufzeichnungskammer auf einen Schieber einbauen, der es ermöglicht, es zwischen dem Standard-Stereoskop zu bewegen, um die Oozyte und das Mikroskop zu platzieren, um die Fluoreszenzmessungen durchzuführen (Abbildung 2c ).
HINWEIS: Die Geometrie der Kammer für aufgeschnittene Oozyten ist nicht geeignet, den normalen Tra zu verwendenLicht für die Beleuchtung während der Manipulation. Daher wird eine "Schwanenhals" Halogenlampe mit rotem Filter verwendet, um seitlich von oben zu beleuchten. Der Kondensator des Mikroskops kann entfernt werden, um Platz für die Elektrophysiologiekammer zu senken. - Verbinden Sie ein Photodioden-Erkennungssystem mit dem C-Mount-Ausgang des Fluoreszenzmikroskops (Abbildung 2a ). Verbinden Sie die Photostromauslesung mit einem zweiten Eingangskanal im digitalen Signalprozessor (DSP, Analog / Digital - Digital / Analog - Wandler).
- Verwenden Sie eine 100 W, 12 V Halogenlampe als Lichtquelle für die Fluoreszenzanregung.
HINWEIS: Alternativ können Hg-Brenner verwendet werden, müssen aber in der Intensität reduziert werden, um ein zu schnelles Photobleichen während der Aufnahmen zu verhindern. LED-Beleuchtung wird nur empfohlen, wenn die jeweiligen LEDs im Erregungsbereich ( zB ~ 350 nm für Anap) eine signifikante Intensität aufweisen. Die meisten weißen LEDs erreichen nicht weit in das UV-Spektrum. - InsErt einen elektrisch gesteuerten Verschluss zwischen Anregungslichtquelle und Mikroskop und verbinden seine Steuerung (typischerweise TTL-Puls) mit einem digitalen Ausgang des DSP. Zeit der TTL-Puls in der Aufnahmesoftware (siehe Herstellerdokumentation), so dass der Verschluss vor Beginn der Aufnahme ~ 100 ms öffnet. Auf diese Weise stört jede Vibration während des Öffnungsvorgangs die Aufnahme nicht. Die Zeit hängt von der Geschwindigkeit und der Vibration des Verschlusses ab. Beenden Sie den Puls 5ms vor dem Ende der Aufnahme, wie in Abbildung 4 gezeigt . Auf diese Weise wird auch der Wert für die Gesamtfluoreszenz aufgezeichnet.
- Setzen Sie einen geeigneten Filterwürfel (Erregungsfilter, dichroitischer Spiegel und Emissionsfilter) in den Filterwürfelrevolver ein. Für Anap verwenden Sie Ex: 377/50 nm Bandpass, dichroitische 409 nm Langpass und Em: 470/40 nm Bandpass.
- Befolgen Sie die Vorbereitungsschritte für die aufgeschnittene Oozytenspannungsklemme, wie zuvor beschrieben und visualisiert 18 (Agarbrückenpräparation, Montage der Oozyte, Saponinpermeabilisierung). Allerdings arbeiten unter rotem Licht zu jeder Zeit zu vermeiden, Bleichen der Fluorophor vor Aufnahmen. Beim Aufstellen der Oozyte ist darauf zu achten, dass der Tierpol nach oben zeigt. Die Pigmentierung unter der Tierpolmembran schützt vor der aus dem Cytosol stammenden Selbstfluoreszenz und reduziert daher die Hintergrundfluoreszenz.
- Schieben Sie die Kammer auf das Mikroskop und fokussieren Sie mit einem 4X-Objektiv.
- Die Oozyte mit der spannungsempfindlichen V1-Elektrode (3 M KCl) auflösen, auf das 40X-Wasser-Immersion (NA 0.8 - 0.9) -Objekt umschalten. Konzentriere dich auf den nach oben weisenden Tierpfosten.
- Schalte das rote Licht aus. Wählen Sie den richtigen Filterwürfel aus, indem Sie den Filterwürfelrevolver drehen und der optische Ausgangsport mit der Photodiode verbunden ist. Schalte den Heiligenschein einEn-Lampe mit höchster Intensität und schalten Sie kurz den Auslöser für 2-5 s offen, um die Hintergrund-Fluoreszenzintensität zu lesen, die von der Eizelle stammt. Bei der beschriebenen Einstellung sollte der Wert etwa 50-200 pA für Anap sein.
- Schalten Sie die Klemme ein, drehen Sie den Bad- / Schutzschalter auf aktiv und stellen Sie das Membranpotential (V1 - V2) auf das Befehlspotential ein, indem Sie den Drehknopf auf der Kopfstufe drehen.
- Wählen Sie Haltepotential, Schrittprotokoll, Anzahl und Länge der Impulse etc. in der Aufnahmesoftware. Aufzeichnung von spannungsabhängigen Strömen und Anap-Fluoreszenz-Intensitäten.
- Um zwei Standorte im selben Protein gleichzeitig zu überwachen, mutieren Sie eine extrazelluläre und zugängliche Aminosäure in Cystein und entfernen Sie andere Cysteine, um eine spezifische Markierung mit Thiol-Chemie zu gewährleisten.
- Führen Sie Schritt 2.1-2.5 aus.
- Vor VCF-Aufnahmen inkubieren Oozyten in 5 μM TMR-Maleimid in Markierungslösung für 15 min (oder andere Farbstoffe)Mit nicht überlappenden Spektren im Vergleich zu Anap).
- Die Oozyten mit Etikettierlösung dreimal waschen, um überschüssigen Farbstoff zu entfernen.
- Führen Sie Schritt 4.1-4.6 aus.
- Setzen Sie einen geeigneten Filterwürfel für TMR (Erregungsfilter, dichroitischer Spiegel und Emissionsfilter) in den Filterwürfelrevolver ein. Wechseln Sie in den TMR-Filterwürfel, indem Sie den Filterrevolver drehen.
- Lesen Sie die Hintergrundfluoreszenz für TMR wie für Anap in Schritt 4.4 beschrieben.
HINWEIS: Die Etikettierung mit Thiol-Chemie führt zu einer hohen Hintergrundfluoreszenz durch unspezifische Markierung in der Membran. Daher kann die TMR-Hintergrundfluoreszenz den Verstärker (> 2.000 pA) sättigen. In diesem Fall nicht die Lichtintensität verringern, sondern einfach die Hintergrundfluoreszenz subtrahieren, indem der Photodiode ein Offsetstrom hinzugefügt wird. In handelsüblichen Systemen wird das "Sample-and-Hold" -Funktion auf dem Detektorsystem verwendet. Beachten Sie den Hintergrund-Fluoreszenzwert (mit einem 10X Neutral-Dichte-Filter) in einem Labor jAllnal, da dieser Wert nicht aufgezeichnet wird (Sättigung). - Aufzeichnung von spannungsabhängigen Strömen und TMR-Fluoreszenzintensitäten gleichzeitig wie in Schritt 4.6.
2. Oozytenvorbereitung und Injektion

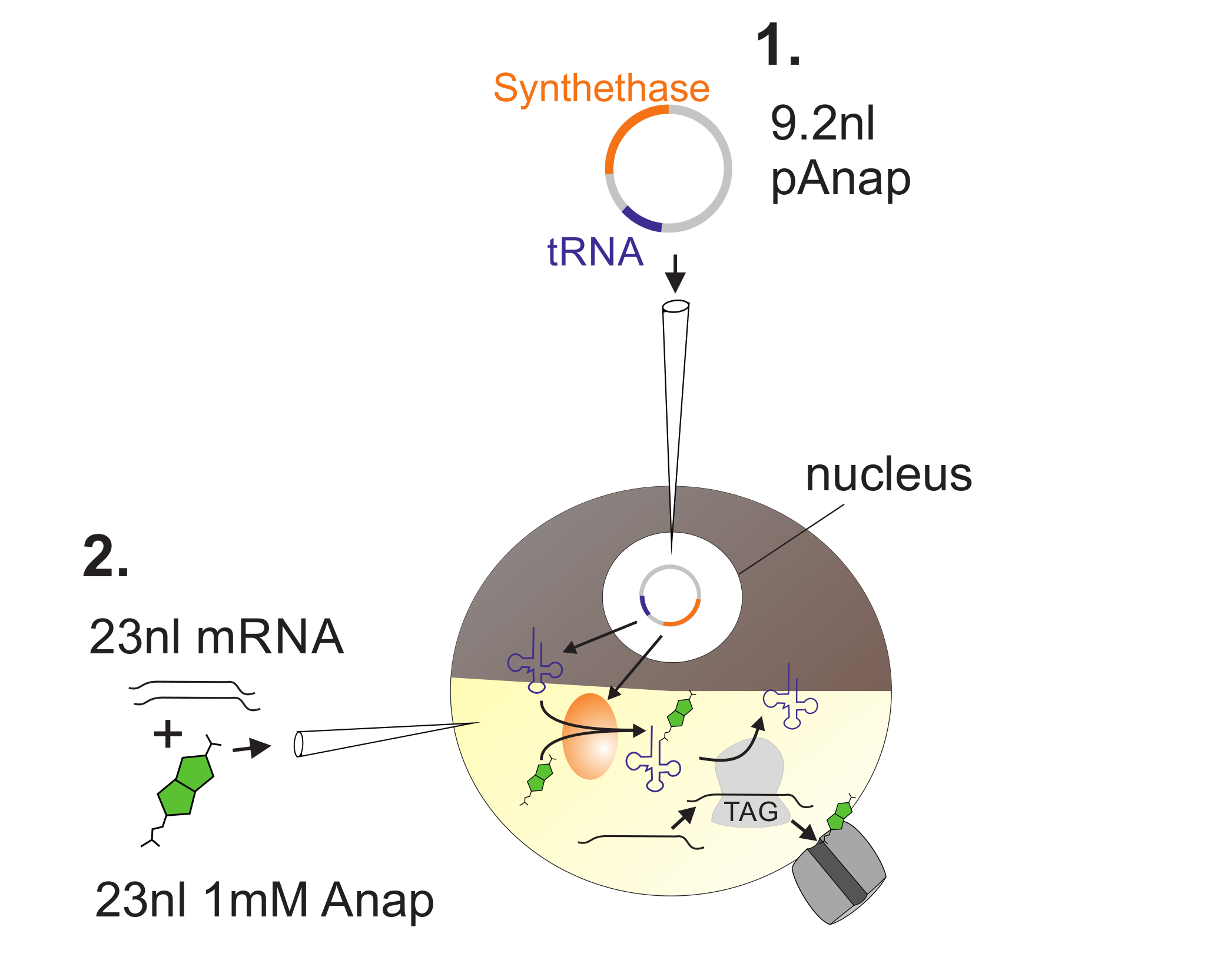
Abbildung 2 : Darstellung der DNA- und RNA-Injektion in Xenopus -Oozyten zur Anap-Inkorporation.
Zuerst wird pAnap in den Kern der Xenopus- Oozyte injiziert ( 1 ). Nach 6-24 h werden Anap und Kanal-RNA in den Pflanzenpol ( 2 ) coinjiziert. Anap wird mit der tRNA, die ein Amber-Stop-Anti-Codon trägt, orthogonal aminoacyliert, durch die Aminosäure-tRNA-Synthetase, die durch pAnap codiert wird. Auf diese Weise werden die aminoacylierten Anap-tRNAs vom Ribosom am eingesetzten Bernstein-Stop-Codon in der Channe erkanntL RNA, was zur Unterdrückung des Stopcodons und der Insertion von Anap führt. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
3. VCF-Setup

Abbildung 3 < / Strong> : VCF-Setup. ( A ) Seitenansicht des VCF-Setups zeigt den Lichtweg im Mikroskop. Der Filterwürfel enthält einen Erregungsfilter, einen dichroitischen Spiegel und einen Emissionsfilter. ( B ) Ausgewählte Oozytenkammerabmessungen sind 3,4 cm für den oberen Kammerradius (1), 5,5 cm für die untere Kammerlänge (2), 1,4 cm für die untere Kammerbreite (3) und 1,7 cm für die mittlere Kammerbreite (4). ( C ) Vorderansicht des VCF-Setups. Das erste Okular auf der linken Seite ist für die Montage der Oozyte in der abgeschnittenen Spannungskammer und für die Permeabilisierung. Dann wird die Kammer unter dem Mikroskop im zweiten Okular nach rechts geschoben. Hier wird die V1-Elektrode unter Verwendung des 4X-Objektivs in die Oozyte eingeführt und die Fluoreszenz wird mit dem Wasser-Immersions-40X-Objektiv aufgezeichnet. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
5. Zweifarbige VCF
Ergebnisse
Fig. 4 zeigt ein Beispiel von VCF-Aufzeichnungen, die aus einer Oozyten-Expression von Shaker-Kanälen mit schneller Inaktivierung entfernt wurden (IR), L382stop-W434F in Gegenwart von pAnap und Anap. Die W434F-Mutation blockiert die ionischen Kaliumströme, was es ermöglicht, die transienten Gatterladungsverschiebungen (Torströme) zu messen. Die gleichzeitige Aufzeichnung von Torströmungen (obere Spur) und Anap-Fluoreszenzintensitätsänderungen (untere Spur) bei Dep...
Diskussion
Die in vivo- Aminoacylierung von tRNAs, die kontinuierlich zusammen mit der tRNA-Synthetase transkribiert werden, ermöglicht es, hohe Expressionsniveaus für Fluoreszenzmessungen zu erhalten. Für eine effiziente fUAA-Inkorporation ist es entscheidend, dass pAnap korrekt in den Zellkern injiziert wird. Aufgrund der Ungewissheit der genauen Lage des Kerns wird erwartet, dass 10-40% der DNA-Injektionen versagen, was zu nicht-exprimierenden (oder leckexprimierenden) Oozyten führt. Daher ist es wichtig, den Ausdr...
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
PAnap war ein freundliches Geschenk von Dr. Peter Schultz (Scripps Research Institute). Diese Arbeit wurde von den kanadischen Instituten für Gesundheitsforschungsstipendien MOP-102689 und MOP-136894 (an RB) und der kanadischen Stiftung für Innovation Grant 950-225005 finanziert.
Materialien
| Name | Company | Catalog Number | Comments |
| Solutions | |||
| Barth's solution | |||
| NaCl | Sigma-Aldrich | S7653 | 90 mM |
| KCl | Fisher Scientific | BP366-500 | 3 mM |
| MgSO4 | Sigma-Aldrich | M-9397 | 0.82 mM |
| CaCl2 | Sigma-Aldrich | C-7902 | 0.41 mM |
| Ca(NO3)2 | Sigma-Aldrich | C-1396 | 0.33 mM |
| HEPES | Sigma-Aldrich | H4034 | 5 mM |
| NaOH hydrate | BDH | BDH7225-4 | pH 7.6 |
| Penicilin | Invitrogen | 15140122 | 100 U/mL |
| Streptomycin | Invitrogen | 15140122 | 100 µg/mL |
| Kanamycin | Invitrogen | 15160054 | 10 mg/100mL |
| Horse Serum (HS) | Invitrogen | 16050122 | 5% |
| SOS Standard Oocyte Solution | |||
| NaCl | Sigma-Aldrich | 746398 | 102 mM |
| KCl | Sigma-Aldrich | 746436 | 3 mM |
| MgCl2 | Sigma-Aldrich | M9272 | 1 mM |
| HEPES | Sigma-Aldrich | H4034 | 5 mM |
| External recording solution | |||
| N-methyl-D-glucamine (NMDG) | Alfa Aesar | L14282 | 115 mM |
| HEPES | Sigma-Aldrich | H4034 | 10 mM |
| Calcium hydroxide | Sigma-Aldrich | 239232 | 2 mM |
| MES hydrate | Sigma-Aldrich | 258105 | pH 7.2 |
| Internal recording solution | |||
| N-methyl-D-glucamine (NMDG) | Alfa Aesar | L14282 | 115 mM |
| HEPES | Sigma-Aldrich | H4034 | 10 mM |
| Ethylenediamine Tetraacetic Acid (EDTA) | Fisher Scientific | E478-500 | 2 mM |
| MES hydrate | Sigma-Aldrich | 258105 | pH 7.2 |
| Labeling solution | |||
| KOH | Fisher Scientific | P250-1 | 115 mM |
| HEPES | Sigma-Aldrich | H4034 | 10 mM |
| Calcium hydroxide | Sigma-Aldrich | 239232 | 2 mM |
| MES hydrate | Sigma-Aldrich | 258105 | pH 7.2 |
| TMR stock solution | |||
| Tetramethylrhodamine-5-maleimide (TMR) | Molcular Probes by Life Technologies | T6027 | 5 mM in DMSO |
| Anap stock solution | |||
| Anap | ABZENA (TCRS) | Custom synthesis TCRS-170 | 1 mM in nuclease-free water and 1% NaOH 1 N |
| Name | Company | Catalog Number | Comments |
| Material/Equipment | |||
| pAnap | Addgene | 48696 | |
| High Performance Oocyte Clamp | Dagan Corporation | CA-1B | |
| Gpatch Acquisition software | Department of Anesthesiology, University of California, Los Angeles | ||
| Analysis software | Department of Anesthesiology, University of California, Los Angeles | ||
| Recording Chamber | Custom machined | ||
| Photo diode detection system | Dagan Corporation | PhotoMax-200/PIN | |
| Electrical shutter driver | UNIBLITZ | VCM-D1 |
Referenzen
- Mannuzzu, L. M., Moronne, M. M., Isacoff, E. Y. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271 (5246), 213-216 (1996).
- Kalstrup, T., Blunck, R. Dynamics of internal pore opening in K(V) channels probed by a fluorescent unnatural amino acid. Proc Natl Acad Sci U S A. 110 (20), 8272-8277 (2013).
- Xiao, H., Schultz, P. G. At the Interface of Chemical and Biological Synthesis: An Expanded Genetic Code. Cold Spring Harb Perspect Biol. 8 (9), (2016).
- Blunck, R. Chapter 9. Handbook of Ion Channels. , 113-133 (2015).
- Chatterjee, A., Guo, J., Lee, H. S., Schultz, P. G. A genetically encoded fluorescent probe in mammalian cells. J Am Chem Soc. 135 (34), 12540-12543 (2013).
- DeBerg, H. A., Brzovic, P. S., Flynn, G. E., Zagotta, W. N., Stoll, S. Structure and Energetics of Allosteric Regulation of HCN2 Ion Channels by Cyclic Nucleotides. J Biol Chem. 291 (1), 371-381 (2016).
- Shen, B., et al. Genetically encoding unnatural amino acids in neural stem cells and optically reporting voltage-sensitive domain changes in differentiated neurons. Stem Cells. 29 (8), 1231-1240 (2011).
- Aman, T. K., Gordon, S. E., Zagotta, W. N. Regulation of CNGA1 Channel Gating by Interactions with the Membrane. J Biol Chem. 291 (19), 9939-9947 (2016).
- Haddad, G. A., Blunck, R. Mode shift of the voltage sensors in Shaker K+ channels is caused by energetic coupling to the pore domain. J Gen Physiol. 137 (5), 455-472 (2011).
- Batulan, Z., Haddad, G. A., Blunck, R. An intersubunit interaction between S4-S5 linker and S6 is responsible for the slow off-gating component in Shaker K+ channels. J Biol Chem. 285 (18), 14005-14019 (2010).
- Kusch, J., et al. How subunits cooperate in cAMP-induced activation of homotetrameric HCN2 channels. Nat Chem Biol. 8 (2), 162-169 (2012).
- Lee, H. S., Guo, J., Lemke, E. A., Dimla, R. D., Schultz, P. G. Genetic incorporation of a small, environmentally sensitive, fluorescent probe into proteins in Saccharomyces cerevisiae. J Am Chem Soc. 131 (36), 12921-12923 (2009).
- Summerer, D., et al. A genetically encoded fluorescent amino acid. Proc Natl Acad Sci U S A. 103 (26), 9785-9789 (2006).
- Kalstrup, T., Blunck, R. Reinitiation at non-canonical start codons leads to leak expression when incorporating unnatural amino acids. Sci Rep. 5, 11866 (2015).
- Liu, H., Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 (2008).
- Beckert, B., Masquida, B. Synthesis of RNA by in vitro transcription. Methods Mol Biol. 703, 29-41 (2011).
- Goldin, A. L. Maintenance of Xenopus laevis and oocyte injection. Methods Enzymol. 207, 266-279 (1992).
- Rudokas, M. W., Varga, Z., Schubert, A. R., Asaro, A. B., Silva, J. R. The Xenopus oocyte cut-open vaseline gap voltage-clamp technique with fluorometry. J Vis Exp. (85), (2014).
- Zhao, J., Blunck, R. The isolated voltage sensing domain of the Shaker potassium channel forms a voltage-gated cation channel. Elife. 5, (2016).
- Posson, D. J., Ge, P., Miller, C., Bezanilla, F., Selvin, P. R. Small vertical movement of a K+ channel voltage sensor measured with luminescence energy transfer. Nature. 436 (7052), 848-851 (2005).
- Chanda, B., Asamoah, O. K., Blunck, R., Roux, B., Bezanilla, F. Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature. 436 (7052), 852-856 (2005).
- Taraska, J. W., Puljung, M. C., Zagotta, W. N. Short-distance probes for protein backbone structure based on energy transfer between bimane and transition metal ions. Proc Natl Acad Sci U S A. 106 (38), 16227-16232 (2009).
- Baker, B. J., et al. Genetically encoded fluorescent sensors of membrane potential. Brain Cell Biol. 36 (1-4), 53-67 (2008).
- Miranda, P., et al. State-dependent FRET reports calcium- and voltage-dependent gating-ring motions in BK channels. Proc Natl Acad Sci U S A. , (2013).
- Sisido, M., Ninomiya, K., Ohtsuki, T., Hohsaka, T. Four-base codon/anticodon strategy and non-enzymatic aminoacylation for protein engineering with non-natural amino acids. Methods. 36 (3), 270-278 (2005).
- Hohsaka, T., Ashizuka, Y., Murakami, H., Sisido, M. Five-base codons for incorporation of nonnatural amino acids into proteins. Nucleic Acids Res. 29 (17), 3646-3651 (2001).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten